
Abstract
Stunted shrimp caused by Penaeus monodon densovirus (PmDNV) infection is one of the main problems leading to a significant economic loss in Thailand. To control this pandemic disease, a double-stranded-RNA-mediated virus-specific gene silencing approach was applied to inhibit viral replication. In this study, two dsRNAs corresponding to the non-structural protein (ns1) and the structural protein (vp) genes of PmDNV were synthesized and introduced into shrimp haemolymph prior to viral challenge. After allowing viral replication for two weeks, the suppression effect by each dsRNA was evaluated by semi-quantitative PCR and compared with the control. A reduction of PmDNV in shrimp treated with each dsRNA was observed. In contrast, a high level of viral infection was detected in the control group (NaCl). Based on a limited sample number, we reached the tentative conclusion that virus-specific dsRNA can inhibit PmDNV replication, in which the dsRNA-ns1was more effective than the dsRNA-vp.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the past few years, the shrimp culturing industry has encountered a tremendous economic loss, which is mainly from viral infectious diseases. Among the causative pathogens, Penaeus monodon densovirus (PmDNV) (formerly hepatopancreatic parvovirus or HPV) has been reported to be associated with a slow growth rate and stunted appearance of shrimp [11]. Although it does not cause an abrupt mortality crisis of cultured shrimp as do WSSV and YHV-associated outbreaks, the infected shrimp have the same value as the dead ones. This problem leads to the significant reduction of shrimp production and a consequent loss of profit. At present, there is no available treatment for this disease.
The recently described phenomenon of RNA interference (RNAi) provides a powerful means for silencing gene expression in a sequence-specific manner [12]. Moreover, it has been shown to be applicable to viral protection in a number of organisms, including shrimp, through the introduction of exogenous, specific double-stranded RNA (dsRNA). Previously, published data have demonstrated that the replication of both RNA [21, 22, 25] and DNA viruses [3, 13, 19] was effectively inhibited by dsRNAs corresponding to their viral gene targets. Consequently, the shrimp mortality was significantly diminished when compared to shrimp without treatment with dsRNA, in which case all of the shrimp were dead [3, 18, 21, 24].
PmDNV is a non-enveloped virus with an icosahedral-shaped particle [14]. It contains a linear single-stranded DNA genome with typical palindromic termini [6]. According to a report of the complete sequence of a Thai isolate [20], it contains three large open reading frames (ORFs): two non-structural protein genes (ns1 and ns2) and one structural protein gene (vp). The ns1 protein of parvoviruses is known to have multiple functions, including ATP-dependent site-specific DNA binding and nicking and also helicase activity, which is essential for viral replication [10, 16, 24]. Meanwhile, the phospholipase A2 activity of the vp protein has been reported to be critical for viral infectivity [7]. However, the biological function of ns2 during the parvovirus life cycle is still obscure [9, 15].
In this study, we determined the effectiveness of dsRNA for the inhibition of PmDNV in P. monodon. Two dsRNAs specifically targeted to genes that are essential for viral infection, ns1 and vp, were synthesized in bacterial cells and subsequently injected into shrimp followed by viral challenge. The suppression of PmDNV by virus-specific dsRNA was then evaluated by semi-quantitative PCR analysis.
Materials and methods
Shrimp specimens
The 300-mg juvenile P. monodon was used for all experiments in this study. The post-larva-stage shrimps (P15–20), obtained from a hatchery farm, were reared in a 500-L tank containing artificial sea water at 10 parts per thousand (ppt) salinity with aeration until they reached juvenile stage with the appropriate size. The shrimps were fed daily with a shrimp pellet diet.
Virus source
Hepatopancreas isolated from naturally PmDNV-infected shrimp was utilized for viral infection via the oral route. To quantify the amount of virus, the level of PmDNV in each hepatopancreas was determined by PCR. Only hepatopancreas samples with equal amounts of virus were then selected for further experimental feeding. The tissue was kept at −20°C until required.
Stem-loop RNA expression plasmid construction
For virus-specific dsRNA: Two regions of the PmDNV genome corresponding to the ns1 and vp genes shown in Fig. 1a were selected for dsRNA targeting. In order to minimize misfolding of RNA during the dsRNA production, the Clone Manager program was used for scanning any repeats throughout the regions of ns1 and vp before selection. Moreover, a survey of the specificity of the selected region using the BLAST program was also performed to minimize the off-target effect. PCR products (400 and 600 bp) of each targeted gene were amplified using Vent DNA polymerase and the specific primer pair shown in Table 1 (primers C and D of each gene for the 400-bp product and A and B of each gene for the 600-bp product). These two fragments were then cloned into the pET17b vector in an inverted orientation, directed by restriction enzymes flanking the fragments (NdeI and EcoRV for 600 bp, EcoRV and XhoI for 400 bp) (Fig. 1b). PCR and restriction enzyme analysis were then used to screen the recombinant clones. Furthermore, the nucleotide DNA sequence of the insert was confirmed by automated DNA sequencing. By this cloning strategy, the transcribed RNA can form a stem-loop structure with a 400-bp stem and a 200-bp loop due to complementary binding of the RNA ends.

Double-stranded RNA production. a Schematic diagram of dsRNA-targeted DNA regions of the PmDNV genome. Each gray arrow represents a particular gene [ns2 (nt 216–1,502), ns1 (nt 1,487–3,226), and vp (nt 3,642–6,098)], lying below a line representing the PmDNV genome. The regions targeted by dsRNA-ns1 (nt 1,547–2,170) and dsRNA-vp (nt 3,689–4,303) are shown in gray boxes, while the open arrows show tentative promoters of each gene. The scale bar relative to the full length of the PmDNV genome (6,321 bp) is shown below the diagram. b Schematic diagram of stem-loop RNA expression plasmid construction. Primers A + B and C + D were used for amplification of a 600-bp PCR fragment (PCR I) and a 400-bp fragment (PCR II) of each virus-specific gene. The restriction enzyme sites allowing directional cloning are indicated at the 5′ end of each primer. The shaded arrow and dotted line represent an inverted repeat of the target and the spacer between the inverted repeats, respectively. c dsRNAs verification by RNase treatment. The integrity of each dsRNA targeted to the ns1 gene (ns1), vp gene (vp) or gfp gene (gfp) was confirmed by incubation with RNase A (A) and RNase III (III), which specifically digest ssRNA and dsRNA, respectively. The expected sizes of dsRNA-ns1 and dsRNA-vp are 425 bp and 409 bp, respectively. The untreated dsRNAs (U) and ssRNA (ss) were used as a control. Approximately 50 ng of dsRNA was loaded in each lane of a normal 1× TBE 2% agarose gel. M 100-bp DNA ladder
For non-related dsRNA (green fluorescent protein, gfp): The recombinant plasmid was kindly provided by Dr. Witoon Tirasophon, Mahidol University. The cloning strategy was similar to that for specific dsRNA except that pET3a was used as a cloning vector instead of pET17b, XbaI was used for joining the two fragments, and NdeI was used for inserting them into the plasmid vector [25].
Double-stranded RNA production
dsRNA was expressed in Escherichia coli HT115 according to the protocol of Ongvarrasopone et al. [17]. An overnight bacterial culture in LB medium with antibiotic was diluted with fresh medium and grown at 37°C until an OD600 of 0.4 was achieved. The expression of dsRNA was induced by adding isopropyl-β-D-thiogalactopyranoside (IPTG), and the culture was further incubated for 4 h. The bacterial cells were then harvested by centrifugation at 6,000×g for 5 min at 4°C. The cell pellet was resuspended in 0.1% sodium dodecyl sulphate (SDS) at a ratio of 50 μl per OD unit. To lyse the cells, the cell suspension was boiled for 2 min. Next, the bacterial single-stranded RNAs (ssRNAs) and the loop region (200 bp) of the expressed RNA were eliminated by incubation with RNaseA (1 μg RNaseA per 1 OD cells) in the reaction buffer (300 mM sodium acetate, 10 mM Tris–Cl, pH 7.5) at 37°C for 30 min. The remaining dsRNA (400 bp) was subjected to purification by TRI Reagent (Molecular Research Center) following the manufacturer’s protocol. Finally, the concentration of each dsRNA was estimated by gel electrophoresis using a standard DNA marker.
Verification of dsRNA
To ensure the integrity of the synthesized dsRNA before introducing it into shrimp, treatment with RNaseA (specific for ssRNA) and RNaseIII (specific for dsRNA) was carried out. An equal amount of dsRNA was digested separately with RNaseA (0.01 μg RNaseA per 2 μg dsRNA) and RNaseIII (0.5 units RNaseIII per 2 μg dsRNA) in the reaction buffer for RNaseA (300 mM sodium acetate, 10 mM Tris–Cl) and for RNaseIII (10 mM Tris–Cl, 0.1 mM CaCl2, and 2.5 mM MgCl2), respectively, at 37°C for 5 min. The patterns of the digested RNAs were then determined by gel electrophoresis with a standard size marker.
Experimental conditions
For infectivity of PmDNV
In order to determine the infectivity profile of PmDNV in shrimp, the 300-mg shrimp were kept in individual Petri dishes (90 mm × 15 mm) in 10 ppt artificial seawater. Thirty-six hours pre-fasting, shrimp were fed 3 times with PmDNV-infected hepatopancreas (approximately 10% of their body weight per meal) at roughly 12-h intervals. Subsequently, the shrimp were fed daily with shrimp pellet diet up to two weeks. Sampling of 2–3 shrimp at 4, 8, and 14 days after the first feeding was performed to investigate production of virus in the hepatopancreas by PCR analysis.
For viral inhibition
According to the infectivity test, shrimp of the same size, kept in individual Petri dishes, were injected with approximately 750 ng of each virus-specific dsRNA (ns1 and vp) 24 h before viral infection by oral feeding as described above. To maintain the effect of dsRNA during the experiment, another dsRNA administration was carried out on day 5 after the first injection. Thereafter, the shrimp were fed daily with a shrimp pellet diet. Two weeks after oral feeding with PmDNV-infected tissue, the hepatopancreas of each shrimp was isolated for further DNA extraction. The inhibitory effect of the delivered dsRNA was then evaluated by semi-quantitative PCR.
DNA extraction
Total DNA was extracted from hepatopancreas tissues (or as indicated) using TRI Reagent (Molecular Research Center). Samples of 50–100 mg of tissue were ground in 1 ml TRI Reagent. The DNA in the interphase and organic phase was precipitated using absolute ethanol. The DNA pellet was washed with 0.1 M trisodium citrate and 75% ethanol and then resuspended in sterile distilled water. To increase solubilization, the DNA pellet was heated at 65°C for 10 min. The absorbance at 260 nm of each DNA sample was measured in order to calculate its concentration.
Sample analysis
Semi-quantitative PCR
The total amount of extracted DNA (approximately 600 ng) was used for amplification of the virus-specific gene (vp) using the primers listed in Table 1 (primers 1 and 2 for the infectivity and inhibition experiment and primers 3 and 4 for the tissue distribution test). To determine the relative amount of virus in the samples, PCR of an internal control gene (shrimp beta actin) was included (primers 1 and 2 for the infectivity and inhibition experiment and primers 3 and 4 for the tissue distribution test). The temperature profile for PCR amplification was as follows; 94°C for 2 min, denaturation at 94°C for 10 s, annealing at 55°C for 30 s, and extension at 72°C for 1 min. After 20 cycles (35 cycles for the tissue distribution experiment), the reaction was held at 72°C for another 5 min. The PCR product was analyzed by agarose gel electrophoresis.
Statistical analysis
The data were analyzed statistically using GraphPad software. One-way ANOVA followed by Tukey–Kramer multiple comparison test was used for analysis. The data are presented as mean ± standard error (standard error of the mean, SEM).
Results
Double-stranded RNA verification
To test whether RNA was double-stranded, the RNA extracted from bacterial cells was incubated with RNaseA and RNaseIII, which specifically cleaved ssRNA and dsRNA, respectively. The result showed that all synthesized RNA was digested by RNaseIII but not RNaseA, indicating that it was double-stranded RNA (Fig. 1c). In contrast, the control single-stranded RNA (ss) was completely digested with RNaseA. This clearly demonstrated that the synthesized dsRNAs (ns1, vp and gfp) were suitable for injection into shrimp to trigger the anti-virus pathway.
Localization of PmDNV in various tissues of shrimp
To date, the distribution of this virus in the tissues of shrimp has not been investigated. In this study, the localization of PmDNV in various organs of shrimp (hepatopancreas, gill, pleopod, periopod and muscle) was determined. Semi-quantitative PCR of the specific viral vp gene was performed by using an equal amount of DNA template extracted from each tissue. By normalization with the β-actin control, it could be seen that PmDNV was predominantly in the hepatopancreas (Fig. 2). Much less was noted in the periopod, gill, pleopod and muscle. Based on this information, tissue from the hepatopancreas was used for all subsequent experiments.

Tissue distribution of PmDNV in a naturally infected shrimp. Equal amounts of total DNA extracted from various tissues (H hepatopancreas, G gill, Pl pleopods, P periopods and M muscle) of each naturally PmDNV-infected shrimp were subjected to PCR analysis to determine the viral localization. The product was amplified separately using virus-specific primers (PmDNV, vp) and host beta-actin primers (actin). Negative (−) and positive (+) controls were included. Numbers at the top indicate individual shrimp. The size of the PCR product was compared with the 100-bp DNA marker (M)
Experimental infection of PmDNV in shrimp
In order to establish an infectivity profile, shrimp were fed with PmDNV-infected hepatopancreas and randomly selected for testing at three different time points (4, 8, and 14 days after the first feeding). Viral replication was then assayed by PCR. A reduction in virus levels was observed at day 8 compared with total virus uptake at day 4. The amount of virus then increased significantly at day 14 (Fig. 3). This indicates that the infection of shrimp with PmDNV can be performed experimentally, and viral replication can also be investigated over a time period of up to 14 days.

Infectivity profile of PmDNV in shrimp after feeding with hepatopancreas of PmDNV-infected shrimp. The 300-mg shrimp were fed three times (approximately 30 mg each meal, at 12-h intervals) with PmDNV-infected hepatopanceas. Shrimp were picked randomly and sacrificed at 4 (Day4), 8 (Day8), and 14 (Day14) days post-feeding. Total DNA extracted from hepatopancreas of each shrimp was used as a template for semi-quantitative PCR using the viral specific primers (PmDNV) and the internal control actin primers (actin). Each shrimp is indicated by a number at the top of the gel, while M is the 100-bp marker. Negative (−) and positive (+) controls are included
Suppression of PmDNV replication by dsRNA
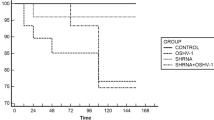
To test the efficacy of dsRNA for inhibition of viral replication, long dsRNA (approximately 400 bp) targeted to viral genes, including non-structural (ns1) and structural protein genes (vp), were introduced into the shrimp’s haemolymph followed by PmDNV challenge by feeding. The amount of virus in each shrimp that received virus-specific dsRNA was analyzed and compared with the control (without dsRNA) and non-specific dsRNA (gfp). Fourteen days post-infection, viral DNA in the hepatopancreas of each shrimp was monitored by PCR, co-amplified with a shrimp actin gene for normalization (Fig. 4b). A high level of viral infection was observed in the control group. The reduction of PmDNV in shrimp treated with dsRNA-ns1 was more pronounced than with dsRNA-vp and significantly different from the control (no dsRNA) (P = 0.004) and non-related dsRNA (dsRNA-gfp) (P = 0.02) (Fig. 4c). These results demonstrate that the virus-specific dsRNA can suppress the replication of PmDNV in shrimp, with dsRNA-ns1 being more potent than the dsRNA-vp.

Treatment with dsRNA for inhibition of PmDNV in shrimp. The 300-mg shrimp were injected two times with 750 ng dsRNA specific either for the ns1 gene or vp gene or the unrelated gfp gene (as indicated in the diagram) before challenging with PmDNV by oral feeding with infected tissue. In the control group, shrimp were treated with NaCl instead of dsRNA. Fourteen days after the first administration of dsRNA, the hepatopancreas of each shrimp was collected for DNA extraction, and further PCR analysis was performed. a The diagram represents the experimental condition. b Semi-quantitative PCR of PmDNV DNA. The amount of PmDNV in an individual shrimp was determined by multiplex PCR using virus-specific primers (vp) together with the host control gene (actin) primers for normalization. Lanes 1–4 represent individual shrimp receiving no dsRNA; 5–8 dsRNA-ns1; 9–11, dsRNA-vp; and 12–15, dsRNA-gfp. N is the normal shrimp. Negative (−) and positive (+) controls are included, with the 100-bp DNA marker in lane M. c Relative amount of PmDNV normalized with actin. Data are shown as means of duplicate experiments with error bar. Statistical significance was analyzed by ANOVA test. P values, * less than 0.05 and ** less than 0.01
Discussion
This study showed for the first time that the replication of PmDNV in shrimp can be effectively inhibited by the administration of virus-specific long dsRNA. Our findings are in agreement with a number of previous studies demonstrating dsRNA-mediated viral suppression in shrimp [3, 13, 19, 21]. It has been documented that silencing the expression of an essential viral replication protein gene can significantly inhibit infection with that particular virus. According to the strength of the inhibitory effect, the different gene targets of dsRNA provided different degrees of effect for viral protection. The non-structural protein gene has been shown to be more potent than the structural protein gene [19, 21]. In the case of PmDNV, the most effective target was probably the multi-functional ns1 protein gene. The ns1 protein has been reported to be involved in the early step of parvovirus infection via the binding and nicking of the double-stranded replicative form (RF) of DNA for generating the single-stranded DNA. This then further facilitates the replication process [2, 4, 5]. In addition to this, the ns1 of parvovirus minute virus of mice (MVM) has been shown to regulate the expression of the late protein gene (vp) through promoter activation [8]. Because of these essential functions, the ns1 of PmDNV may represent a potential target for further development of an anti-PmDNV approach.
As described recently by several research groups [3, 13, 15, 25], the antiviral response in shrimp triggered by long dsRNA occurs through two pathways: sequence-dependent RNAi and sequence-independent innate immunity. In the case of nonspecific inhibition from dsRNA-gfp or other irrelevant dsRNAs, the protection was not high and could be observed at a low level of viral infection, whereas it was overwhelmed by heavy infection. Our results show that a moderate inhibition was also found in the dsRNA-gfp-treated shrimp group (Fig. 4b, lane 14–15). This suggests that shrimp could be partially protected from PmDNV infection by nonspecific dsRNA. This might be due to the slow rate of its infection (approximately 2 weeks after receiving viruses as shown in Fig. 3); therefore, nonspecific inhibition could be effective enough to block PmDNV infection.
This report is the first to show successful infection of PmDNV in juvenile Penaeus monodon (300 mg body weight). Interestingly, shrimp infected by feeding with virus-infected hepatopancreas had a specific infectivity profile. The amount of virus declined after initial infection but then rose significantly at a later time point. This phenomenon may be due to the cellular composition of the hepatopancreas. It is widely known that parvoviral replication depends on cellular factors associated with the S phase of the cell cycle to convert its ssDNA genome into a double-stranded RF for further genome amplification [5]. There are four main epithelial cell types present in the hepatopancreas, E (embryonic), F (fibrilar), B (blister-like), and R (absorptive) cells [1]. Of these, only the E cells show mitotic activity. Hence, it is possible that a large number of virus particles initially entered into all of the types of epithelial cells during the feeding period, but only the dividing E cells, which are a small proportion (less than 10%) of the total hepatopancreatic epithelial cells [26], were able to support viral replication. On the other hand, the other differentiated epithelial cells (F, B, and R cells) would have eventually been disintegrated and extruded from the tubule epithelium, releasing a digestive enzyme together with the viral particles into the lumen of hepatopancreatic tubule [23]. The virus would then later be excreted out via feces [18]. This explains the significant reduction of PmDNV at day 8 compared to the initial uptake. Subsequently, propagation of the virus was promoted due to division of the infected E cells. On the basis of this finding, approximately 14 days should be the appropriate experimental time course to assess PmDNV replication in 300-mg juvenile shrimp.
In conclusion, an effective tool for inhibition of PmDNV replication in shrimp using virus-specific dsRNA has been demonstrated in this study. However, several aspects, such as therapeutic effect as well as methodology for continuous delivery of dsRNA into shrimp, have to be further resolved in order to apply this RNAi technology for controlling this disease in the shrimp farm.
References
Al-Mohanna SY, Nott JA (1989) Functional cytology of the hepatopancreas of Penaeus semisulcatus (Crustacea: Decapoda) during the moult cycle. Mar Biol 101:535–544
Astell CR, Chow MB, Ward DC (1985) Sequence analysis of the termini of virion and replicative forms of minute virus of mice DNA suggests a modified rolling hairpin model for autonomous parvovirus DNA replication. J Virol 54:171–177
Attasart P, Kaewkhaw R, Chimwai C, Kongphom U, Namramoon O, Panyim S (2009) Inhibition of white spot syndrome virus replication in Penaeus monodon by combined silencing of viral rr2 and shrimp PmRab7. Virus Res 145:127–133
Baldauf AQ, Willwand K, Mumtsidu E, Nuesch JP, Rommelaere J (1997) Specific initiation of replication at the right-end telomere of the closed species of minute virus of mice replicative-form DNA. J Virol 71:971–980
Bashir T, Horlein R, Rommelaere J, Willwand K (2000) Cyclin A activates the DNA polymerase delta-dependent elongation machinery in vitro: a parvovirus DNA replication model. Proc Natl Acad Sci USA 97:5522–5527
Bonami JR, Mari J, Poulos BT, Lightner DV (1995) Characterization of hepatopancreatic parvo-like virus, a second unusual parvovirus pathogenic for penaeid shrimps. J Gen Virol 76:813–817
Canaan S, Zadori Z, Ghomashchi F, Bollinger J, Sadilek M, Moreau ME, Tijssen P, Gelb MH (2004) Interfacial enzymology of parvovirus phospholipases A2. J Biol Chem 279:14502–14508
Christensen J, Cotmore SF, Tattersall P (1995) Minute virus of mice transcriptional activator protein NS1 binds directly to the transactivation region of the viral P38 promoter in a strictly ATP-dependent manner. J Virol 69:5422–5430
Cotmore SF, D’Abramo AM Jr, Carbonell LF, Bratton J, Tattersall P (1997) The NS2 polypeptide of parvovirus MVM is required for capsid assembly in murine cells. Virology 231:267–280
Cotmore SF, Tattersall P (1994) An asymmetric nucleotide in the parvoviral 3′ hairpin directs segregation of a single active origin of DNA replication. EMBO J 13:4145–4152
Flegel TW, Thamavit V, Pasharawipas T, Alday-Sanz V (1999) Statistical correlation between severity of hepatopancreatic parvovirus infection and stunting of farmed black tiger shrimp (Penaeus monodon). Aquaculture 174:197–206
Hannon GJ (2002) RNA interference. Nature 418:244–251
Kim CS, Kosuke Z, Nam YK, Kim SK, Kim KH (2006) Protection of shrimp (Penaeus chinensis) against white spot syndrome virus (WSSV) challenge by double-stranded RNA. Fish Shellfish Immunol 23:242–246
Lightner DV, Redman RM (1985) A parvo-like virus disease of penaeid shrimp. J Invertebr Pathol 45:47–53
Naeger LK, Cater J, Pintel DJ (1990) The small nonstructural protein (NS2) of the parvovirus minute virus of mice is required for efficient DNA replication and infectious virus production in a cell-type-specific manner. J Virol 64:6166–6175
Nuesch JPF, Cotmore CF, Tattersall P (1995) Sequence motifs in the replicator protein of parvovirus MVM essential for nicking and covalent attachment to the viral origin: identification of the linking tyrosine. Virology 209:122–135
Ongvarrasopone C, Roshorm Y, Panyim S (2007) A simple and cost effective method to generate dsRNA for RNAi studies in invertebrates. Sci Asia 33:35–39
Pantoja CR, Lightner DV (2001) Detection of hepatopancreatic parvovirus (HPV) of penaeid shrimp by in situ hybridization at the electron microscope level. Dis Aquat Org 44:87–96
Robalino J, Bartlett T, Shepard E, Prior S, Jaramillo G, Scura E, Chapman RW, Gross PS, Browdy CL, Warr GW (2005) Double-stranded RNA induces sequence-specific antiviral silencing in addition to nonspecific immunity in a marine shrimp: convergence of RNA interference and innate immunity in the invertebrate antiviral response? J Virol 79:13561–13571
Sukhumsirichart W, Attasart P, Boonsaeng V, Panyim S (2006) Complete nucleotide sequence and genomic organization of hepatopancreatic parvovirus (HPV) of Penaeus monodon. Virology 346:266–277
Tirasophon W, Roshorm Y, Panyim S (2005) Silencing of yellow head virus replication in penaeid shrimp cells by dsRNA. Biochem Biophys Res Commun 334:102–107
Tirasophon W, Yodmuang S, Chinnirunvong W, Plongthongkum N, Panyim S (2007) Therapeutic inhibition of yellow head virus multiplication in infected shrimps by YHV-protease dsRNA. Antiviral Res 74:150–155
Vogt G (1994) Life-cycle and functional cytology of the hepatopancreatic cells of Astacus astacus (Crustacea, Decapoda). Zoomorphology 114:83–101
Wilson GM, Jindal HK, Yeung DE, Chen W, Astell CR (1991) Expression of minute virus of mice major nonstructural protein in insect cells: purification and identification of ATPase and helicase activities. Virology 185:90–98
Yodmuang S, Tirasophon W, Roshorm Y, Chinnirunvong W, Panyim S (2006) YHV-protease dsRNA inhibits YHV replication in Penaeus monodon and prevents mortality. Biochem Biophys Res Commun 341:351–356
Zilli L, Schiavone R, Scordella G, Zonno V, Verri T, Storelli C, Vilella S (2003) Changes in cell type composition and enzymatic activities in the hepatopancreas of Marsupenaeus japonicus during the moulting cycle. J Comp Physiol 173:355–363
Acknowledgments
We thank Dr. Katie H Smith for her grammatical correction. Our appreciation is expressed to Mr. Banjong Nisapawanit and Mr. Prasert Fugtang-on for their kindness to provide shrimp (post-larva) and the infected shrimp, respectively. We are very grateful to Miss Chanikarn Boonchoy, Miss Suparp Hongthong, Mrs. Payong Sukprasert, Mrs. Pensri Hongthong, and Mr. Wanlop Chinnirunvong for their technical assistance. This work is supported by Thailand Research Fund (DBG 5080005 to S.P.), Commission on Higher Education (CHE) and Mahidol University research grant. P.A. was supported by TRF-CHE grant (MRG4980036).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Attasart, P., Kaewkhaw, R., Chimwai, C. et al. Inhibition of Penaeus monodon densovirus replication in shrimp by double-stranded RNA. Arch Virol 155, 825–832 (2010). https://doi.org/10.1007/s00705-010-0649-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-010-0649-5