
Abstract
Bats are an important natural reservoir for a variety of viral pathogens, including polyomaviruses (PyVs). The aims of this study were: (i) to determine which PyVs are present in bats in Indonesia and (ii) to analyze the evolutionary relationships between bat PyVs and other known PyVs. Using broad-spectrum polymerase chain reaction (PCR)-based assays, we screened PyV DNA isolated from spleen samples from 82 wild fruit bats captured in Indonesia. Fragments of the PyV genome were detected in 10 of the 82 spleen samples screened, and eight full-length viral genome sequences were obtained using an inverse PCR method. A phylogenetic analysis of eight whole viral genome sequences showed that BatPyVs form two distinct genetic clusters within the proposed genus Orthopolyomavirus that are genetically different from previously described BatPyVs. Interestingly, one group of BatPyVs is genetically related to the primate PyVs, including human PyV9 and trichodysplasia spinulosa-associated PyV. This study has identified the presence of novel PyVs in fruit bats in Indonesia and provides genetic information about these BatPyVs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polyomaviruses (PyVs) are small DNA viruses with a genome of approximately 5 kbp. They belong to the family Polyomaviridae. The viral genome is composed of three parts: the noncoding control region (NCCR), the early region, and the late region. The NCCR controls bidirectional transcription from both the early and late promoters and also contains the origin of viral DNA replication. The early region contains genes coding for regulatory proteins, known as tumor antigens, including a large T-antigen (T-Ag) and a small t-antigen (t-Ag). The late region encodes the structural proteins VP1, VP2, and VP3. Some primate PyVs also encode a small highly basic protein known as agnoprotein.
With the development of molecular diagnostic techniques, new PyVs have been identified recently in various animals [7, 16, 22, 24]. The Polyomaviridae Study Group of the International Committee on Taxonomy of Viruses (ICTV) has recommended that family Polyomaviridae be divided into three genera [7]. The proposed genus of avian-hosted PyVs is named Avipolyomavirus, and the divergence of members of this genus from the mammalian-hosted PyVs is based on both biological and genomic differences [3, 7]. The two proposed genera of mammalian-hosted PyVs are designated Wukipolyomavirus and Orthopolyomavirus, and these genera are divided based on nucleotide sequence divergence [3, 7]. The genus Wukipolyomavirus includes four human viruses: WU polyomavirus (WUPyV), KI polyomavirus (KIPyV), human polyomavirus 6 (HPyV6), and human polyomavirus 7 (HPyV7), whereas the proposed genus Orthopolyomavirus includes a variety PyVs from primates, humans, cattle, rodents, and bats. Some of these PyVs cause subclinical infections with life-long persistence in their natural immunocompetent hosts, but they can also reactivate and cause diseases in immunocompromised hosts [9, 10, 24].
Various PyV genomes have been detected in bats [3, 15, 22], which are known to harbor and transmit a variety of emerging viruses to other mammals [4, 12, 14]. The bat PyV (BatPyV) and primate PyV groups tend to form monophyletic groups in phylogenetic trees [22]. One group of BatPyVs is genetically related to the human Merkel cell PyV, which has been implicated in the pathogenesis of Merkel cell carcinoma [22]. These findings led us to hypothesize that BatPyVs may be important for the maintenance and transmission of primate PyVs, including human PyVs. In this study, to verify our hypothesis, we investigated the presence of PyVs in bats in Indonesia using molecular sequencing and phylogenetic analysis.
Materials and methods
Sample collection and DNA extraction
A total of 88 fruit bat specimens were collected from the Republic of Indonesia during 2012 and 2013. After euthanasia, a complete necropsy was performed on the captured bats. Specimens including the spleen were obtained and stored on ice in the field and were later transferred to −80 °C storage before further processing. Our research was performed in accordance with the ethical guidelines of the Animal Care and Use Committee of the Animal Teaching Hospital, Bogor Agricultural University. The protocols used were approved by the Animal Care and Use Committee of Veterinary Teaching Hospital, Bogor Agricultural University (permit number 05-2010 RSHP-IPB). All samples were exported with the permission of the Directorate General of Livestock and Animal Health Services, Ministry of Agriculture, Republic of Indonesia. DNA was isolated from each spleen using a QIAamp DNA Mini Kit (QIAGEN, Valencia, CA, USA) or DNAzol Reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions.
PyV PCR and identification of bat species
The total DNA isolated from bat spleen tissues (n = 88) was screened for the presence of PyV DNA using a nested, broad-spectrum, polymerase chain reaction (PCR) method with consensus degenerate primers targeted against a conserved region within the VP1 gene, using high-fidelity Taq DNA polymerase (Roche Diagnostics, Indianapolis, IN, USA) [6, 16, 24]. PCR products were visualized after electrophoresis using ethidium-bromide-stained 2 % agarose gels, and positive bands of the expected size were purified using a QIAquick Gel Extraction Kit (QIAGEN). The purified PCR products were sequenced in both directions, using the PCR primers and a BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies). After sequencing, a homology search was performed using BLAST. The species of fruit bats were determined based on their morphological characteristics and sequence analysis of their mitochondrial 16S rRNA and mitochondrial cytochrome b genes [1, 19].
Complete genome of PyV
Ten of the samples of PyV obtained from the fruit bats were selected for full-genome sequencing. We applied two strategies: direct PCR amplification and rolling-circle amplification (RCA). The RCA was performed on samples that had a low yield upon PCR amplification using an illustra TempliPhi DNA Amplification Kit (GE Healthcare UK Ltd, Buckinghamshire, England) [23]. The PCR primer sets were designed from the nucleotide sequence of VP1 as described above. The PCRs were performed using PrimeSTAR GXL DNA Polymerase (Takara Biochemical, Berkeley, CA, USA). The PCR products were cloned into the pUC118 vector using a Mighty Cloning Kit (Takara Biochemical), and sequences were determined using an Ion PGM Sequencing 400 Kit (Life Technologies). The resulting reads were assembled de novo using CLC genomics 6.0. We obtained at least 400,000 reads (mean read length, 284 bp) for each BatPyV. Gaps and low-coverage regions were sequenced manually using the Sanger method. Prediction of splicing sites was performed using the NetGene2 server (http://www.cbs.dtu.dk/services/NetGene2/) [2, 5] and NNSplice server (http://www.fruitfly.org/seq_tools/splice.html) [17].
Phylogenetic analysis
The amino acid sequences associated with the following reference virus genes were obtained from GenBank: BatPyV2a (AT7), BatPyV2b (R266), BatPyV2c (A504), BatPyV3a (A1055), BatPyV3a (B0454), BatPyV4a (R104), BatPyV4b (C1109), BKPyV, LPyV, BPyV, ChPyV, GggPyV1, GTM203 PteronotusPyV, HaPyV, HPyV9, HPyV12, JCPyV, KY156 Otomops PyV2, KY157 Otomops PyV1, KY270 Eidolon PyV1, KY336 Cardioderma PyV, KY369 Miniopterus PyV, and KY397 Chaerephon PyV1, MasPyV, MCPyV, MPtV, MPyV, MyoPyV, OraPyV1, OraPyV2, PtvPyV1a, PtvPyV2a, SV12, SV40, SqPyV, TSPyV and BPyV-2 (see Supplementary Table S1 for accession numbers). The selected viruses are the reported BatPyVs and are considered reference strains of the genus Orthopolyomavirus by the ICTV [11]. Multiple sequence alignments of the amino acid sequences of LT-Ag and VP1 were carried out using MEGA5 [21]. Bayesian phylogenetic analysis was performed using MrBayes software version 3.2.2 [18] with the WAG amino acid substitution model. The resulting trees were visualized with FigTree software, version 1.4.
Results
Detection of polyomavirus DNA in the spleens of fruit bats
We collected spleens from 88 fruit bats belonging to four different species (Acerodon celebensis, Pteropus vampyrus, Pteropus sp., and Dobsonia moluccensis) from three different districts (Paguyaman District, Surabaya District, and Yogyakarta District) in Indonesia. DNA was extracted from the spleens of these fruit bats and screened using a nested broad-spectrum PCR technique to detect the PyV VP1 region [6, 16, 24]. PCR products, approximately 250 bp in length, were detected in ten fruit bat samples (11.4 %), including one from a Pteropus vampyrus specimen captured in Surabaya District and five from Dobsonia moluccensis, two from Acerodon celebensis, and two from Pteropus sp. specimens captured in Paguyaman District. The PCR products were analyzed using BLAST searches, which showed that all PCR products had at least a 76 % nucleotide sequence identity to the VP1 region of PyV sequences previously deposited in the GenBank database.
Viral genome analysis of BatPyVs
We attempted to identify the viral genomes of the 10 samples that were positive for the PyV VP1 region, using inverse primer sets that were designed based on the nucleotide sequence of the VP1 fragments. We amplified most of the viral genome, 5,000 bp, from each of five of the DNA samples. The other five DNA samples were used as a template for RCA [23], from which we successfully amplified the whole viral genome from three of these samples using an inverse PCR method. We failed to amplify the full PyV genome from the remaining two samples. In total, we managed to determine the complete viral genome sequences from eight of the samples. According to the ICTV, to establish a new PyV species, less than 81 % nucleotide sequence identity to other known PyVs is required, as determined through isolation of the whole genome and nucleotide sequencing [7]. Pairwise nucleotide sequence identities were calculated for comparisons with known reference PyV genome sequences (Table 1). Each of the eight genomes showed <81 % nucleotide sequence identity to the known reference PyVs, indicating that they represent novel PyV species. Because BatPyV1, BatPyV2, BatPyV3, and BatPyV4 have been reported previously [3, 15], these eight novel viruses were named BatPyV5a (detected in Dobsonia moluccensis specimens captured in Paguyaman District), BatPyV5b-1 (detected in Pteropus vampyrus specimens captured in Surabaya District), BatPyV5b-2 (detected in Acerodon celebensis specimens captured in Paguyaman District), BatPyV6a (detected in Acerodon celebensis specimens captured in Paguyaman District), BatPyV6b (detected in Dobsonia moluccensis specimens captured in Paguyaman District), BatPyV6c (detected in Dobsonia moluccensis specimens captured in Paguyaman District), BatPyV6d-1 (detected in Pteropus sp. specimens captured in Paguyaman District), and BatPyB6d-2 (detected in Pteropus sp. specimens captured in Paguyaman District). BatPyV6d-1 and BatPyV6d-2 had a 99.6 % nucleotide identity to each other, and BatPyV5b-1 shared 88.8 % nucleotide identity with BatPyV5b-2, suggesting that these sub-types belonged to the same strain. All viral genomes had the typical PyV genome organization, comprising one strand coding for regulatory proteins (t-Ag and T-Ag) and the other for structural proteins (VP1 and VP2/3) (Table 2).
Genome organization of BatPyVs
ORFs present within the genome of PyVs are separated by NCCR into regulatory protein coding regions and structural protein coding regions [22]. Repeats of the DNA element GAGGC and its reverse complement GCCTC within the NCCR are regarded as T-Ag-binding sites, as shown previously [8]. The novel BatPyVs harbor four to six copies of these T-Ag-binding elements, and BatPyV6b possessed a palindromic octamer (Fig. 1), which is an overlapping bidirectional T-Ag-binding element (GAGGCCTC) [8]. BatPyV6a, BatPyV6c, BatPyV6d-1, and BatPyV6d-2 had no GCCTC complements, and none of the novel BatPyVs had two repeats of GAGGC followed by a palindromic repeat of the GCCGTC complement (Fig. 1). The number and arrangement of T-Ag-binding elements in the novel BatPyVs were different from those of TsPyV, OraPyV1, and previously known BatPyVs. Interestingly, a high degree of similarity in the NCCR of the BatPyVs from cluster D (BatPyV5a, BatPyV5b-1, and BatPyV5b-2), HPyV9, and LPyV was shown, even though the NCCR is the most variable region of the PyV genome.

Sequence alignment of the noncoding control region of selected PyVs. DNA sequences from the BatPyVs discovered in this study were compared with the NCCR of the other known PyVs using the Clustal W program, with additional manual adjustments. The novel BatPyVs are boxed within a dotted line. T-Ag-binding elements (GAGGC or the complement GCCTC) are shaded in gray (GAGGC) and boxed (GCCTC)
The T-Ag of PyVs exhibits a high degree of conservation within its functional domains, including the J domain and the retinoblastoma protein (Rb)-binding motif [8, 13]. As for TSPyV, HpyV9, LPyV, and OraPyV1, all of the T-Ags from the novel BatPyVs possessed a J domain with the highly conserved motif HPDKGG (Fig. 2). The T-Ag from BatPyVs of cluster D (BatPyV5a, BatPyV5b-1, and BatPyV5b-2) lacked an LXCXE Rb-binding motif, whereas the other novel BatPyVs contained this motif (Fig. 2).

Protein sequence alignment of the T-Ag N-terminal portions. The alignment was done using the Clustal W program, with additional manual adjustments. The regions shaded in gray regions indicate the J domain (HPDKGG) and the Rb-binding domain (LXCXE)
Phylogenetic analysis
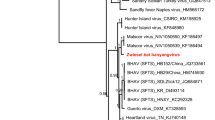
Phylogenetic analysis was performed on the basis of the aligned amino acid sequences of the LT-Ag and VP1 to investigate the evolutionary relationships of the novel BatPyV to other known PyVs (Fig. 3). To construct phylogenetic tree, we excluded members of the proposed genera Avipolyomavirus and Wukipolyomavirus because of their significant heterogeneity. It has been reported that there are three distinct well-supported genetic clusters of BatPyVs (A, B, and C) [3]. Based on the LT-Ag and VP1 phylogenies (Fig. 3), the novel BatPyVs formed two distinct genetic clusters: (i) BatPyV5a, BatPyV5b-1, and BatPyV5b-2 (cluster D); and (ii) BatPyV6a, BatPyV6b, BatPyV6c, BatPyV6d-1, and BatPyV6d-2 (cluster E). From the LT-Ag phylogeny, the BatPyVs from cluster E were related to PyVs from a variety of species, including monkeys (SqPyV), rodents (MPtV and MasPyV), and bats (MyoPyV). None of the BatPyVs from cluster D showed a relationship to known BatPyVs, whereas BatPyVs from cluster D were related to TSPyV and OraPyV1.

Phylogenetic analysis based on the amino acid sequence of LT-Ag and VP1 of PyVs. Lineages leading to BatPyVs identified in this study are shaded in gray. Phylogenetic analysis was performed using MrBayes software version 3.2.2, with percentages indicated at the nodes. GenBank accession numbers for the viruses used in the tree are shown in parentheses. Scale bars are in units of nucleotide substitutions per site
Discussion
In this study, we have identified eight novel PyV genomes in fruit bats from Indonesia. Phylogenetic analysis indicated that these viruses are divided into two clusters (cluster D and E). Cluster D BatPyVs are related to the primate PyVs, such as TSPyV and OraPyV1. Previous reports have shown that BatPyV clusters B and C and primate PyVs have relevance, as they tended to form monophyletic groups in phylogenetic trees [3, 22]. These relationships between BatPyVs and primate PyVs suggest that bats and primates may be closely tied in the PyV infectious cycle. TSPyV has been identified in human trichodysplasia spinulosa, which is a rare skin disease exclusively found in severely immunocompromised hosts [9, 20, 23]. The genome of the novel BatPyVs described herein contains common ORFs of known PyVs, such as T-Ag, t-Ag, VP1, and VP2/3, but not agnoprotein. The T-Ag of PyVs is a multifunctional protein composed of several functional domains [16]. The T-Ag interacts with molecular chaperones and Rb family proteins that regulate the cell cycle to replicate viral genome [8, 13]. The cluster D BatPyVs lack an Rb-binding motif (LXCXE), but they have a molecular chaperone-binding motif (the J domain: HPDKGG). The introns of the LT-Ag mRNAs from the cluster D BatPyVs contain the canonical dinucleotides GT and AG for splice donor and splice acceptor sites, respectively (Supplementary Table S2). The Rb-binding motif was not found in the region upstream of the splice acceptor sites. However, because splicing sites were analyzed using a splice-site prediction program, they need to be confirmed by analyzing mRNA in PyV-infected cells.
We found eight novel BatPyV genomes in spleen tissue from fruit bats using broad-spectrum PCR, but there were no noticeable gross pathological findings in the tissues of these fruit bats. Although an infection with any of these eight novel BatPyVs is likely to be asymptomatic in a healthy individual, as for other PyVs, this clearly requires further study.
In conclusion, we detected eight novel PyV genomes in fruit bats in Indonesia. This is the first study to identify the viral genome of PyV in fruit bats in Indonesia. Our findings are consistent with previous reports that transmission of PyV occurs between bats and primates [3, 22]. Further epidemiological investigations are needed to determine the extent of PyV genetic variation in various mammalian species.
References
Almeida FC, Giannini NP, DeSalle R, Simmons NB (2011) Evolutionary relationships of the old world fruit bats (Chiroptera, Pteropodidae): another star phylogeny? BMC Evol Biol 11:281
Brunak S, Engelbrecht J, Knudsen S (1991) Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol 220:49–65
Fagrouch Z, Sarwari R, Lavergne A, Delaval M, de Thoisy B, Lacoste V, Verschoor EJ (2012) Novel polyomaviruses in South American bats and their relationship to other members of the family Polyomaviridae. J Gen Virol 93:2652–2657
Halpin K, Young PL, Field HE, Mackenzie JS (2000) Isolation of Hendra virus from pteropid bats: a natural reservoir of Hendra virus. J Gen Virol 81:1927–1932
Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouzé P, Brunak S (1996) Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res 24:3439–3452
Johne R, Enderlein D, Nieper H, Müller H (2005) Novel polyomavirus detected in the feces of a chimpanzee by nested broad-spectrum PCR. J Virol 79:3883–3887
Johne R, Buck CB, Allander T, Atwood WJ, Garcea RL, Imperiale MJ, Major EO, Ramqvist T, Norkin LC (2011) Taxonomical developments in the family Polyomaviridae. Arch Virol 156:1627–1634
Johnson EM (2010) Structural evaluation of new human polyomaviruses provides clues to pathobiology. Trends Microbiol 18:215–223
Kazem S, van der Meijden E, Kooijman S, Rosenberg AS, Hughey LC, Browning JC, Sadler G, Busam K, Pope E, Benoit T, Fleckman P, de Vries E, Eekhof JA, Feltkamp MC (2012) Trichodysplasia spinulosa is characterized by active polyomavirus infection. J Clin Virol 53:225–230
Khalili K, Stoner GL (2004) Human polyomaviruses: molecular and clinical perspectives. Wiley, New York
King AMQ, Adams MJ, Lefkowitz EJ, Carstens EB (2012) Virus taxonomy: classification and nomenclature of viruses: Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier
Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Wong SS, Leung SY, Chan KH, Yuen KY (2005) Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci USA 102:14040–14045
Leendertz FH, Scuda N, Cameron KN, Kidega T, Zuberbühler K, Leendertz SA, Couacy-Hymann E, Boesch C, Calvignac S, Ehlers B (2011) African great apes are naturally infected with polyomaviruses closely related to Merkel cell polyomavirus. J Virol 85:916–924
Luby SP, Rahman M, Hossain MJ, Blum LS, Husain MM, Gurley E, Khan R, Ahmed BN, Rahman S, Nahar N, Kenah E, Comer JA, Ksiazek TG (2006) Foodborne transmission of Nipah virus, Bangladesh. Emerg Infect Dis 12:1888–1894
Misra V, Dumonceaux T, Dubois J, Willis C, Nadin-Davis S, Severini A, Wandeler A, Lindsay R, Artsob H (2009) Detection of polyoma and corona viruses in bats of Canada. J Gen Virol 90:2015–2022
Orba Y, Kobayashi S, Nakamura I, Ishii A, Hang’ombe BM, Mweene AS, Thomas Y, Kimura T, Sawa H (2011) Detection and characterization of a novel polyomavirus in wild rodents. J Gen Virol 92:789–795
Reese MG, Eeckman FH, Kulp D, Haussler D (1997) Improved splice site detection in Genie. J Comput Biol 4:311–323
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Sasaki M, Setiyono A, Handharyani E, Rahmadani I, Taha S, Adiani S, Subangkit M, Sawa H, Nakamura I, Kimura T (2012) Molecular detection of a novel paramyxovirus in fruit bats from Indonesia. Virol J 9:240
Scuda N, Hofmann J, Calvignac-Spencer S, Ruprecht K, Liman P, Kühn J, Hengel H, Ehlers B (2011) A novel human polyomavirus closely related to the african green monkey-derived lymphotropic polyomavirus. J Virol 85:4586–4590
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tao Y, Shi M, Conrardy C, Kuzmin IV, Recuenco S, Agwanda B, Alvarez DA, Ellison JA, Gilbert AT, Moran D, Niezgoda M, Lindblade KA, Holmes EC, Breiman RF, Rupprecht CE, Tong S (2013) Discovery of diverse polyomaviruses in bats and the evolutionary history of the Polyomaviridae. J Gen Virol 94:738–748
van der Meijden E, Janssens RW, Lauber C, Bouwes Bavinck JN, Gorbalenya AE, Feltkamp MC (2010) Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog 6:e1001024
Yamaguchi H, Kobayashi S, Ishii A, Ogawa H, Nakamura I, Moonga L, Hang’ombe BM, Mweene AS, Thomas Y, Kimura T, Sawa H, Orba Y (2013) Identification of a novel polyomavirus from vervet monkeys in Zambia. J Gen Virol 94:1357–1364
Acknowledgments
This study was supported by the Japan Initiative for Global Research Network of Infectious Diseases (J-GRID) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kobayashi, S., Sasaki, M., Nakao, R. et al. Detection of novel polyomaviruses in fruit bats in Indonesia. Arch Virol 160, 1075–1082 (2015). https://doi.org/10.1007/s00705-015-2349-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2349-7