
Abstract
Toll-like receptor 3 (TLR3) is a critical component of the innate immune system against viral infection and controls the activation of adaptive immunity. The role of TLR3 in Marek’s disease virus (MDV) infection is not clear. In this study, we found that the abundance of TLR3 mRNA was significantly higher in chicken embryo fibroblast cells (CEF) infected with MDV than in a control group. Activated TLR3 signaling via TLR3 ligand stimulation inhibited replication of the RB1B strain of MDV in CEF cells. In contrast, CEF cells transfected with TLR3 siRNA promoted RB1B infection and replication. However, treatment with other TLR ligands, whether stimulatory (LPS, imiquimod and CpG) or inhibitory (TLR2/4 inhibitor and/or MyD88 inhibitor), had little effect on RB1B infection and replication. In addition, we found that the expression trend of TLR3 mRNA in RB1B-infected CEF cells was similar to that of mdv1-mir-M4-5p (a functional ortholog of oncogenic miR-155 encoded by MDV). Inconsistent with this, the TLR3 protein level was sharply reduced in RB1B-infected CEF cells at 96 hpi, while there was an at least 200-fold increase in miR-M4-5p at the same time point. Additionally, CEF cells transfected with an mdv1-mir-M4-5p mimic promoted RB1B infection and replication, while an mdv1-mir-M4-5p inhibitor inhibited RB1B infection and replication. Similar results were observed in CEF cells transfected with a gga-miR-155 mimic or inhibitor. These findings suggest that TLR3 and MDV-encoded miRNAs might be involved in MDV infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Toll-like receptor 3 (TLR3) is a critical component of the innate immune system against viral infections that controls the activation of adaptive immunity [3, 27]. TLR3 strongly enhances antigen-specific CD8+ T-cell responses and promotes antigen cross-priming against virus-infected cells [5, 28, 32]. TLR3 innate immunity is involved in immunity to several herpesviruses, including Kaposi’s sarcoma-associated herpesvirus (KSHV) [35], herpes simplex virus (HSV) [5] and Marek’s disease virus (MDV) [11]. The TLR3 pathway is upregulated by KSHV during primary infection [35]. Moreover, activation of the TLR3-TRIF pathway enhances KSHV replication and transcription activator (RTA) protein expression, and in turn, KSHV RTA degrades TRIF, thus blocking innate immunity [2, 21]. TLR3 is required for the generation of CD8 T-cell immunity to HSV-1, and intrinsic immunity to HSV-1 has been shown to be impaired in human iPSC-derived TLR3-deficient CNS cells [5, 17]. Recent evidence suggests a role for TLR3 in MDV infection, and MDV may interfere with T-cell immunity by TLR3 signaling [11]. TLR3 has been shown to be upregulated in chicken lungs infected with MDV [1]. In response to MDV infection, TLR3 expression was enhanced in chicken spleens at 5 dpi [8]. Another study revealed that a TLR3 ligand enhanced the efficacy of the HVT vaccine, improved protection against MDV, and hindered tumor development in chickens [25].
MDV is an oncogenic alphaherpesvirus that provides a natural model for lymphomas, particularly with respect to the pathogenesis and immune control of virus-induced lymphoma [24]. To date, 26 MDV-1 miRNAs have been identified [9], and they are located in the oncogene MEQ cluster and the latency-associated transcript (LAT) cluster. Some of these miRNAs have been reported to contribute to viral latency and oncogenesis. One MDV-encoded miRNA, miR-M3, was shown to suppress apoptosis by targeting Smad2 in the transforming growth factor beta (TGFb) signaling pathway [36], and another MDV-encoded miRNA, mdv1-miR-M7-5p, was implicated in the establishment and maintenance of latency by targeting the immediate-early genes ICP4 and ICP27 [31]. In particular, mdv1-miR-M4, a functional ortholog of oncogenic miR-155, was shown to have an essential role in the induction of MD lymphomas [41]. Expression analysis of miR-M4 confirmed its high expression in MD tumors, and an miR-M4 null mutant RB1B MDV-1 virus was unable to transform infected T-cells. Another report showed that miR-M4 is an important potential regulator but is not essential for the development of lymphomas induced by highly virulent MDV [38]. These facts suggest that MDV-encoded miRNAs are likely to be critical regulators of pathways associated with the lytic/latent switch and viral transformation.
In a previous study, we found that miR-155 can directly bind to the coding region of TLR3 and negatively regulate TLR3 expression [12]. Moreover, mdv1-miR-M4 can inhibit TLR3 expression. It was confirmed that miR-M4 has an essential role in the induction of MD lymphomas and that it has a high level of expression in MD tumors [41]. The miR-M4 null mutant RB1B MDV-1 cannot transform infected T cells. TLR3 is expressed in T cells, and this has implications for antiviral defense and tumor suppression. Thus, we infer that TLR3 may be involved in MDV infection. Here, we investigated the influence of TLR3 on RB1B infection and replication.
Materials and methods
Reagents
The TLR2 ligand ultrapure E. coli 0111:B4 peptidoglycan (PGN-EB), the TLR3 ligand synthetic analogue of dsRNA poly (I:C) with a high molecular weight, the TLR4 ligand ultrapure E. coli 0111:B4 lipopolysaccharide (LPS-EB), and the TLR7 ligand small synthetic antiviral molecule imiquimod (R837) were all from InvivoGen (California, USA). Lipofectamine RNAiMax transfection reagent was from Invitrogen (Life Technologies, MD, USA).
The gga-mir-155 and mdv1-mir-M4-5p mimics, inhibitors and controls were synthesized by Ribobio (Guangzhou RiboBio Co., Ltd, Guangzhou, China). Small interfering RNA (siRNA) and Lipofectamine RNAiMax transfection reagent were purchased from Invitrogen (Life Technologies, MD, USA). Stealth RNAi™ siRNAs specific for chicken TLR3 and negative control Stealth RNAi™ siRNA were designed and synthesized by Invitrogen (Life Technologies, MD, USA). The sequences of the Stealth siRNAs as follows: Chicken TLR3_Stealth_218, 5′-CCGAGUACAGCAAUCUGAUUUACUU-3′ and 5′-AAGUAAAUCAGAUUGCUGUACUCGG-3′; Chicken TLR3_stealth_482, 5′-CAGCAAAUUUAGGAUUGCAGCAACA-3′ and 5′-UGUUGCUGCAAUCCUAAAUUUGCUG-3′; Chicken TLR3_stealth_2507, 5′-GAGACUCCAUCAUACUGAUCUUUCU-3′ and 5′-AGAAAGAUCAGUAUGAUGGAGUCUC-3′.
Cells and virus
Primary chicken embryo fibroblast cells (CEF) were prepared from 10-day-old specific-pathogen-free (SPF) embryos obtained from Merial Vital (Laboratory Animal Technology CO., Ltd., Beijing, China), and secondary CEF cells were used for virus infection or other experiments. RB1B strains of highly virulent MDV were maintained in the laboratory.
Virus infection
Secondary CEF cells were seeded in 6-well plates in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies/GIBCO, MD, USA) supplemented with 5 % fetal bovine serum (FBS) at 37 °C (5 % CO2 and 95 % humidity). After 24 hours, the cells were infected with strain RB1B at a multiplicity of infection (MOI) of 0.1. Each virus was placed into three wells and collected at 8, 24, 72, 96 and 120 hours postinfection (hpi).
TLR stimulation and inhibition
Secondary CEF cells were seeded in 6-well plates in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies/GIBCO, MD, USA) supplemented with 5 % fetal bovine serum (FBS) at 37 °C (5 % CO2 and 95 % humidity) and incubated for 16-24 h in medium containing select TLR ligands or inhibitors. After incubation, the supernatants were collected, clarified and stored, and the cells were washed and used for infection or additional tests.
Oligonucleotide transfection
Oligonucleotide transfection was performed with Lipofectamine RNAiMax transfection reagent according to the manufacturer’s instructions. siRNAs were diluted 10 times to a final concentration of 200 nM.
RNA extraction and quantitative real-time PCR
Total RNA was extracted using an miRNeasy Mini Kit (QIAGEN, Hilden, Germany), and mature miRNAs were reverse transcribed using a miScript II RT Kit (QIAGEN, Hilden, Germany) and amplified using a miScript SYBR Green PCR Kit (QIAGEN, Hilden, Germany). miScript Primer Assays (QIAGEN, Hilden, Germany) were used for gga-mir-155 (assay ID: MSC0003997), mdv1-mir-M4 (assay ID: MSC0003997), and gga-mir-21 (assay ID: MSC0003998) in 96-well plates (Life Technologies, MD, USA) on an ABI 7500 real-time PCR system (Life Technologies, MD, USA). Gene expression was calculated relative to small ncRNAU6 levels. Real-time PCR was performed on the MDV and other host genes (the primer sequences are provided in Table 1) as reported previously [10, 11], and gene expression levels were normalized to chicken 18S.
Immunoblot analysis
Cells (5 × 105 to 10 × 105) were lysed in Cell Lysis Buffer (10X) (9803, Cell Signaling Technologies, MA, USA) with protease inhibitors added. The immunoblot experiment was performed as described previously [12]. The samples were loaded with 5× denaturing sample buffer and separated by 12 % SDS-PAGE. The proteins were transferred to polyvinylidene difluoride membranes using standard techniques and were subsequently analyzed by immunoblotting with relevant antibodies. The blots were developed using chemiluminescence (protein simple, Fluorchem E FE0605). The monoclonal antibody used to detect β-actin was from Santa Cruz Biotechnology (sc-47778, Dallas, USA), and the polyclonal antibody against TLR3 was from Novus Biologicals (NBP2-24565, Littleton, USA).
Statistical analysis
Statistical analysis was performed with either the Statistical Package for the Social Sciences (SPSS version 16.0) or Excel GraphPad (Prism 5) software. P-values were determined by paired Student’s t-tests or unpaired tests for normal distributions of at least three independent experiments.
Results
High expression of TLR3 induced by RB1B infection in CEF cells
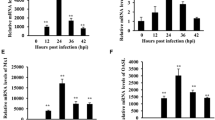
We investigated the dynamic expression patterns of Toll-like receptors (2, 3, 4, 7, 15 and 21) in RB1B-infected CEF cells at 8, 24, 72, 96 and 120 hours postinfection. In infected CEF cells, we found that only TLR3 mRNA expression was abnormally higher than in the uninfected control group, whereas TLR2, TLR4, TLR7, TLR15 and TLR21 showed no change (Fig. 1). The most significant change in TLR3 mRNA expression was observed at 96 hpi (at least a 40-fold change).

Relative expression of Toll-like receptors in RB1B-infected CEF cells. Expression of TLR2A, TLR3, TLR4, TLR7, TLR15 and TLR21 genes in CEF cells at 8, 24, 72, 96 and 120 hours after infection with strain RB1B
Influence of TLR3 on RB1B infection
We found that stimulation of CEF cells with poly(I:C) consistently inhibited MDV infection and replication (Fig. 2A and B), while CEF cells treated with LPS, Imiquimod and CpG behaved similarly to the unstimulated groups. Similar results were observed in plaque assays (Fig. 2C). Next, treatment with inhibitors of other TLR pathways except for TLR3 (TLR2/4 inhibitor and/or MyD88 inhibitor) had little effect on RB1B infection and replication (Fig. 2D). In contrast, we found that, based on viral gene expression (gB and Meq), infection and replication of RB1B virus was enhanced 24 h after MDV infection when CEF cells were transfected with TLR3 siRNA compared to control siRNA (Fig. 2E-G).

Influence of TLR3 on MDV infection and replication. A: Expression of gB in CEF cells stimulated with TLR ligands at 24, 72, 96 and 120 hours after infection with strain RB1B. B: Expression of Meq in CEF cells stimulated with TLR ligands at 24, 72, 96 and 120 hours after infection with strain RB1B. C: Plaque-forming units of RB1B in unstimulated CEF cells. D: Plaque-forming units of RB1B in CEF cells stimulated with TLR ligands. E: Plaque-forming units of RB1B in CEF cells stimulated with TLR ligands. F: Relative expression of viral genes (gB and Meq) in CEF cells treated with an inhibitor. G: Expression of TLR3 in CEF cells transfected with TLR3 siRNA or control siRNA. H: Expression of viral genes (gB and Meq) in CEF cells transfected with TLR3 siRNA or control siRNA. I: Plaque-forming units of RB1B in CEF cells transfected with TLR3 siRNA or control siRNA
mdv1-miR-M4-5p is involved in regulation of TLR3 expression
We observed the expression kinetics of mdv1-miR-M4-5p and TLR3 during RB1B infection. Infection with RB1B resulted in the production of IFN-β and increased expression of gga-miR-155 from 8 hpi to 120 hpi, but it did not affect the expression of gga-miR-21 (a control miRNA). However, mdv1-miR-M4-5p showed a different expression trend in MDV-infected CEF cells; mdv1-miR-M4-5p was sharply downregulated from 96 hpi to 120 hpi, while the virus level continued to increase (Fig. 3A-F). Notably, TLR3 mRNA levels were also lower from 96 hpi to 120 hpi, similar to those of mdv1-mir-M4-5p. Inconsistent with the change in TLR3 mRNA levels, we found that the expression of TLR3 gene at the protein level was sharply reduced in RB1B-infected CEF cells at 96 hpi (Fig. 3G). In addition, we also observed that the TLR3 protein level was clearly increased at 72 hpi, following the change in TLR3 mRNA levels.

Control of TLR3 by MDV encoding miR-155. Relative expression of gB (A), Meq (B), mdv1-miR-M4-5p (C), gga-miR-155 (D), IFN-β (E) or TLR3 (F) in CEF cells at 8, 24, 72, 96 and 120 hours after infection with strain RB1B. G: Expression level of TLR3 protein in CEF cells at 72 and 96 hours after infection with strain RB1B. H-I: Expression of gB and Meq in CEF cells stimulated with gga-miR-155 or a mdv1-miR-M4-5p mimic at 24 or 48 hours after infection with strain RB1B. J-K: Expression of gB and Meq in CEF cells stimulated with gga-miR-155 or mdv1-miR-M4-5p inhibitor at 24 or 48 hours after infection with strain RB1B
Next, we transfected CEF cells with an mdv1-mir-M4-5p mimic or mimic control and then infected CEF cells with RB1B 48 hours after transfection. This treatment enhanced the expression levels of gB and Meq (indicating virus replication) 24 and 48 hours after infection (Fig. 3H and I). In contrast, this effect was impaired in CEF cells transfected with an mdv1-mir-M4-5p inhibitor, which significantly suppressed replication of the virus (Fig. 3J and K). Similar results were observed in CEF cells transfected with a gga-miR-155 mimic or inhibitor (Fig. 3H-3K).
Discussion
In a previous study, we showed that MDV miR-M4-5p targeted the TLR3 coding region, which resulted in the downregulation of the TLR3 protein [12]. In the present study, we found that only TLR3 mRNA expression was abnormally higher than in the control group; activation of TLR3 with poly(I:C) consistently suppressed RB1B infection and replication, but inhibition of TLR3 enhanced infection. It has demonstrated that TLR3 is upregulated upon infection in CEF cells and that TLR3 stimulation inhibits MDV replication. Although CEFs are not the natural target cells of MDV in vivo, the effects observed in MDV-infected CEF might reflect the host response to virulent MDV infection. However, this should be investigated for B and T cells infected with MDV in the future.
TLR3 is critical for establishing an immune response to viral infection. TLR3 deficiency in human CNS cells affects intrinsic immunity and the generation of CD8 T-cell immunity to HSV-1 infection [39]. Recent evidence revealed a role for TLR3 in MDV infection. In response to MDV infection, the expression of TLR3 was enhanced in the chicken lung [1], spleen [8] and thymus [11] during early virus infection. A TLR3 ligand enhanced the efficacy of the HVT vaccine, improved protection against MDV, and hindered tumor development in chickens [25]. We confirmed that the activation of TLR3 inhibits replication and infection by MDV. On the basis of published studies and the findings of this study, we conclude that TLR3 immunity interferes with MDV viral replication and infection.
In addition to its involvement in antiviral immunity, TLR3 plays a key role in tumor suppression [16]. Several studies have indicated that the tumor suppressor p53 positively regulates TLR3 transcription by binding to the p53 site in the TLR3 promoter, and TLR3 induction is p53 dependent in several tumor cell lines [26, 29, 33, 34]. Obviously, p53 is an obstacle to the induction of MD tumors that must be overcome. A study showed that the oncogenic MEQ protein can directly inhibit the expression and transcriptional activity of p53 [6]. MEQ could suppress the host anti-tumor immune responses by inhibiting p53 and thus interfere with TLR3 transcription. In fact, deletion of the Meq gene significantly decreased MDV-mediated immune suppression in chickens [18]. Therefore, we infer that the expression of TLR3 can hinder MDV-induced T-cell lymphoma transformation and could regulate survival of the virus. This hypothesis was supported by a report [15] that the function of TLR3 in lymphoid tissues was impaired when the infection entered the tumor transformation phase.
In this study, we found that TLR3 inhibition by RNA interference technology promoted MDV infection and replication. Inhibition of the TLR3 pathway was observed in the MDV-infected chicken thymus at 21 dpi; moreover, there was a positive correlation between the downregulation of CD4, CD8 and TLR3 signals in response to MDV infection [11]. Avoiding the host immune response is particularly important if MDV is to establish a lifelong latent infection, and a sufficient number of latently infected cells must be generated to reliably induce lymphoma [24]. UL49.5 is involved in the downregulation of MHC class I molecules in MDV-infected cells, while deletion of the Meq gene significantly decreases MDV-mediated immune suppression in chickens [13, 18]. Viral miRNAs are not immunogenic and thus might offer a better option for attenuating the antiviral immune responses [4, 23]. Seed sites for mdv1-miR-M4, mdv1-miR-M8 and mdv1-miR-M11 were found in the TLR3 coding region, suggesting that MDV has at least three miRNA-based mechanisms to regulate TLR3 expression. Among these miRNAs, mdv1-miR-M4 was highly expressed in MDV-induced tumors, which suggests that it is an important potential regulator that could affect the occurrence of MD tumors [38] and TLR3 expression.
In a previous study, we demonstrated that mdv-miR-M4 can suppress TLR3 immunity by targeting the coding region of the TLR3 gene [12]. In this study, we also found that the expression of TLR3 gene at the protein level was sharply reduced in infected CEF cells at 96 hpi. In contrast, miR-M4-5p was increased at least 200-fold at this time point. MDV-miR-M4 is a functional homolog of oncogenic miR-155, which has been reported to rescue the oncogenic function in a miR-M4-deleted virus [41]. This viral miRNA dominates the expression profile in tumor samples (72 % of MDV1 miRNAs are mdv1-miR-M4). Interestingly, overexpression of miR-155 was found in most tumors, while downregulation of miR-155 was seen in MDV-transformed cell lines and MD tumors [22, 37]. The downregulation of miR-155 in MDV-transformed tumor cell lines was not permanent, as these cells could be induced to express miR-155 [41]. Downregulation of miR-155 was also found in KSHV-transformed tumors that express high levels of miR-K12-11, the functional miR-155 ortholog encoded by KSHV [30]. In contrast, EBV induced miR-155 expression in transformed cell lines and tumors but did not encode an miR-155 ortholog [14, 19, 20]. These observations suggest that different oncogenic herpesviruses have evolved alternative strategies that either supply miR-155-like activities by encoding functional orthologs of miR-155 or induce the expression of cellular miR-155 for viral tumorigenesis [7, 20, 40]. The high expression level of mdv1-miR-M4 leads us to conclude that it can act in the role of gga-miR-155. In this study, we observed that an mdv1-mir-M4-5p mimic promoted the infection and replication of MDV in CEF cells, while the cells were suppressed by an mdv1-mir-M4-5p inhibitor. The same results were observed in CEF cells transfected with a gga-miR-155 mimic or inhibitor. In view of these observations, we suggest that mdv-miR-M4 is primarily responsible for the suppression of TLR3 immunity.
In conclusion, our study shows that activation of TLR3 can inhibit MDV infection and replication, while TLR3 knockdown by RNAi promotes infection and replication. We also observed that mdv1-miR-M4 has a specific effect on TLR3 expression and MDV infection and replication. Overexpression of mdv1-miR-M4 promotes MDV infection and replication, while MDV infection is inhibited by an mdv1-miR-M4 inhibitor. Although we do not yet understand how MDV controls miR-155 expression, it is most likely associated with a feedback regulatory mechanism of TLR3 immunity that will require further research.
References
Abdul-Careem MF, Haq K, Shanmuganathan S, Read LR, Schat KA, Heidari M, Sharif S (2009) Induction of innate host responses in the lungs of chickens following infection with a very virulent strain of Marek’s disease virus. Virology 393:250–257
Ahmad H, Gubbels R, Ehlers E, Meyer F, Waterbury T, Lin R, Zhang L (2011) Kaposi sarcoma-associated herpesvirus degrades cellular Toll-interleukin-1 receptor domain-containing adaptor-inducing beta-interferon (TRIF). J Biol Chem 286:7865–7872
Beutler B (2004) Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430:257–263
Cullen BR (2009) Viral and cellular messenger RNA targets of viral microRNAs. Nature 457:421–425
Davey GM, Wojtasiak M, Proietto AI, Carbone FR, Heath WR, Bedoui S (2010) Cutting edge: priming of CD8 T cell immunity to herpes simplex virus type 1 requires cognate TLR3 expression in vivo. J Immunol 184:2243–2246
Deng X, Li X, Shen Y, Qiu Y, Shi Z, Shao D, Jin Y, Chen H, Ding C, Li L, Chen P, Ma Z (2010) The Meq oncoprotein of Marek’s disease virus interacts with p53 and inhibits its transcriptional and apoptotic activities. Virol J 7:348
Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, Braich R, Manoharan M, Soutschek J, Ohler U, Cullen BR (2007) A viral microRNA functions as an orthologue of cellular miR-155. Nature 450:1096–1099
Heidari M, Sarson AJ, Huebner M, Sharif S, Kireev D, Zhou H (2010) Marek’s disease virus-induced immunosuppression: array analysis of chicken immune response gene expression profiling. Viral Immunol 23:309–319
Hicks JA, Liu HC (2013) Current state of Marek’s disease virus microRNA research. Avian Dis 57:332–339
Hu X, Qin A, Qian K, Shao H, Yu C, Xu W, Miao J (2012) Analysis of protein expression profiles in the thymus of chickens infected with Marek’s disease virus. Virol J 9:256
Hu X, Xu W, Qin A, Wu G, Qian K, Shao H, Ye J (2014) Marek’s disease virus may interfere with T cell immunity by TLR3 signals. Vet Res Commun 38:149–156
Hu X, Ye J, Qin A, Zou H, Shao H, Qian K (2015) Both microRNA-155 and virus-encoded MiR-155 ortholog regulate TLR3 expression. PLoS ONE 10:e0126012
Jarosinski KW, Hunt HD, Osterrieder N (2010) Down-regulation of MHC class I by the Marek’s disease virus (MDV) UL49.5 gene product mildly affects virulence in a haplotype-specific fashion. Virology 405:457–463
Jiang J, Lee EJ, Schmittgen TD (2006) Increased expression of microRNA-155 in Epstein–Barr virus transformed lymphoblastoid cell lines. Genes Chromosomes Cancer 45:103–106
Jie H, Lian L, Qu LJ, Zheng JX, Hou ZC, Xu GY, Song JZ, Yang N (2013) Differential expression of Toll-like receptor genes in lymphoid tissues between Marek’s disease virus-infected and noninfected chickens. Poultry Sci 92:645–654
Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, Appukuttan B, Gibbs D, Yang Z, Kariko K, Ambati BK, Wilgus TA, DiPietro LA, Sakurai E, Zhang K, Smith JR, Taylor EW, Ambati J (2008) Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 452:591–597
Lafaille FG, Pessach IM, Zhang SY, Ciancanelli MJ, Herman M, Abhyankar A, Ying SW, Keros S, Goldstein PA, Mostoslavsky G, Ordovas-Montanes J, Jouanguy E, Plancoulaine S, Tu E, Elkabetz Y, Al-Muhsen S, Tardieu M, Schlaeger TM, Daley GQ, Abel L, Casanova JL, Studer L, Notarangelo LD (2012) Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 491:769–773
Li Y, Sun A, Su S, Zhao P, Cui Z, Zhu H (2011) Deletion of the Meq gene significantly decreases immunosuppression in chickens caused by pathogenic Marek’s disease virus. Virol J 8:2
Linnstaedt SD, Gottwein E, Skalsky RL, Luftig MA, Cullen BR (2010) Virally induced cellular microRNA miR-155 plays a key role in B-cell immortalization by Epstein–Barr virus. J Virol 84:11670–11678
Lu F, Weidmer A, Liu CG, Volinia S, Croce CM, Lieberman PM (2008) Epstein–Barr virus-induced miR-155 attenuates NF-kappaB signaling and stabilizes latent virus persistence. J Virol 82:10436–10443
Meyer F, Ehlers E, Steadman A, Waterbury T, Cao M, Zhang L (2013) TLR-TRIF pathway enhances the expression of KSHV replication and transcription activator. J Biol Chem 288:20435–20442
Morgan R, Anderson A, Bernberg E, Kamboj S, Huang E, Lagasse G, Isaacs G, Parcells M, Meyers BC, Green PJ, Burnside J (2008) Sequence conservation and differential expression of Marek’s disease virus microRNAs. J Virol 82:12213–12220
Murphy E, Vanicek J, Robins H, Shenk T, Levine AJ (2008) Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc Natl Acad Sci USA 105:5453–5458
Osterrieder N, Kamil JP, Schumacher D, Tischer BK, Trapp S (2006) Marek’s disease virus: from miasma to model. Nat Rev Microbiol 4:283–294
Parvizi P, Mallick AI, Haq K, Haghighi HR, Orouji S, Thanthrige-Don N, St Paul M, Brisbin JT, Read LR, Behboudi S, Sharif S (2012) A Toll-like receptor 3 ligand enhances protective effects of vaccination against Marek’s disease virus and hinders tumor development in chickens. Viral Immunol 25:394–401
Rivas C, Aaronson SA, Munoz-Fontela C (2010) Dual role of p53 in innate antiviral immunity. Viruses 2:298–313
Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R (2001) Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2:947–950
Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljestrom P, Reis e Sousa C (2005) Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 433:887–892
Shatz M, Menendez D, Resnick MA (2012) The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res 72:3948–3957
Skalsky RL, Samols MA, Plaisance KB, Boss IW, Riva A, Lopez MC, Baker HV, Renne R (2007) Kaposi’s sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol 81:12836–12845
Strassheim S, Stik G, Rasschaert D, Laurent S (2012) mdv1-miR-M7-5p, located in the newly identified first intron of the latency-associated transcript of Marek’s disease virus, targets the immediate-early genes ICP4 and ICP27. J Gen Virol 93:1731–1742
Tabiasco J, Devevre E, Rufer N, Salaun B, Cerottini JC, Speiser D, Romero P (2006) Human effector CD8+ T lymphocytes express TLR3 as a functional coreceptor. J Immunol 177:8708–8713
Taura M, Eguma A, Suico MA, Shuto T, Koga T, Komatsu K, Komune T, Sato T, Saya H, Li JD, Kai H (2008) p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol Cell Biol 28:6557–6567
Taura M, Fukuda R, Suico MA, Eguma A, Koga T, Shuto T, Sato T, Morino-Koga S, Kai H (2010) TLR3 induction by anticancer drugs potentiates poly I:C-induced tumor cell apoptosis. Cancer Sci 101:1610–1617
West J, Damania B (2008) Upregulation of the TLR3 pathway by Kaposi’s sarcoma-associated herpesvirus during primary infection. J Virol 82:5440–5449
Xu S, Xue C, Li J, Bi Y, Cao Y (2011) Marek’s disease virus type 1 microRNA miR-M3 suppresses cisplatin-induced apoptosis by targeting Smad2 of the transforming growth factor beta signal pathway. J Virol 85:276–285
Yao Y, Zhao Y, Smith LP, Lawrie CH, Saunders NJ, Watson M, Nair V (2009) Differential expression of microRNAs in Marek’s disease virus-transformed T-lymphoma cell lines. J Gen Virol 90:1551–1559
Yu ZH, Teng M, Sun AJ, Yu LL, Hu B, Qu LH, Ding K, Cheng XC, Liu JX, Cui ZZ, Zhang GP, Luo J (2014) Virus-encoded miR-155 ortholog is an important potential regulator but not essential for the development of lymphomas induced by very virulent Marek’s disease virus. Virology 448:55–64
Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L, Leonard J, Hamilton C, Lebon P, Heron B, Vallee L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova JL (2007) TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527
Zhao Y, Yao Y, Xu H, Lambeth L, Smith LP, Kgosana L, Wang X, Nair V (2009) A functional MicroRNA-155 ortholog encoded by the oncogenic Marek’s disease virus. J Virol 83:489–492
Zhao Y, Xu H, Yao Y, Smith LP, Kgosana L, Green J, Petherbridge L, Baigent SJ, Nair V (2011) Critical role of the virus-encoded microRNA-155 ortholog in the induction of Marek’s disease lymphomas. PLoS Pathog 7:e1001305
Acknowledgments
This research was supported by the National Natural Science Foundation of China (31472192) and the Priority Academic Program Development of Jiangsu Higher Education Institutions and the Science.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hu, X., Zou, H., Qin, A. et al. Activation of Toll-like receptor 3 inhibits Marek’s disease virus infection in chicken embryo fibroblast cells. Arch Virol 161, 521–528 (2016). https://doi.org/10.1007/s00705-015-2674-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2674-x