
Abstract
Recombination has been recognized as a major mechanism of evolution in enteroviruses. The Echovirus 30 (E-30) strain Gior was sequenced and phylogenetically compared to all available E-30 sequences to detect recombination events between the 5΄UTR and VP1 genomic regions. The comparison of phylogenetic trees of the 5΄UTR and VP1 revealed incongruences concerning strains, lineages and sub-lineages. Comparative analysis of 62 E-30 sub-genomic sequences revealed six different recombination events that almost all occurred in the same region, having a start point in the 3΄end of the 5΄ UTR and end point in VP4. The only exception was the sub-lineage of Gior for which both borders of recombination were located in the 5΄UTR. These results describe for the first time recombination events in this region in circulating EV-B strains, revealing the exact points of these recombination events, highlighting the impact of such events on the evolution and epidemiology of enteroviruses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Echovirus 30 (E-30) is one of the most commonly isolated enterovirus serotypes from acute infections and has been recognized as the main cause of viral aseptic meningitis, associated with several meningitis outbreaks worldwide [2, 11, 15, 19, 21, 38, 39, 41]. Enteroviruses (EV) that infect humans can be taxonomically classified within the Picornavirus family and according to the degree of their genetic relatedness, are classified into four species, EV-A – D. Echovirus 30 is a member of enteroviruses species B (EV-B), along with 57 other enterovirus serotypes: coxsackie viruses B1-B6 (CV-B1 to CV-B6), CV-A9, echovirus 1 (E-1), E-2 to E-7, E-9, E-11 to E-21, E-24 to E-27, E-29 to E-33, enterovirus 69 (EV-69), EV-73 to EV-75, EV-77 to EV-88, EV-93, EV-97, EV-98, EV-100, EV-101, EV-106 and EV-107.
Enteroviruses consist of a non-enveloped virus capsid, formed by the four capsid proteins VP1–VP4, which enclose a single-stranded, positive-sense RNA genome of about 7.5 kb. Apart from the capsid proteins, the genome also encodes the seven non-structural 2A to 2C and 3A to 3D viral proteins. The open reading frame (ORF) is flanked by two untranslated regions, the 5’ UTR and the 3’ UTR that play a crucial role in translation and replication of the viral genome. The 5΄ UTR is about 750 nt in length and contains highly structured secondary elements (designated as domains I-VII, dI-dVII). The cloverleaf (CL) structure is formed by domain dI and is essential for virus replication, whereas the Internal Ribosome Entry Site (IRES) is formed by domains dII-VI and directs translation of the genome. The CL and IRES structures are separated by a pyrimidine-rich region named spacer 1, while the IRES is separated from the AUG codon by domain dVII and a sequence of about 100nt known as spacer 2.
The prototype strain of E-30 (strain Bastianni) was isolated in 1958 and since then interesting molecular epidemiology features have occurred, regarding the epidemic profile of circulating viruses, characterized by sequential displacement among multiple genetic variants. Based on VP1 phylogeny, recent E-30 strains form five lineages that have succeeded one another [4]. These five lineages can be further divided into 10 lineages in 3D and their emergence was correlated with recombination events [32, 33, 35]. The likelihood of recombination in the non-capsid coding region was correlated with VP1 divergence and the recombination frequency matched the epidemic periodicity of 5-6 years [33]. Strains of E-30 have frequently been implicated in recombination events either as acceptors or donors of genetic material [5, 9, 25, 28, 30].
Recombination events are observed in the non-structural regions, mainly in the P2 region [30, 35], which is thought to be a particular hotspot in EV-B. Inter-serotypic genetic exchanges within the capsid coding region are rarely documented, mainly in polioviruses [7, 8, 17, 22]; there is only one event concerning non-polio enteroviruses [9]. Concerning the 5΄ end of the genome, phylogenetic incongruences have been observed between the 5΄ UTR and the structural regions [43, 45], highlighting this region as a hotspot for recombination, mainly in polioviruses [18], but to our knowledge there is no nucleotide sequence and recombination analysis for this region in non-polio enteroviruses. Moreover, inter-serotypic recombinations within the 5΄ UTR are thought to be very rare, with only one report concerning a vaccine-derived poliovirus 2 (VDPV 2) [1].
A previous study by our group was based on RFLP analysis of the 5΄ UTR genomic region of clinical enterovirus strains isolated in Greece. In this analysis, the E-30 strain Gior presented incongruences in the topology of the 5΄ UTR and VP1 phylogenetic trees, indicating a putative recombination event [26]. In order to explain the phylogenetic behavior of strain Gior and to investigate how extensive recombination is in E-30 enteroviruses between the 5΄UTR and VP1 region, full genome sequencing of the strain was performed. In the present study, a phylogenetic analysis of the 5΄ UTR and VP1 of all available E-30 sequences was performed in order to investigate recombination events in the genomic region spanning 5΄ UTR-VP1. A similarity plot analysis of the available 5΄ UTR-VP2 sequences of the E-30 strains was also conducted, showing the presence of recombination events within the 5΄ UTR.
Materials and methods
The strain Gior was isolated from stool samples collected during an aseptic meningitis outbreak in 2001 and was initially characterized by seroneutralization assays and by partial sequencing of VP1 genomic region [20]. The isolate stock has been maintained in our laboratory in Rd cells.
Genome amplification
Viral RNA was extracted from 100μl of infected cell cultures using the guanidine thiocyanate extraction protocol [12]. The extracted RNA was reverse-transcribed into cDNA as previously described [24]. The primers that were used for the amplification of the viral genome are presented in the table in Online Resource 1. New primers were designed based on the sequences of Gior with the aid of Primer3 software, obtained on-line from the Whitehead Institute (http://www.genome.wi.mit.edu/genomesoftware/other/).
The PCR mixture for each tube comprised: 3μl cDNA from Gior isolate, 1µl of each primer at a concentration of 25pmol/µl, 5µl 10x PCR reaction buffer, 5µl dNTPs 10mM, 2,5 units Paq5000 DNA polymerase (Stratagene) and double-distilled nuclease-free water up to a final volume of 50µl/tube. The PCR thermal conditions for primer pairs used in previous studies were carried out as described in reference articles referenced in Online Resource 1. For the primer pairs Gior1051F-Gior1778R, Gior2227F-Gior3193R and Gior4878F-Gior6018R, designed as part of this study, 40 cycles of denaturation (95 °C for 20sec), annealing (52 °C for 20sec) and extension (72 °C for 20, 25 and 30 sec respectively) were used. Each PCR reaction started with an initial denaturation step at 95 °C for 2 min, and ended with a final extension at 72 °C for 5 min.
All PCR products were purified with Nucleospin Gel and PCR Clean-up kit (Macherey-Nagel, Duren, Germany) and were sequenced at CeMIA SA (Larissa, Thessaly, Greece) using the primers used for amplification of each product.
Nucleotide sequence accession number
The almost complete genomic sequence of isolate Gior (from nucleotide 1 to nucleotide 7180) was deposited in GenBank under the accession number KY131965.
Sequence analysis
The 5΄ UTR and VP1 nucleotide sequences of multiple E-30 strains was retrieved in FASTA format by NCBI blast, using as a query sequence the respective sequences from the newly sequenced strain Gior, with Blast E-value cutoff 1e-10. Many other enterovirus sequences were retrieved as well, indicating that the cutoff was sufficient to retrieve all E-30 sequences that existed in the database.
Two multiple sequence alignments, for the 5΄ UTR and VP1 nucleotide regions, respectively, were performed using the ClustalW algorithm implemented in MEGA v6.06 [40]. Each of the alignments consisted of a mixture of sequences of varying lengths. The region that was sufficient in length and also included a large number of sequence fragments was selected for phylogenetic analysis. For the 5΄ UTR, the selected region spanned nucleotides 148nt to 498nt, and was based on the E-30 prototype strain Bastianni. For VP1, the selected region spanned 2614nt to 2889nt, and again was based on the Bastianni strain. Next, the two lists were compared and only those E-30 strains that had a sequence fragment in both the 5΄UTR and VP1 alignments were selected. This final filtering step resulted in 85 strains with a complete sequence fragment in both the 5΄UTR (from 148 to 498nt) and VP1 (from 2614 to 2889nt) alignments.
Phylogenetic trees were reconstructed with MEGA v6.06 using the maximum likelihood estimation method and the reliability of the trees was determined by bootstrap analysis with 1000 replicates.
Sub-genomic sequences 946nt in length (from 150 to 1091 nt) that cover the partial 5΄ UTR (601nt), VP4 (209 nt) and the 5΄ end of VP2 (135nt) from all available E-30 sequences (62 strains) were used for the detection of recombination events. Similarity plot and bootscanning analyses were performed with the SimPlot (3.5 version) and T-RECs [42] software, using a sliding window size of 180nt and a step of 20 nt. With the exception of the recombinant strains, all other E-30 strain sequences were grouped based on the 5΄UTR and VP1 phylogenetic clusters, in order to detect recombination events concerning lineages or sub-lineages. The strains and group of strains used in SimPlot as well as the mean distance within the groups are presented in Online Resource 2.
Results
Phylogeny of VP1
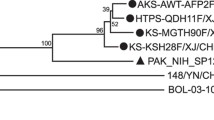
The phylogenetic tree representing the partial VP1 sequences is presented in Figure 1b. The 85 strains analyzed in the present study clustered into seven lineages (c to i) wherein lineages e and f were further divided into six sub-lineages: e.C0 to e.C2 and f.C3 to f.C5, respectively. The four Greek strains Kal, Han and Kar and the newly sequenced strain Gior, isolated from the same epidemic of aseptic meningitis in 2001, were classified in separate genetic clusters. The strains Kal, Han and Kar grouped in the e.C1 cluster with strains circulating in Europe between 1994-2001 while the strain Gior clustered with the epidemic strains isolated from France between 2000-2001 in the f.C4 sub-lineage. Blast and nucleotide analysis of all other parts of the genome of the Gior strain revealed high nucleotide identity (97-99%) with the strain CF2575-00, representative of the f.C4 sub-lineage, which is a recombinant in the 2C genomic region [35], carrying the same recombination event (data not shown).

Phylogenetic trees for (a) the 5΄UTR and (b) VP1 genomic regions of E-30 strains. The names of the strains include the country and the year of isolation. The lineages are indicated by the same color and the strains of the same sub-lineage are indicated by the same graphic
The lineages and sub-lineages in the present study correspond to VP1 lineages c to h, classified by Bailly et al., 2009 [4]. The prototype E-30 strain Bastianni represents lineage c. Cluster d is represented by strains 89T2090 and 91TLC isolated in France in 1989 and 1991 respectively and strain 1491net87 isolated in The Netherlands in 1987. The sub-lineage e.C0 contains strains that circulated in France between 1991-1992 and in 1981 and the strain /p/Roma99 isolated in Italy in 1999. The e.C1 sub-lineage is represented by E-30 strains isolated in Europe in 1994-2001, Australian strains from 2005-2007 and the strains Kor08-ECV30 and ECV30/GX10/05 isolated in South Korea and China in 2008 and 2010 respectively. Four French strains isolated in 1997 along with Australian ones from 2006-2007 form the e.C2 sub-lineage. The lineage f is divided into three sub-lineages (f.C3-C5) with f.C3 represented by French strains from 1996-2000 and the Italian strain /dr/Roma97 from 1999. The sub-lineages f.C4 and f.C5 contain strains mainly isolated in France between 2000-2001 and in 2005, respectively. The lineage g is represented by two strains, both isolated in Taiwan in 2001 (TW/2513/01 and TW/3182/01) and the lineage h is formed by three Chinese strains isolated in 2003 and 2010 and the Ukrainian strain EV30-14125-00 from 2000. Finally, the two strains isolated in Switzerland in 2006 (1167438) and 2009 (1167438_phMC) form a separate group, the newly designated group i.
Phylogeny of the 5΄UTR
Within the 5΄UTR phylogenetic tree, represented in Figure 1a, the majority of the E-30 strains grouped in the lineages and sub-lineages defined by the VP1 phylogenetic tree, although the lineages and sub-lineages dispersed differently. Within the 5΄ UTR the E-30 sequences form three major clusters. One represented e.C0 to e.C2, f.C3 and d lineages, one represented f.C4, f.C5, g and h lineages, while the three strains of the e.C1 lineage (ECV30/GX10/05, Kor08-ECV30 and 200.4715) separated from the other strains of e.C1 forming a separate cluster (e.C1r).
Aside from the above separation of the three strains from the e.C1 sub-lineage, several segregations are obvious in the 5΄ UTR tree topology. The two Switzerland strains of the i lineage (100% nucleotide identity in VP1), are split into two clades in the 5΄ UTR analysis with 89,2% nucleotide identity. The e.C0 sub-lineage is segregated into three groups in the 5΄UTR analysis, one comprising the strains isolated in France in 1991 (e.C0 I), one from the strains CF495-92, CF1347-92 and the strain p/Roma99 (e.C0 II), and the third one representing the strain CF298-81 (e.C0 III). Additionally, the sub-groups f.C4-f.C5 are separated from the f.C3 despite their phylogenetic relationship in the VP1 phylogenetic tree.
Detection of recombinations
The above incongruences in topology between the phylogenetic trees for the 5΄ UTR and VP1 were further analyzed for recombination events by similarity plot analysis. Sixty-two E-30 sequences of 946nt in length available in GenBank and covering the partial 5΄ UTR (601nt), VP4 (209 nt) and the 5΄ of VP2 (135nt) were included in this analysis. Consensus E-30 sequences for each lineage and sub-lineage were grouped together under the name of a representative (Online Resource 2) and each group was compared to all other E-30 groups. The results of the similarity plot analysis for the E-30 groups that were found to be recombinant are presented in Figure 2, while those with no recombination events are presented in Online resource 3.

Similarity plot analysis for the recombinations of a) the CF298-81 strain, b) the e.C1r lineage, c) the f.C 4- f.C5 sub-lineages, d) the g lineage, e) the h lineage and f) the strains of the i lineage
The incongruences between the VP1 and 5΄UTR phylogenetic trees concerning: the strain CF298-81 of the e.C0 sub-lineage; the three strains ECV30/GX10/05, Kor08-ECV30 and 200.4715 of the e.C1 lineage; the f.C 4- f.C5 subgroups and finally the Switzerland strains of the i lineage, were confirmed by the similarity plot analysis, see figures 2a to 2c and 2f respectively. Except for these strains, two additional cases of a steep dip in the SimPlot were observed, concerning the groups g and h (figures 2d and 2e respectively), suggesting that these particular genomic regions are probably coming from other serotypes. In addition, as shown in figure 2, most of the cases have a SimPlot dip in the same region at the 5΄ UTR-VP4 junction, with the recombination start point in the 5΄UTR and end point in VP4.
Analysis of the recombinants
The hypothesis for inter-serotypic recombination events in these E-30 groups or strains was further confirmed by blast analysis of the focused region against NCBI sequences and also by comparison of phylogenetic trees for the 5΄ UTR and VP1 that included 437 sequences from several serotypes of EV-B (Online resources 4 and 5). Nevertheless, the different serotypes identified by blast could not be included in the above similarity plot analysis, because frequently only a small fragment of the sequence identified as a possible donor was recovered. Consequently, the recombinant groups or strains were studied separately in order to provide more information about the genetic donors.
The e.C1r group was found to be a recombinant in the studied region with the possible donor being the E-7 strain E7_08-10-2005 (accession number FJ796983). As is shown in Figure 3a, similarity plot and nucleotide analysis revealed not only the donor but also the exact point for the recombination event. The recombination is located from 643nt in the 5΄ UTR to 833nt in VP4. The sub-groups f.C4-f.C5 were also found to be recombinant, carrying a 5΄ UTR most similar to that of the CV-B4 strain CBV4_THA/2009/2012 (accession number KP062991) (figure 3b). Due to the short sequence of the proposed donor available in GenBank, only the end point of this insertion could be defined (figure 3b) which was located at 693nt in the E-30 genome. Another recombination event that has been well characterized is that of lineage g (representative strain TW/2513/01). As is shown in figure 3c, the lineage g carries a recombination in the 5΄ end of its genome with the possible donor being the CV-B4 strain 10508 (accession number AF160118). Due to the short available sequence for the CV-B4 strain 10508 only the end point of this insertion, described in figure 3c, was located; position 768nt in the E-30 genome, within the VP4 genomic region. The E-30 strain 1167438 of the i lineage is probably also implicated in a recombination event with CV-B1 strains, but the absence of sequences of sufficient length, made depicting this difficult in SimPlot. The CF298-81 isolate of the e.C0 sub-lineage was probably the product of an intra-serotypic recombination event (data not shown), while the donor for the recombinant group h could not be recognized.

Similarity plot and sequence analyses describing the recombinations and the recombination junctions of: a) the strain Kor08-ECV30, b) the strain Gior and c) the strain TW/2513/01. In the similarity plot analysis, the parental E-30 strains are depicted with grey, the inter-serotypic donors are depicted with red, while a blue color is used for strains of other serotypes. Concerning the sequence alignments for the recombination junctions, similarities between the recombinant strains and the E-30 parental strains are highlighted in green, while those between the recombinants and the donors are highlighted in yellow (color figure online)
Finally, the exact points of recombination in the 5΄ UTR described above all located at the 3΄end of the 5΄ UTR, outside the main secondary structures of the cloverleaf and IRES. As is shown in Figure 4, the start point of recombination for the strain Kor08-ECV30 (e.C1r group) located at the end of the secondary structure domain dVII, while the end point for recombination of strains Gior (f.C4- f.C5 sub-lineages) and TW/2513/01 (lineage g) located in the Spacer 2 region of the 5΄ UTR and VP4 respectively.

A nucleotide sequence alignment of CV-B3 strain Nancy, E-30 strain Bastianni, and the recombinant strains Kor08-ECV30, Gior and TW/2513/01. The recombination points are depicted in reference to the domains dVI-dVII and spacer 2 at the end of the 5΄ UTR, as described for CBV3 [3]. The sequences of the donors are highlighted in yellow. The start point of the recombinations for the strains Gior and TW/2513/01 are unknown
Discussion
Enteroviruses, like all RNA viruses can evolve at very high rates due to the lack of proofreading activity in their polymerases (responsible for causing a high mutation rate in these viruses) as well as because of recombination events. In the last two decades recombination has been described as a major mechanism of evolution in all four enterovirus species A-D [5, 16, 24, 27, 29, 34]. Extensive studies by phylogenetic analysis of different genomic regions and complete genome sequencing have suggested that recombination events are localized mainly in the non-capsid regions of an enterovirus genome [6, 23, 35], while recombination events in the capsid coding regions have been considered to occur very rarely [7, 9, 17, 22] and even more rarely in the 5΄ UTR [1]. Nevertheless, discrepancies between the phylogenetic trees for the 5΄UTR and capsid coding regions [30, 45], have led to the hypothesis that the 5΄UTR-VP4 junction can serve as a recombination hotspot for enteroviruses, however this region has so far under-explored. In a previous study [26], a discrepancy between the 5΄UTR and VP1 phylogenetic trees for the E-30 strain Gior aroused suspicions that recombination events may have taken place between these two regions. In the present study the complete genome sequence of the strain Gior was obtained and compared phylogenetically (both the 5΄ UTR and VP1 regions) with all available E-30 sequences in order to investigate the presence of recombination events in the genomic region spanning 5΄ UTR-VP1.
Phylogenetic analysis of the VP1 and 5΄ UTR genomic regions clustered the strain Gior with E-30 epidemic strains isolated from France in 2000-2001 in the f.C4 sub-lineage. Blast and nucleotide analysis of all other parts of the genome of the strain Gior revealed high nucleotide identity (97-99%) with the strain CF2575-00, representative of f.C4 sub-lineage, which is recombinant in the non-capsid genomic region [35]. This resemblance to the recombinant strain CF2575-00 implies that our strain carries the same recombination event in the 2C genomic region, a theory that was supported by similarity plot analysis in the present study (data not shown).
Comparison of the phylogenetic trees representing the 5΄ UTR and VP1 genomic regions revealed incongruences concerning: strain CF298-81 of the e.C0 sub-lineage; strains ECV30/GX10/05, Kor08-ECV30 and 200.4715 of the e.C1 sub- lineage; and two Switzerland strains of the i lineage, as well as the topology of subgroups f.C 4-f.C5 which separated from f.C3 in 5΄ UTR. These phylogenetic discordances implied recombination events between the 5΄ UTR and VP1 genomic regions. Similarity plot analysis not only confirmed the presence of the above recombinations but also revealed two others, concerning lineages g and h. Although the phylogenetic tree for the 5΄ UTR was constructed on only part of the sequence used for similarity plot analysis, all the recombinant clades phylogenetically separated from the non-recombinants. The recombinants placed in the lower part of the tree with the stain CF298-81, carrying the intra-serotypic recombination, serving as the border of this phylogenetic segregation.
Previous studies that compared phylogenetic trees representing the 5΄ UTR and VP1 genomic regions of enteroviruses had highlighted the discordances in the phylogeny of these two regions, implying the occurrence mainly of intra- [6, 14, 37, 43] and inter-serotypic recombination events [45]. The first evidence for inter-serotypic recombination in the 5΄ UTR came from the complete genome sequencing of a Sabin 2 strain, which was found to share a part of its 5΄ UTR with an EV-C enterovirus [1]. The results of the present study describe for the first time recombination events in this region among circulating EV-B viruses.
Although it was originally believed that recombination events at the 5΄ UTR were unlikely to occur, the present study detected and analyzed six different recombination events at the 5΄ UTR-VP4 region in E-30. Recombination has been recognized as a major mechanism of enterovirus evolution and several studies demonstrated that the different serotypes recombine their genomes at different rates [33, 34]. Indeed, the recombination frequency in the non-capsid coding region tightly correlated with VP1 divergence and E-30 has been characterized as one of the serotypes that recombine frequently, with an estimated recombination half-life of 3.1 years [33]. The presence of genomic exchanges in the 5΄ end of E-30 genomes also possibly associates with the high divergence in VP1, but this hypothesis requires a larger number of E-30 complete genome sequences.
According to the similarity plot analysis, almost all the recombination events revealed in the present study occurred at the same region, having a start point in the 3΄end of the 5΄ UTR and an end point in the VP4 genomic region, leading to the conclusion that this region may serve as a hot spot for recombination in enteroviruses in general. Due to the high nucleotide identity with the donors, in the cases of strains Kor08-ECV30 (e.C1r group), Gior (f.C4- f.C5 sub-groups) and TW/2513/01 (lineage g), the exact sequence of some of the recombination breakpoints was identified. Two of them located within the 5΄ UTR, outside the cloverleaf and IRES secondary structures, one at the end of dVII structure and the other in the Spacer 2 region. The 3΄ end of the 5΄ UTR was also identified as the breakpoint for the only previous recombination in the 5΄UTR described for enteroviruses so far; in the Sabin 2 strain [1]. The dVII-spacer 2 region has been recognized as a hot-spot for in vitro genetic exchanges within the 5΄ UTR between polioviruses and non-polio enteroviruses [36], probably because this region can tolerate modifications without phenotypic changes. The CL and IRES domains seem to be protected from recombination events, probably due to their exceptional role in translation and replication of the viral genome.
The exact role and impact of recombination in the emergence of enteroviruses with altered properties is still not well understood. In a recent study, the exceptional virulence and rapid spread of a CV-A9 strain isolated from members of a family with signs of meningitis, was ascribed to 5΄UTR – 5΄ end of VP4 sequences that originated from E-11/EV-75 strains [10]. Keeping in mind the role of the 5΄ UTR in viral translation and RNA synthesis, and its essential interactions with viral proteins, such as 2B, 2BC, 3A, and 3D as well as host proteins, such as polypyrimidine tract-binding protein and poly(rC) binding protein [31], a recombinant 5΄ UTR has to be efficient in interactions with the rest of the viral genome. Surprisingly, aside from the Greek strain Gior which was found to be a double recombinant, all the other strains that were found to be recombinants at the 5΄ end of their genome were also recombinants in their non structural regions (except for the strains of genogroup i and the e.C1r sub-genogroup, for which there is no information about recombination) [13, 35, 44]. In accordance with the present study, the majority of enterovirus strains that carry a 5΄ UTR belonging to a different serotype as a product of recombination, are also double recombinants in the 3΄ part of their genome, reviewed in [27]. To our knowledge, only one enterovirus strain has been reported so far with its 5΄ UTR originating from another serotype with no other recombination in the rest of its genome [10]. These observations may lead to the hypothesis that the 5΄ UTR of enteroviruses has to be in congruence with the non-structural proteins and thus recombination at the 5΄end can trigger recombination at a 3΄ part of the genome.
In conclusion, the 5΄ UTRs of enteroviruses have all the prerequisites for recombination, such as highly conserved sequences and RNA secondary structures. The present study indicates that recombination is not as rare in the 5΄ UTR as was originally believed, but the detection of such recombination events is difficult due to the limited number of complete genome sequences and the high conservation of this specific region between enteroviruses. As more complete genome sequence data from circulating enteroviruses become available, the role of recombination on the evolutionary pathways of enteroviruses will be elucidated. Understanding the mechanism and tempo of enterovirus evolution is of great importance for elimination of such elements that offer selective advantages and better adaptation.
References
Adu F, Iber J, Bukbuk D, Gumede N, Yang SJ, Jorba J, Campagnoli R, Sule WF, Yang CF, Burns C, Pallansch M, Harry T, Kew O (2007) Isolation of recombinant type 2 vaccine-derived poliovirus (VDPV) from a Nigerian child. Virus Res 127:17–25
Antona D, Leveque N, Chomel JJ, Dubrou S, Levy-Bruhl D, Lina B (2007) Surveillance of enteroviruses in France, 2000-2004. Eur J Clin Microbiol Infect Dis 26:403–412
Bailey JM, Tapprich WE (2007) Structure of the 5’ nontranslated region of the coxsackievirus b3 genome: Chemical modification and comparative sequence analysis. J Virol 81:650–668
Bailly JL, Mirand A, Henquell C, Archimbaud C, Chambon M, Charbonne F, Traore O, Peigue-Lafeuille H (2009) Phylogeography of circulating populations of human echovirus 30 over 50 years: nucleotide polymorphism and signature of purifying selection in the VP1 capsid protein gene. Infect Genet Evol 9:699–708
Bailly JL, Mirand A, Henquell C, Archimbaud C, Chambon M, Regagnon C, Charbonne F, Peigue-Lafeuille H (2011) Repeated genomic transfers from echovirus 30 to echovirus 6 lineages indicate co-divergence between co-circulating populations of the two human enterovirus serotypes. Infect Genet Evol 11:276–289
Bessaud M, Joffret ML, Holmblat B, Razafindratsimandresy R, Delpeyroux F (2011) Genetic relationship between cocirculating Human enteroviruses species C. PLoS One 6:e24823
Blomqvist S (2003) Characterization of a recombinant type 3/type 2 poliovirus isolated from a healthy vaccinee and containing a chimeric capsid protein VP1. J Gen Virol 84:573–580
Blomqvist S, Savolainen-Kopra C, Paananen A, El Bassioni L, El Maamoon Nasr EM, Firstova L, Zamiatina N, Kutateladze T, Roivainen M (2010) Recurrent isolation of poliovirus 3 strains with chimeric capsid protein Vp1 suggests a recombination hot-spot site in Vp1. Virus Res 151:246–251
Bouslama L, Nasri D, Chollet L, Belguith K, Bourlet T, Aouni M, Pozzetto B, Pillet S (2007) Natural recombination event within the capsid genomic region leading to a chimeric strain of human enterovirus B. J Virol 81:8944–8952
Butsch R, Tapparel C, Keller P, Herzog K, Krause M, Wunderli W, Kaiser L, Bossart W (2013) Family outbreak of an infection with a recombinant Coxsackie A virus in eastern Switzerland. Infection 41:231–235
Cabrerizo M, Echevarria JE, Gonzalez I, de Miguel T, Trallero G (2008) Molecular epidemiological study of HEV-B enteroviruses involved in the increase in meningitis cases occurred in Spain during 2006. J Med Virol 80:1018–1024
Casas I, Powell L, Klapper PE, Cleator GM (1995) New method for the extraction of viral RNA and DNA from cerebrospinal fluid for use in the polymerase chain reaction assay. J Virol Methods 53:25–36
Chen GW, Huang JH, Lo YL, Tsao KC, Chang SC (2007) Mosaic genome structure of echovirus type 30 that circulated in Taiwan in 2001. Arch Virol 152:1807–1817
Chevaliez S, Szendroi A, Caro V, Balanant J, Guillot S, Berencsi G, Delpeyroux F (2004) Molecular comparison of echovirus 11 strains circulating in Europe during an epidemic of multisystem hemorrhagic disease of infants indicates that evolution generally occurs by recombination. Virology 325:56–70
Chomel JJ, Antona D, Thouvenot D, Lina B (2003) Three ECHOvirus serotypes responsible for outbreak of aseptic meningitis in Rhone-Alpes region, France. Eur J Clin Microbiol Infect Dis 22:191–193
Combelas N, Holmblat B, Joffret ML, Colbere-Garapin F, Delpeyroux F (2011) Recombination between poliovirus and coxsackie A viruses of species C: a model of viral genetic plasticity and emergence. Viruses 3:1460–1484
Dedepsidis E, Pliaka V, Kyriakopoulou Z, Brakoulias C, Levidiotou-Stefanou S, Pratti A, Mamuris Z, Markoulatos P (2008) Complete genomic characterization of an intertypic Sabin 3/Sabin 2 capsid recombinant. FEMS Immunol Med Microbiol 52:343–351
Joffret ML, Jegouic S, Bessaud M, Balanant J, Tran C, Caro V, Holmblat B, Razafindratsimandresy R, Reynes JM, Rakoto-Andrianarivelo M, Delpeyroux F (2012) Common and diverse features of cocirculating type 2 and 3 recombinant vaccine-derived polioviruses isolated from patients with poliomyelitis and healthy children. J Infect Dis 205:1363–1373
Kapusinszky B, Szomor KN, Farkas A, Takacs M, Berencsi G (2010) Detection of non-polio enteroviruses in Hungary 2000-2008 and molecular epidemiology of enterovirus 71, coxsackievirus A16, and echovirus 30. Virus Genes 40:163–173
Kottaridi C, Bolanaki E, Siafakas N, Markoulatos P (2005) Evaluation of seroneutralization and molecular diagnostic methods for echovirus identification. Diagn Microbiol Infect Dis 53:113–119
Kottaridi C, Bolanaki E, Mamuris Z, Stathopoulos C, Markoulatos P (2006) Molecular phylogeny of VP1, 2A, and 2B genes of echovirus isolates: epidemiological linkage and observations on genetic variation. Arch Virol 151:1117–1132
Kyriakopoulou Z, Kottaridi C, Dedepsidis E, Bolanaki E, Levidiotou-Stefanou S, Markoulatos P (2006) Molecular characterization of wild-type polioviruses isolated in Greece during the 1996 outbreak in Albania. J Clin Microbiol 44:1150–1152
Kyriakopoulou Z, Dedepsidis E, Pliaka V, Mastorakos P, Stamati A, Pratti A, Levidiotou-Stefanou S, Markoulatos P (2010) Molecular identification and full genome analysis of an echovirus 7 strain isolated from the environment in Greece. Virus Genes 40:183–192
Kyriakopoulou Z, Dedepsidis E, Pliaka V, Tsakogiannis D, Pratti A, Levidiotou-Stefanou S, Markoulatos P (2010) Full-genome sequence analysis of a multirecombinant echovirus 3 strain isolated from sewage in Greece. J Clin Microbiol 48:1513–1519
Kyriakopoulou Z, Pliaka V, Tsakogiannis D, Ruether IG, Komiotis D, Gartzonika C, Levidiotou-Stefanou S, Markoulatos P (2012) Genome analysis of two type 6 echovirus (E6) strains recovered from sewage specimens in Greece in 2006. Virus Genes 44:207–216
Kyriakopoulou Z, Tsolis K, Pliaka V, Tsakogiannis D, Ruether IG, Gartzonika C, Levidiotou-Stefanou S, Markoulatos P (2013) Combined 5’ UTR RFLP analysis and VP1 sequencing for epidemic investigation of enteroviruses. Arch Virol 158:103–111
Kyriakopoulou Z, Pliaka V, Amoutzias GD, Markoulatos P (2015) Recombination among human non-polio enteroviruses: implications for epidemiology and evolution. Virus Genes 50:177–188
Lindberg AM, Andersson P, Savolainen C, Mulders MN, Hovi T (2003) Evolution of the genome of Human enterovirus B: incongruence between phylogenies of the VP1 and 3CD regions indicates frequent recombination within the species. J Gen Virol 84:1223–1235
Lukashev AN (2005) Role of recombination in evolution of enteroviruses. Rev Med Virol 15:157–167
Lukashev AN, Lashkevich VA, Ivanova OE, Koroleva GA, Hinkkanen AE, Ilonen J (2005) Recombination in circulating Human enterovirus B: independent evolution of structural and non-structural genome regions. J Gen Virol 86:3281–3290
Martinez-Salas E, Pacheco A, Serrano P, Fernandez N (2008) New insights into internal ribosome entry site elements relevant for viral gene expression. J Gen Virol 89:611–626
McWilliam Leitch EC, Bendig J, Cabrerizo M, Cardosa J, Hyypia T, Ivanova OE, Kelly A, Kroes AC, Lukashev A, MacAdam A, McMinn P, Roivainen M, Trallero G, Evans DJ, Simmonds P (2009) Transmission networks and population turnover of echovirus 30. J Virol 83:2109–2118
McWilliam Leitch EC, Cabrerizo M, Cardosa J, Harvala H, Ivanova OE, Kroes AC, Lukashev A, Muir P, Odoom J, Roivainen M, Susi P, Trallero G, Evans DJ, Simmonds P (2010) Evolutionary dynamics and temporal/geographical correlates of recombination in the human enterovirus echovirus types 9, 11, and 30. J Virol 84:9292–9300
McWilliam Leitch EC, Cabrerizo M, Cardosa J, Harvala H, Ivanova OE, Koike S, Kroes AC, Lukashev A, Perera D, Roivainen M, Susi P, Trallero G, Evans DJ, Simmonds P (2012) The association of recombination events in the founding and emergence of subgenogroup evolutionary lineages of human enterovirus 71. J Virol 86:2676–2685
Mirand A, Henquell C, Archimbaud C, Peigue-Lafeuille H, Bailly JL (2007) Emergence of recent echovirus 30 lineages is marked by serial genetic recombination events. J Gen Virol 88:166–176
Muslin C, Joffret ML, Pelletier I, Blondel B, Delpeyroux F (2015) Evolution and Emergence of Enteroviruses through Intra- and Inter-species Recombination: Plasticity and Phenotypic Impact of Modular Genetic Exchanges in the 5’ Untranslated Region. PLoS Pathog 11:e1005266
Oberste MS, Penaranda S, Pallansch MA (2004) RNA recombination plays a major role in genomic change during circulation of coxsackie B viruses. J Virol 78:2948–2955
Richter J, Koptides D, Tryfonos C, Christodoulou C (2006) Molecular typing of enteroviruses associated with viral meningitis in Cyprus, 2000–2002. J Med Microbiol 55:1035–1041
Siafakas N, Markoulatos P, Levidiotou-Stefanou S (2004) Molecular identification of enteroviruses responsible for an outbreak of aseptic meningitis; implications in clinical practice and epidemiology. Mol Cell Probes 18:389–398
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Thoelen I, Lemey P, Van Der Donck I, Beuselinck K, Lindberg AM, Van Ranst M (2003) Molecular typing and epidemiology of enteroviruses identified from an outbreak of aseptic meningitis in Belgium during the summer of 2000. J Med Virol 70:420–429
Tsimpidis M, Bachoumis G, Mimouli K, Kyriakopoulou Z, Robertson DL, Markoulatos P, Amoutzias GD (2017) T-RECs: rapid and large-scale detection of recombination events among different evolutionary lineages of viral genomes. BMC Bioinform 18:13
van der Sanden S, van Eek J, Martin DP, van der Avoort H, Vennema H, Koopmans M (2011) Detection of recombination breakpoints in the genomes of human enterovirus 71 strains isolated in the Netherlands in epidemic and non-epidemic years, 1963–2010. Infect Genet Evol 11:886–894
Zhang T, Du J, Xue Y, Su H, Yang F, Jin Q (2013) Epidemics and frequent recombination within species in outbreaks of human enterovirus B-associated hand, foot and mouth disease in Shandong China in 2010 and 2011. PLoS One 8:e67157
Zhou F, Wang Q, Sintchenko V, Gilbert GL, O’Sullivan MV, Iredell JR, Dwyer DE (2014) Use of the 5’ untranslated region and VP1 region to examine the molecular diversity in enterovirus B species. J Med Microbiol 63:1339–1355
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by research grants of the Postgraduate Program “Toxicology”, of the University of Thessaly, School of Health Sciences, Department of Biochemistry & Biotechnology (grant number 5050).
Conflict of interest
All authors declare that they have no conflicting or dual interests
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Tim Skern.
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2017_3600_MOESM2_ESM.doc
Online resource 2: Table with the strains and groups of strains used in Similarity Plot. The mean distances within the groups for the region from150 to1090 nt are presented. (DOC 107 kb)
705_2017_3600_MOESM3_ESM.ppt
Online resource 3: Similarity Plot analysis for the non-recombinant E-30 groups a) e.C0 I, b)e.C0 II, c) e.C1 sub-lineage, d) e.C2 sub-lineage, e) f.C3 sub-lineage and f) d lineage. (PPT 349 kb)
705_2017_3600_MOESM4_ESM.pdf
Online resource 4: Phylogenetic trees for 5΄UTR (left) and VP1(right) genomic regions of 437 strains of E30 and other EV-B serotypes. The serotype, the name and the country of origin of each strain is presented for each sequence. (PDF 105 kb)
705_2017_3600_MOESM5_ESM.xls
Online resource 5: Annotation file for the strains included in phylogenetic analysis of online resource 4. For each sequence is presented the name of strain, the accession number, the serotype, the date and the country of origin. (XLS 157 kb)
Rights and permissions
About this article

Cite this article
Kyriakopoulou, Z., Amoutzias, G.D., Dimitriou, T.G. et al. Intra- and inter-serotypic recombinations in the 5΄ UTR-VP4 region of Echovirus 30 strains. Arch Virol 163, 365–375 (2018). https://doi.org/10.1007/s00705-017-3600-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3600-1