
Abstract
Here, we report the circulation of highly related virulent Newcastle disease viruses (NDV) in Bulgaria and Ukraine from 2002 until 2013. All of these NDV isolates have the same virulence-associated cleavage site (“113RQKR↓F117”), and selected ones have intracerebral pathogenicity index values ranging from 1.61 to 1.96. These isolates are most closely related to viruses circulating in Eastern Europe, followed by viruses isolated in Asia during the same period of time. Interestingly, the majority of the viruses were isolated from backyard poultry, suggesting the possibility of a “domestic” or “urban” cycle of maintenance. The molecular characterization of the nucleotide sequence of the complete fusion protein gene of the studied viruses suggests continued circulation of virulent NDV of sub-genotype VIId in Eastern Europe, with occasional introductions from Asia. Furthermore, the high level of genetic similarity among those isolates suggests that the NDV isolates of sub-genotype VIId from Bulgaria and Ukraine may have been part of a broader epizootic process in Eastern Europe rather than separate introductions from Asia or Africa. The continuous monitoring of backyard poultry flocks for the presence of circulating virulent NDV strains will allow early identification of Newcastle disease outbreaks.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Newcastle disease virus (NDV) is one of the most important pathogens of poultry worldwide [2, 26]. Newcastle disease (ND) is caused by virulent strains of avian paramyxovirus 1 (APMV-1, synonymous to NDV). APMV-1 belongs to the family Paramyxoviridae, genus Avulavirus of the order Mononegavirales [23]. The disease devastates poultry flocks with up to 100 % mortality and also causes significant economic losses from trade restrictions imposed on countries and regions where outbreaks have occurred [6, 26]. Based on genomic analysis, APMV-1 isolates are classified into two main evolutionary related groups – class I and class II [11]. Employing the coding sequence of the fusion protein gene (F gene) or the complete genome sequence, NDV isolates can be further classified into genotypes [13]. Currently, there are 18 genotypes in class II (genotypes I to XVIII) and a single genotype in class I (genotype 1), with some genotypes further divided into sub-genotypes [9, 12, 13, 41, 42].
Viruses of class II genotype VII are responsible for the fourth ND panzootic, which began around 1985 in Southeast Asia, spread to Africa, Europe and South America, and continues today [16, 21, 22, 26, 37, 51]. Since 2011, reports have documented that some of the newly emerged viruses of genotype VII can cause mortality in poorly vaccinated poultry [39, 50]. Furthermore, viruses of genotype VII have been frequently isolated from wild and domestic waterfowl [17, 20, 46], with documented cases of clinical disease in geese, which have been shown previously to be resistant to morbidity [17, 26, 48].
Bulgaria and Ukraine are wintering sites for many wild-bird species and are also located within the largest wild-bird migration system – the Black Sea-Mediterranean flyway. Some areas of this flyway overlap with other major migration systems (e.g., Central and East Asian-Australasian flyways, West Asian-East African flyway, East Atlantic flyway) [31]. Additionally, Bulgaria and Ukraine are geographically located on main trade routes between Asia and Europe. The continuous expansion of the poultry industry worldwide and international trade of poultry products [38] is reflected in Bulgaria and Ukraine with these two countries having well-developed poultry farming industries. As a result of established good vaccination practices, ND outbreaks on industrial poultry farms have not occurred for almost a decade in those countries. However, the backyard poultry population is still considerably large and widely distributed. Not all backyard birds are vaccinated, and biosecurity management is scarce or non-existent. This provides opportunity for virulent ND viruses to infect and cause clinical disease after the introduction in these birds [14, 29].
Previous studies of virulent NDV (vNDV) isolates from Bulgaria and Ukraine have been performed employing mainly serological and conventional virological approaches. Molecular analyses have also been performed, but these have been based on limited genetic information (partial F gene sequences) or, in some cases, targeting low-virulence NDV from wild birds [1, 10, 16, 18, 29, 34]. Here, we report the circulation of highly related virulent viruses from 2002 until 2013 in Bulgaria and Ukraine and of one isolate from Russia. The epidemiology of ND was evaluated on the basis of phylogenetic analysis of the complete coding sequence of the fusion protein gene of these isolates.
Materials and methods
Sample collection and virus isolation
Information for the isolates studied in this work, including location, bird species, presence of clinical signs, husbandry practice, vaccination history, and type of samples, is presented in Table 1. For the samples from Bulgaria, sampled swabs were placed in tubes with the following transport medium: medium 199 supplemented with 10,000 IU of penicillin, 10 mg of streptomycin, 0.250 mg of gentamicin and 1 mg of oxytetracycline per ml, chilled at 4 °C and transported to the lab for immediate testing. Swab medium (0.2 ml) from each sample was inoculated into five 9 to 11-day-old embryonating chicken eggs (ECEs) from NDV- and AIV-free layers using standard methods as described previously [3]. For the Ukrainian samples, immediately after sampling, the swabs were placed in tubes with transport medium (Hank’s balanced salt solution containing 0.5 % lactalbumin, 10 % glycerol, and 200 U penicillin, 0.200 mg streptomycin, 100 U polymyxin, 0.250 mg gentamicin, and 50 U nystatin per ml). Organ suspensions (10 % w/v) were also prepared using the same medium. Samples were chilled at 4 °C, transported to the lab and stored at -70 °C for 1 to 4 weeks until analyzed. Swab medium or suspension supernatant (0.2 ml) from each sample was inoculated into five 9 to 11-day-old specific-pathogen-free (SPF) embryonating chicken eggs using standard methods as described previously [3]. For the Russian sample, the virus isolation procedure was similar to the one described for the Ukrainian sample with the difference that NDV- and AIV-free ECEs were used instead of SPF. For all samples, the allantoic fluids from inoculated ECEs were harvested and subjected to a hemagglutination assay (HA) using the microtiter method. All of the hemagglutinating samples were confirmed to be APMV-1 by hemagglutination inhibition (HI) assay [32].
Virus propagation
Twenty-one isolates (Table 1) were submitted to the Southeast Poultry Research Laboratory of the USDA in Athens, GA, USA. Viruses were propagated in 9 to 11-day-old SPF embryonating chicken eggs from the SEPRL SPF white leghorn flock [3].
Intracerebral pathogenicity index (ICPI) assay
An intracerebral pathogenicity index (ICPI) assay was conducted on 14 viruses (selection was based on phylogenetic analysis) following established procedures [32]. The ICPI test for isolate chicken/Ukraine/Ivano-Frankivsk/58/2007 was performed in Ukraine following the same procedures.
RNA isolation, PCR amplification and sequencing
RNA from each isolate was extracted from allantoic fluids using TRIzol LS (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. One-step reverse transcription PCR (SuperScript® III One-Step RT-PCR System with Platinum® Taq DNA Polymerase, Life Technologies, Carlsbad, CA, USA) was used to convert and amplify the extracted RNA as described previously [28] using primers described by Miller et al. (4008F/4994R; 4715F/5637R; 5410F/6332R) [27]. All PCR products were subjected to electrophoresis in a 1% agarose gel (0.5X TBE). The DNA bands were excised and purified using a QuickClean II Gel Extraction Kit (GenScript, Piscataway, NJ, USA). Nucleotide sequencing and assembly were performed as described previously by Miller et al. [28].
Collection of sequences
Complete fusion protein gene (F gene) coding sequences (n = 1458) of all available class II NDV isolates were downloaded from GenBank (National Center for Biotechnology Information) (available as of January 2015, http://www.ncbi.nlm.nih.gov/genbank) and aligned using ClustalW [45].
Evolutionary and phylogenetic analysis
Phylogenetic analysis was performed using MEGA6 software (MEGA, version 6) [44]. Preliminary analysis was performed to infer the evolutionary history with 1458 sequences available in GenBank (data not shown). A smaller group (including the most closely related viruses, n = 82) (Table S1) was parsed from the initial dataset and further analyzed. Determination of the best-fit substitution model was performed using MEGA6, and the goodness-of-fit for each model was measured by corrected Akaike information criterion (AICc) and Bayesian information criterion (BIC) [44]. The final tree was constructed using the maximum-likelihood method based on the general time-reversible model as implemented in MEGA 6, with 500 bootstrap replicates [30]. For all analyses, the codon positions included were 1st + 2nd + 3rd + noncoding, and all positions containing gaps and missing data were eliminated. The Roman numerals presented for each sequence in the phylogenetic tree represent the genotype of the isolate, followed by the GenBank accession number, host name (if available), country of isolation, strain designation, and year of isolation.
The estimates of average evolutionary distances were inferred using MEGA6. Analyses were conducted using the maximum composite likelihood model [43]. The rate variation among sites was modeled with a gamma distribution (shape parameter = 4). Previously described criteria [13] based on the phylogenetic topology and evolutionary distances between different taxonomic groups were used to determine genotypes and sub-genotypes.
Accession numbers
The complete F gene sequences (n = 21) of virulent NDV obtained in this study were submitted to GenBank and are available under the accession numbers KJ914673, KU295450 to KU295455, KU710269 to KU710281 and KU726619.
Results
Virus recovery and ICPI test
Twenty-one NDV isolates were studied in the present work. In Bulgaria and Ukraine, national monitoring programs (active surveillance) are implemented, and both industrial and backyard poultry are monitored for the presence of virulent Newcastle disease viruses. Eleven of the viruses in this study were isolated in Bulgaria and were representative for all 33 NDV outbreaks (sub-genotype VIId) that were identified during the described period. Selection was done based on phylogenetic analysis of the 374-nucleotide region of the fusion protein gene (data not shown). However, our data on the prevalence of NDV genotypes in Ukraine between 2002 and 2013 is limited to the viruses studied here. Most of the samples in the present study (n = 18) were recovered from unvaccinated backyard chickens that showed respiratory, neurological and/or enteric signs. The viruses were recovered from six regions in Bulgaria, six regions in Ukraine and one region in Russia in 2006–2009, 2002–2013 and 2007, respectively (Table 1). Biological assessment of the pathogenicity of selected isolates was performed by ICPI test in 1-day-old SPF chickens. All of the viruses tested using the ICPI assay yielded values ranging from 1.61 to 1.93 (Table 1), which characterizes them as virulent NDV based on the OIE standards [32] and is in agreement with the observed clinical signs. ICPI values above 1.5 are indicative of velogenic NDV [3].
Nucleotide sequencing and distance analysis
To determine the genetic characteristics of the NDV isolated in Bulgaria, Ukraine and Russia, we performed sequencing and distance analysis of these isolates. The sequencing analysis predicted fusion protein cleavage sites that contained three basic amino acids at positions 113–116 and a phenylalanine residue at position 117 (“113RQKR↓F117”) in all isolates. Such a cleavage site is specific for virulent viruses based on criteria utilized by OIE to assess virulence of NDV isolates [32], and these results are in agreement with the data obtained in the ICPI test.
All isolates were analyzed in order to determine the evolutionary distances among them and between them and other ND viruses. Phylogenetic analysis of the complete fusion coding regions classified all of the isolates studied here as class II sub-genotype VIId. Their genetic distance when compared to the rest of the sub-genotype VIId isolates was in the range of 1.7 to 2.9 %. Fourteen of the isolates obtained in Bulgaria and Ukraine between 2003 and 2013 were very similar among themselves, and the mean nucleotide sequence identity within this group was 99.4 %. The nearest related isolates (99.3-99.7 % genetic identity) were dove/Serbia/749/2007 and sparrowhawk/Serbia/1038/2007. The 14 viruses were also genetically close (98.6-98.7 % genetic identity) to isolates obtained from poultry and wild birds in China between 2002 and 2011 (chicken/China/SHY06/2006, chicken/China/SDSG01/2011, chicken/China/SD2/2007, Muscovy Duck/China/FP1/02/2002 and mallard/China/Md/CH/LGD/1/2005). Seven of the analyzed viruses isolated between 2002 and 2008 (five Ukrainian, one Bulgarian and one Russian, 99 % identity within the group) were more distant and showed 97.3 % nucleotide sequence identity when compared to the rest of Bulgarian and Ukrainian viruses studied here. The five Ukrainian isolates had high genetic similarity among themselves (99.6 %). The isolates that were genetically closest to these seven isolates were duck/Russia/Adygea/12/2008 and turkey/Israel/832 497/2009, with nucleotide sequence identities of 98.7 % and 98.2 %, respectively.
Phylogenetic analysis
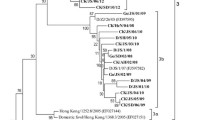
In order to confirm the evolutionary relationship between the NDV isolates studied here and viruses isolated in other geographical regions, a phylogenetic analysis was performed. Other isolates that were closely related to those studied in the present work were selected from the GenBank database and used for a final alignment. The dataset contained 82 complete F gene nucleotide sequences: sub-genotype VIId (n = 37) (including the 21 sequences obtained in this study), the rest of the sub-genotypes in genotype VII (n = 10) and selected isolates representing the rest of the currently classified genotypes of NDV class II (n = 35) (Table S1). A phylogenetic tree based on the complete coding sequences of the fusion protein gene of these 82 NDV isolates is presented in Figure 1. Expectedly, the isolates studied here clustered together with the viruses that showed highest nucleotide sequence identity to them. The fourteen isolates that showed the highest genetic similarity between them (Fig. 1, group 1) formed a monophyletic branch with the two isolates from wild birds in Serbia. They all grouped together with isolates obtained from poultry and wild birds mainly in China during 2001-2013, but also from chickens in Venezuela and Colombia in 2009. The other seven isolates (Fig. 1, group 2) clustered with duck/Russia/Adygea/12/2008 and turkey/Israel/832 497/2009, forming another monophyletic branch in the dendrogram.

Phylogenetic analysis based on the complete nucleotide sequence of the fusion protein gene of isolates representing Newcastle disease virus class II. The evolutionary history was inferred by using the maximum-likelihood method based on general time-reversible model with 500 bootstrap replicates as implemented in MEGA 6 [30]. The tree with the highest log likelihood (-17732.1685) is shown. A discrete Gamma distribution was used to model evolutionary rate differences among sites (4 categories [+G, parameter = 0.6688]). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 39.7514 %). The percentages of trees in which the associated sequences clustered together are shown below the branches (numbers below 60 are not shown). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site.The analysis involved 82 nucleotide sequences with a total of 1651 positions in the final dataset. Bulgarian isolates sequenced in this study are designated with ● in front of the isolate name, Ukrainian isolates are designated with ▲, and the Russian isolate is designated with ■. Evolutionary analysis was conducted in MEGA6 [44]
Discussion
Here, we report the isolation and characterization of virulent Newcastle disease viruses isolated primarily from backyard chickens in Bulgaria and Ukraine and from a pigeon in Ukraine and a wild bird in Russia. Overall, this study allowed the recognition of circulation of vNDV of sub-genotype VIId in Bulgaria and Ukraine during 2002, 2003, 2006-2009, 2011 and 2013 and identification of the most closely related NDV isolates from Serbia and other geographical locations.
We provide here the first complete F gene sequences of NDV of sub-genotype VIId from Bulgaria and Ukraine, confirmed by multiple isolations. In contrast, previous reports describing Newcastle disease outbreaks have identified the circulation of genotypes IV, VI and VIIb in Bulgaria during the 1990s [1, 10, 16]. There were no reports on the genotype distribution of NDV in Ukraine prior to this study. However, partial F gene sequences of viruses of genotypes II and VIIb isolated in the 1990s were available in GenBank database (http://www.ncbi.nlm.nih.gov/genbank). Additionally, prior to 2002, sub-genotypes VIb, VIIa and VIIb were predominantly identified in neighboring Russia and the nearby country of Kazakhstan [7, 18, 36]. No virulent viruses of other genotypes were isolated in Bulgaria during the active surveillance in either commercial or backyard poultry holdings throughout the study period. Our results provide evidence that viruses of sub-genotype VIId replaced the strains that were prevalent in this country before 2002. This conclusion was supported by the results of partial F gene analyses of Bulgarian isolates identified between 2005 and 2007 showing them to be VIId isolates [14, 34]. Similar to our results, after 2002, viruses of sub-genotype VIId were also reported to replace the previously predominant isolates of sub-genotype VIIb in Kazakhstan [7]. Sub-genotype VIId is the predominant group of viruses responsible for most ND outbreaks in Asia since the end of the last century [19]. The more frequent identification of viruses of sub-genotype VIId in Eastern Europe is an example of the western spread of this group of viruses, as closely related strains were also isolated in Romania, Greece and Hungary [5]. Genotype VII represents a large and genetically diverse group of viruses and has been associated with recurrent poultry outbreaks, mainly in the Middle East and Asia [25]. Sporadic events in Africa and South America [1, 7, 40, 49] and the intercontinental spread of recently identified sub-genotype VIIi with panzootic potential [28] demonstrate the global significance and economic importance of these viruses.
Fourteen ND viruses from Bulgaria and Ukraine isolated during 2003, 2006-2009, 2011 and 2013 (Fig. 1, bold) showed a high level of genetic similarity among them and to isolates from wild birds in Serbia in 2007. Such high nucleotide sequence identity (99.3-99.7 %) is indicative of an epidemiological link between the outbreaks or introductions from a common source. These results suggests that these 14 NDV isolates (Fig. 1, group 1) of sub-genotype VIId from Bulgaria and Ukraine may have been part of a broader epizootic process in Eastern Europe rather than separate introductions. This hypothesis also corresponds to the conclusions of Vidanović et al. [46] and Dimitrov [14], as genetically similar viruses have also been recovered from poultry in Macedonia, Serbia and Turkey. The rest of the studied viruses (n = 7, Fig. 1, group 2) were less closely related to these 14 NDV isolates (97.3 % nucleotide sequence identity between the two groups). Considering that both groups of viruses (Fig. 1, groups 1 and 2) were circulating during the same time period, it is not likely that they evolved from each other. It is possible that they represent two separate introduction events of vNDV that evolved elsewhere or that they evolved locally from an unidentified common ancestor introduced earlier in the area.
The means by which these viruses have been maintained (up to 10 years between isolations) is puzzling. The possibilities of maintenance in partially immune poultry or in disease resistant wild birds are viable hypotheses, as most backyard poultry do not receive vaccination. However, it has been suggested that vNDV may be maintained in vaccinated poultry [5, 8, 39], as post-vaccination immunity prevents clinical diseases but not shedding [24, 35]. There are two regularly documented natural reservoirs of virulent NDV (columbids and double-crested cormorants), but they harbor viruses of different genotypes (VI and Va, respectively) [15, 26] and are not likely to be the source of the viruses studied here. The role of other wild birds as a reservoir of virulent NDV of other genotypes is disputable, as evidence supporting this is scarce.
The virulent NDV isolates from Bulgaria, Ukraine and Russia studied here were genetically related to isolates from distant geographical locations in Asia, Israel and Russia. Furthermore, partial nucleotide sequence analysis (374 nt of the fusion protein gene coding sequence) showed a close genetic relationship (98%) of the viruses from Bulgaria and Ukraine to isolates from poultry from Saudi Arabia in 2000 and South Africa in 2004 (data not shown). These findings provide an indication of east-to-west and north-to-south transmission of vNDV. The close genetic relationship between isolates from Bulgaria and Ukraine and those from China, Israel, South Africa and Russia might be explained by migration of wild birds. It has been shown that wild birds play a role in the epidemiology of NDV [4, 47]. Anthropogenic factors such as trade and/or illegal movement of birds and poultry products cannot be excluded as mechanisms of introduction of the studied NDV strains. Although possible, we had no sufficient evidence to support any of these hypotheses. Furthermore, the isolates from wild birds from Serbia were linked to simultaneously ongoing poultry outbreaks in the area [46]. Nevertheless, the presence of ND viruses that are very closely related to isolates from distant geographic locations is supportive of the capacity for high mobility and widespread circulation of this group of viruses.
Overall, our study emphasizes the importance of investigating the epidemiology of NDV and studying the mechanisms for intercontinental spread of the virus. It also underlines the need of continuous monitoring of backyard poultry for the presence of circulating vNDV. Early identification of NDV in backyard poultry followed by recommended control measures [33] could help in the prevention of further spread of new genetic variants. Although vaccination practices help to control the disease in Bulgaria and Ukraine, as evidenced from the absence of outbreaks in industrial poultry holdings, widespread backyard poultry farming, where usually no vaccination is performed, poses a significant threat and provides opportunities for future ND outbreaks.
References
Aldous EW, Mynn JK, Banks J, Alexander DJ (2003) A molecular epidemiological study of avian paramyxovirus type 1 (Newcastle disease virus) isolates by phylogenetic analysis of a partial nucleotide sequence of the fusion protein gene. Avian Pathol 32:239–256
Alexander DJ (1988) NDV—an avian paramyxovirus structure. In: Alexander DJ (ed) Developments in veterinary virology: Newcastle disease. Kluwer, Boston, pp 11–22
Alexander DJ, Swayne DE (1998) Newcastle disease virus and other avian paramyxoviruses. In: Swayne DE, Glisson JR, Jackwood MW, Pearson JE, Reed WM (eds) A laboratory manual for the isolation and identification of avian pathogens. The American Association of Avian Pathologists, Kennett Square, pp 156–163
Alexander DJ, Banks J, Collins MS, Manvell RJ, Frost KM, Speidel EC, Aldous EW (1999) Antigenic and genetic characterisation of Newcastle disease viruses isolated from outbreaks in domestic fowl and turkeys in Great Britain during 1997. Vet Rec 145:417–421
Alexander DJ (2011) Newcastle disease in the European Union 2000 to 2009. Avian Pathol 40:547–558
Beard CW, Hanson RP (1984) Newcastle disease. In: Hofstad MS, Barnes HJ, Calnek BW, Reid WM, Yoder HW (eds) Diseases of poultry. Iowa State University Press, Ames, pp 452–470
Bogoyavlenskiy A, Berezin V, Prilipov A, Usachev E, Lyapina O, Korotetskiy I, Zaitceva I, Asanova S, Kydyrmanov A, Daulbaeva K (2009) Newcastle disease outbreaks in Kazakhstan and Kyrgyzstan during 1998(2000), 2001, 2003. 2004, and 2005 were caused by viruses of the genotypes VIIb and VIId. Virus Genes 39:94–101
Capua I, Dalla PM, Mutinelli F, Marangon S, Terregino C (2002) Newcastle disease outbreaks in Italy during 2000. Vet Rec 150:565–568
Courtney SC, Susta L, Gomez D, Hines NL, Pedersen JC, Brown CC, Miller PJ, Afonso CL (2013) Highly divergent virulent isolates of Newcastle disease virus from the Dominican Republic are members of a new genotype that may have evolved unnoticed for over 2 decades. J Clin Microbiol 51:508–517
Czeglédi A, Herczeg J, Hadjiev G, Doumanova L, Wehmann E, Lomniczi B (2002) The occurrence of five major Newcastle disease virus genotypes (II, IV, V, VI and VIIb) in Bulgaria between 1959 and 1996. Epidemiol Infect 129:679–688
Czeglédi A, Ujvári D, Somogyi E, Wehmann E, Werner O, Lomniczi B (2006) Third genome size category of avian paramyxovirus serotype 1 (Newcastle disease virus) and evolutionary implications. Vir Res 120:36–48
de Almeida RS, Hammoumi S, Gil P, Briand F-X, Molia S, Gaidet N, Cappelle J, Chevalier V, Balanca G, Traore A, Grillet C, Maminiaina OF, Guendouz S, Dakouo M, Samake K, Bezeid OEM, Diarra A, Chaka H, Goutard F, Thompson P, Martinez D, Jestin V, Albina E (2013) New avian paramyxoviruses type I strains identified in Africa provide new outcomes for phylogeny reconstruction and genotype classification. PLoS One 8:76413
Diel DG, da Silva LH, Liu H, Wang Z, Miller PJ, Afonso CL (2012) Genetic diversity of avian paramyxovirus type 1: proposal for a unified nomenclature and classification system of Newcastle disease virus genotypes. Infect Genet Evol 12:1770–1779
Dimitrov KM (2013) Ethiological and molecular-epidemiological studies of Newcastle disease in domestic and wild birds. Dissertation, National Diagnostic and Research Veterinary Medicne Institute, Sofia
Dimitrov KM, Ramey AM, Qiu X, Bahl J, Afonso CL (2016) Temporal, geographic, and host distribution of avian paramyxovirus 1 (Newcastle disease virus). Infect Genet Evol 39:22–34
Herczeg J, Wehmann E, Bragg RR, Dias PMT, Hadjiev G, Werner O, Lomniczi B (1999) Two novel genetic groups (VIIb and VIII) responsible for recent Newcastle disease outbreaks in Southern Africa, one (VIIb) of which reached Southern Europe. Arch Virol 144:2087–2099
Huang Y, Wan HQ, Liu HQ, Wu YT, Liu XF (2004) Genomic sequence of an isolate of Newcastle disease virus isolated from an outbreak in geese: a novel six nucleotide insertion in the non-coding region of the nucleoprotein gene. Brief Report. Arch Virol 149:1445–1457
Korotetskiĭ I, Bogoiavlenskiy A, Prilipov A, Usachev E, Usacheva O, Turgambetova A, Zaĭtseva I, Kydyrmanov A, Shakhvorostova L, Saiatov M (2009) Molecular genetic characteristics of the newcastle disease virus velogenic strains isolated in Russia, Ukraine, Kazakhstan, and Kirghizia. Vop Virusol 55:29–32
Liu H, Wang Z, Son C, Wang Y, Yu B, Zheng D, Sun C, Wu Y (2006) Characterization of pigeon-origin Newcastle disease virus isolated in China. Avian Dis 50:636–640
Liu H, Wang Z, Wang Y, Sun C, Zheng D, Wu Y (2008) Characterization of Newcastle disease virus isolated from waterfowl in China. Avian Dis 52:150–155
Liu XF, Wan HQ, Ni XX, Wu YT, Liu WB (2003) Pathotypical and genotypical characterization of strains of Newcastle disease virus isolated from outbreaks in chicken and goose flocks in some regions of China during 1985–2001. Arch Virol 148:1387–1403
Lomniczi B, Wehmann E, Herczeg J, Ballagi-Pordany A, Kaleta EF, Werner O, Meulemans G, Jorgensen PH, Mante AP, Gielkens AL, Capua I, Damoser J (1998) Newcastle disease outbreaks in recent years in western Europe were caused by an old (VI) and a novel genotype (VII). Arch Virol 143:49–64
Mayo MA (2002) A summary of taxonomic changes recently approved by ICTV. Arch Virol 147:1655–1656
Miller PJ, King DJ, Afonso CL, Suarez DL (2007) Antigenic differences among Newcastle disease virus strains of different genotypes used in vaccine formulation affect viral shedding after a virulent challenge. Vaccine 25:7238–7246
Miller PJ, Decanini EL, Afonso CL (2010) Newcastle disease: evolution of genotypes and the related diagnostic challenges. Infect Genet Evol 10:26–35
Miller PJ, Koch G (2013) Newcastle disease. In: Swayne DE, Glisson JR, McDougald LR, Nolan LK, Suarez DL, Nair V (eds) Diseases of poultry. Wiley-Blackwell, Hoboken, pp 89–138
Miller PJ, Dimitrov KM, Williams-Coplin D, Peterson MP, Pantin-Jackwood MJ, Swayne DE, Suarez DL, Afonso CL (2015) International biological engagement programs facilitate Newcastle disease epidemiological studies. Frontier Public Health 3:235
Miller PJ, Haddas R, Simanov L, Lublin A, Rehmani SF, Wajid A, Bibi T, Khan TA, Yaqub T, Setiyaningsih S, Afonso CL (2015) Identification of new sub-genotypes of virulent Newcastle disease virus with potential panzootic features. Infect Genet Evol 29:216–229
Muzyka D, Pantin-Jackwood M, Stegniy B, Rula O, Bolotin V, Stegniy A, Gerilovych A, Shutchenko P, Stegniy M, Koshelev V, Maiorova K, Tkachenko S, Muzyka N, Usova L, Afonso CL (2014) Wild bird surveillance for avian paramyxoviruses in the Azov-black sea region of Ukraine (2006 to 2011) reveals epidemiological connections with Europe and Africa. Appl Environ Microbiol 80:5427–5438
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, Oxford
Newton I (2010) The migration ecology of birds, 1st edn. Academic Press, New York
OIE (2012) Newcastle disease. Manual of diagnostic tests and vaccines for terrestrial animals: mammals, birds and bees. Biological Standards Commission, World Organization for Animal Health, Paris, pp 555–574
OIE (2016) Terrestrial animal health code. World Organization for Animal Health, Paris
Oreshkova N, Goujhgoulova G, Georgiev G, Dimitrov KM (2009) Molecular approach in investigation of Newcastle disease strains, isolated in Bulgaria during 2005–2007. Trak J Sci 7:69–75
Parede L, Young PL (1990) The pathogenesis of velogenic Newcastle disease virus infection of chickens of different ages and different levels of immunity. Avian Dis 34:803–808
Pchelkina IP, Manin TB, Kolosov SN, Starov SK, Andriyasov AV, Chvala IA, Drygin VV, Yu Q, Miller PJ, Suarez DL (2013) Characteristics of pigeon paramyxovirus serotype-1 isolates (PPMV-1) from the Russian Federation from 2001 to 2009. Avian Dis 57:2–7
Perozo F, Marcano R, Afonso CL (2012) Biological and phylogenetic characterization of a genotype VII Newcastle disease virus from Venezuela: efficacy of field vaccination. J Clin Microbiol 50:1204–1208
Peterson EB, Orden D (2005) Effects of tariffs and sanitary barriers on high-and low-value poultry trade. J Agr Resour Econ 30:109–127
Rehmani SF, Wajid A, Bibi T, Nazir B, Mukhtar N, Hussain A, Lone NA, Yaqub T, Afonso CL (2015) Presence of virulent Newcastle disease virus in vaccinated chickens in farms in Pakistan. J Clin Microbiol 53:1715–1718
Snoeck CJ, Ducatez MF, Owoade AA, Faleke OO, Alkali BR, Tahita MC, Tarnagda Z, Ouedraogo JB, Maikano I, Mbah PO, Kremer JR, Muller CP (2009) Newcastle disease virus in West Africa: new virulent strains identified in non-commercial farms. Arch Virol 154:47–54
Snoeck CJ, Owoade AA, Couacy-Hymann E, Alkali BR, Okwen MP, Adeyanju AT, Komoyo GF, Nakouné E, Le Faou A, Muller CP (2013) High genetic diversity of Newcastle disease virus in poultry in West and Central Africa: cocirculation of genotype XIV and newly defined genotypes XVII and XVIII. J Clin Microbiol 51:2250–2260
Susta L, Hamal KR, Miller PJ, Cardenas-Garcia S, Brown CC, Pedersen JC, Gongora V, Afonso CL (2014) Separate evolution of virulent Newcastle disease viruses from Mexico and Central America. J Clin Microbiol 52:1382–1390
Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA 101:11030–11035
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Vidanovic D, Šekler M, Ašanin R, Milic N, Nišavic J, Petrovic T, Savic V (2011) Characterization of velogenic Newcastle disease viruses isolated from dead wild birds in Serbia during 2007. J Wildl Dis 47:433–441
Wan HQ, Chen LG, Wu LL, Liu XF (2004) Newcastle disease in geese: natural occurrence and experimental infection. Avian Pathol 33:216–221
Wang Y, Duan Z, Hu S, Kai Y, Wang X, Song Q, Zhong L, Sun Q, Wang X, Wu Y (2012) Lack of detection of host associated differences in Newcastle disease viruses of genotype VIId isolated from chickens and geese. Virol J 9:1–15
Wang Z, Liu H, Xu J, Bao J, Zheng D, Sun C, Wei R, Song C, Chen J (2006) Genotyping of Newcastle disease viruses isolated from 2002 to 2004 in China. Ann NY Acad Sci 1081:228–239
Yi J, Liu C, Chen B, Wu S (2011) Molecular characterization of a virulent genotype VIId strain of Newcastle disease virus from farmed chickens in Shanghai. Avian Dis 55:279–284
Yu L, Wang Z, Jiang Y, Chang L, Kwang J (2001) Characterization of newly emerging Newcastle disease virus isolates from the People’s Republic of China and Taiwan. J Clin Microbiol 39:3512–3519
Acknowledgements
This work was supported by the Defense Threat Reduction Agency, BAA project FRCALL12-6-3412-0005, and by the USDA, ARS CRIS Project 6040-32000-064. The authors declare that they have no conflict of interest. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer. This manuscript was improved by comments from an editor and an anonymous reviewer.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dimitrov, K.M., Bolotin, V., Muzyka, D. et al. Repeated isolation of virulent Newcastle disease viruses of sub-genotype VIId from backyard chickens in Bulgaria and Ukraine between 2002 and 2013. Arch Virol 161, 3345–3353 (2016). https://doi.org/10.1007/s00705-016-3033-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-3033-2