
Abstract
Parkinson’s disease (PD) comprises a spectrum of disorders with differing subtypes, the vast majority of which share Lewy bodies (LB) as a characteristic pathological hallmark. The process(es) underlying LB generation and its causal trigger molecules are not yet fully understood. α-Synuclein (α-syn) is a major component of LB and SNCA gene missense mutations or duplications/triplications are causal for rare hereditary forms of PD. As typical sporadic PD is associated with LB pathology, a factor of major importance is the study of the α-syn protein and its pathology. α-Syn pathology is, however, also evident in multiple system atrophy (MSA) and Lewy body disease (LBD), making it non-specific for PD. In addition, there is an overlap of these α-synucleinopathies with other protein-misfolding diseases. It has been proven that α-syn, phosphorylated tau protein (pτ), amyloid beta (Aβ) and other proteins show synergistic effects in the underlying pathogenic mechanisms. Multiple cell death mechanisms can induce pathological protein-cascades, but this can also be a reverse process. This holds true for the early phases of the disease process and especially for the progression of PD. In conclusion, while rare SNCA gene mutations are causal for a minority of familial PD patients, in sporadic PD (where common SNCA polymorphisms are the most consistent genetic risk factor across populations worldwide, accounting for 95% of PD patients) α-syn pathology is an important feature. Conversely, with regard to the etiopathogenesis of α-synucleinopathies PD, MSA and LBD, α-syn is rather a bystander contributing to multiple neurodegenerative processes, which overlap in their composition and individual strength. Therapeutic developments aiming to impact on α-syn pathology should take this fact into consideration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the second most frequent chronic neurodegenerative disease worldwide. PD is clinically characterized by bradykinesia and rigidity, often accompanied by postural instability and resting tremor. It is assumed that most cases have a multifactorial pathogenesis, and numerous genetic and environmental risk factors have been identified (Sian-Hulsmann et al. 2011; Nalls et al. 2014; Kalia and Lang 2015). Its incidence ranges from 8 to 18 per 100,000 persons each year. There are estimations which propose a twofold rise of PD prevalence within the next 20 years (Dorsey et al. 2018). Onset of PD is rare under the age of 50 years and considerably rises after the age of 60 years. The exact cause of PD is not well understood. The term PD actually reflects a super ordinate concept for a disease entity consisting of different subtypes (Przuntek et al. 2004). A certain clinical and neuropathological overlap exists between each of them. However, not all these various PD forms share the neuropathological inaugurated concept of an increased presence of Lewy bodies (LB) in the substantia nigra (SN) as an essential pathological hallmark (Weiner 2008). Presumably, LB generation results from mutated α-synuclein (α-syn), elevated α-syn levels in the brain and/or post-translational modified α-syn, which becomes misfolded and subsequently forms protein aggregates such as oligomers and fibrils. These structures impair nerve cell communication and may spread to healthy neurons (Burke et al. 2008; Sian-Hulsmann et al. 2015). Consequences of LB formation (Fig. 1) are increased oxidative stress, disruption of axonal transport for instance of neurotransmitter vesicles, transcriptional dysregulation, protein sequestration, mitochondrial and synaptic dysfunction, inhibition of the ubiquitin/proteasome system and altered lysosomal clearance via autophagy (Foley and Riederer 1999; Burke et al. 2008; Sian-Hulsmann et al. 2015). Formation of LB is observed in the majority of PD patients, including more than 90% of sporadic PD cases. To date, it is far from clear whether these abnormalities represent a specific process responsible for triggering the early onset of PD. They may also result from an unspecific side reaction of the pathological event cascade responsible for cell death and neurodegeneration (Sian-Hulsmann et al. 2015). Neurochemically, motor symptoms of PD are mainly characterized by a loss of dopamine-synthesizing neurons as well as dopaminergic transporters, mainly in the SN (Uhl 1998). Additionally, chronic neuronal depletion also occurs in other neurotransmitter systems both in the periphery and the brain (Jellinger 2010). Thus, various neuronal cell types degenerate in an individually different and pronounced manner in various brain regions. This phenomenon contributes to the clinical heterogeneity of PD (Przuntek et al. 2004). It is under debate whether this process of slow neuronal dying follows a certain pattern, for instance starting in the gastrointestinal tract, ascending over the brainstem with further spread to other regions of the brain (Brooks 2010; Braak and Del Tredici 2017; Rietdijk et al. 2017; Braak et al. 2003a, b; Jellinger 2010). Other authors meet this theory with criticism (Jellinger 2019).

a Normal neuromelanin-containing neuron of midbrain (substantia nigra). b Early stage of alpha-synuclein aggregation (Lewy body) in midbrain neuron. c Late stage of Lewy body with adhesion to the inner neuronal membrane (midbrain). d Lewy body already leaving the neuromelanin-containing neuron (midbrain) (in part from Sian-Hulsmann et al. (2015)). Immunohistochemical staining with a monoclonal primary antibody directed against alpha-synuclein (Novocastra/NCL-ASYN, clone KM51, dilution 1:1000). Please note the intense staining of the Lewy body halo
Monogenic gene carriers for familial forms of PD have been identified. They only account for about 5–10% of PD patients. Up to date, a total of 23 loci and 19 causative genes have been associated with PD (Lunati et al. 2018; Del Rey et al. 2018). The corresponding genetic animal models of PD did not develop the typical motor PD symptoms of dopaminergic neurodegeneration and pathological SN protein aggregation during aging. Therefore, manifestation of PD probably results from so far unidentified environmental triggers acting on genetically predisposed individuals, possibly in combination with other predisposing and/or exacerbating risk factors, for example pesticide exposure with impaired detoxification mechanisms. Mostly, these investigations regarding environmental epigenetic influences, chronic exposure to toxins and perviously unknown infections did not take into consideration the various, yet not well-characterized subtypes of PD (Alonso-Navarro et al. 2014; Ahmed et al. 2017; Foley and Riederer 2000). Nevertheless animal models of PD, i.e., based on toxin exposure, were developed (Gotz et al. 2004; Jiang and Dickson 2018; Trigo-Damas et al. 2018; Bringmann et al. 1995). They show motoric symptoms of PD. However, the in vitro and most of the in vivo PD-mimicking models do not reflect the chronic progression of slow, sometimes intermittent neuronal death. In conclusion, the pathogenesis of sporadic PD forms is still not known. Manifestation of the disease is probably the consequence of several complementary mechanisms leading to onset of PD as a common end point. This individually different acceleration of neuronal dying is of multifactorial origin, its progression shows no uniform pattern and it can be discontinuous with relapses or constant. The majority of the at-risk population and PD patients themselves tend to avoid early genetic investigations or premotor diagnosis, since a disease-modifying therapeutic approach is not available.
In a clinical setting, heterogeneous onset and a combination of the main motoric symptoms of rigidity, akinesia and resting tremor are looked upon as the main diagnostic features. In clinical practice, a diagnosis is usually made when a substantial number of dopaminergic neurons and their terminals in the SN are lost and motoric symptoms appear. Axial symptoms such as gait (freezing) and balance problems mostly occur later in the course of PD. A wide array of initially unspecific non-motoric symptoms can precede the initially often temporary onset of motoric symptoms against the preexisting personality structure, which may influence the initial awareness (Poewe et al. 1990). Focus on initial non-motor signs of PD and initiatives for earlier detection of PD have gained significant interest to better characterize this so-called “prodromal” or “premotor” interval of PD. To date, no reliable and specific biomarker for an earlier detection of PD has been found. Notable techniques include functional imaging methods for visualization of the emerging dopaminergic deficit. Molecular probes to detect misfolded α-syn and the tracking of its accumulation for potential assessment of PD progression have not yet reached the clinical routine (Berg et al. 2013).
Basic pathology/neurochemistry
Intracellular inclusion bodies in post-mortem brain tissue from sporadic paralysis agitans (PD) patients were first described by Friedrich H. Lewy in 1912. In 1919, at the Salpêtrière, Constantin Trétiakoff reported pathological inclusions that he named ‘corps de Lewy’ in the SN in PD (Holdorff et al. 2013; Engelhardt 2017). Initial studies based on histochemical methods to visualize LB and later on ultra-structural approaches revealed the presence of organized filaments within LB.
However, Lewy pathology is not restricted to the brain and can also be found in the spinal cord and peripheral nervous system. According to Braak’s theory, Lewy pathology starts in the peripheral nervous system and progresses through six different stages within the central nervous system in a caudal-to-rostral direction, correlating with disease progression in PD (Braak et al. 2003a). In the Braak-staging classification, α-syn pathology is not found in the SN region of the midbrain until stage 3, suggesting that early pre-motoric symptoms in PD are caused by Lewy pathology mainly found in the enteric nervous system, olfactory system and brain stem including medulla and pons. However, further studies are needed to explicitly confirm Braak’s staging classification as well as the α-syn spreading theory (Dickson 2012).
Another distinct characteristic of PD pathology is the specific loss of dopaminergic (DA) neurons within the SN pars compacta. It has been shown that the severity of PD motoric manifestations correlates with the reduction of DA neurons and thus striatal dopamine levels. Motoric symptoms in PD respond well to dopamine replacement therapy, which is still one of the standard therapies in PD (Fahn 2003). However, it is worth mentioning that by the time motoric symptoms are pronounced and PD is clinically diagnosed, already 50–60% of DA neurons are lost, resulting in an 80% reduction of striatal dopamine (Pavese and Brooks 2009; Bernheimer et al. 1973). These data have been replaced by the notion that at that time only about 30% of dopaminergic neurons but 50–60% of their terminals have been lost (Cheng et al. 2010). Furthermore, it must be kept in mind that the recent standard therapies—including L-dopa, dopamine agonists, monoamine oxidase-B-inhibitors and deep-brain stimulation—are not curative, since they do not stop the progressive loss of DA neurons. For the development of novel therapeutic targets, a comprehensive understanding of the molecular causes of PD is needed. Although α-syn aggregation has been proposed to have a causal role in the death of DA neurons, its exact relationship to neuronal loss has not been sufficiently elucidated to date. One important aspect in this regard is a better understanding of the impact of aggregated α-syn, which—under physiological conditions—is localized at the presynaptic terminal, promoting synaptic-vesicle fusion and trafficking on cellular pathways. Once formation of oligomers and fibrils result in insoluble forms comprising beta-sheets, an even faster aggregation can be triggered at a lower pH—as found in lysosomal compartments. However, it is not only an ineffective clearance by lysosomal or ubiquitin proteasome systems, but also oxidative stress, which is related to an increased tissue iron content in the SN in PD, further enhancing formation of α-syn aggregates. So far, the cause of the increased tissue iron content in PD is not entirely understood, although it has been described that multiple iron pathways in the brain may play a role (Berg et al. 2001; Sian-Hulsmann et al. 2011; Zecca et al. 2004a; Ndayisaba et al. 2019; Foley and Riederer 2000; Wypijewska et al. 2010), with iron seeming to contribute to the aggregation of α-syn.
Large α-syn aggregates have been shown to interfere with synaptic vesicle motility and organelle axonal transport. Moreover, they disturb neurotransmitter release and contribute to mitochondrial, endoplasmic reticulum and Golgi dysfunction. Aggregated α-syn binds to histone complexes, reduces histone acetylation and causes mitochondrial and synaptic dysfunction (Lardenoije et al. 2015).
Importantly, the normal clearance mechanisms, including autophagy, which degrade damaged organelles and proteins, are also affected by α-syn overexpression and aggregates, likely resulting in a vicious circle perpetuating the toxic effects of the aggregates (Wong and Krainc 2017).
Underscoring these severe effects of α-syn aggregates on cellular function, further valuable information has recently come from genome sequencing as well as genome-wide association studies (GWAS) of PD patients. Sequencing results indicate that not only mutated or excess α-syn, but also genes related to mitochondrial oxidative stress, cellular trafficking and lysosomal function are involved in PD pathology (Chang et al. 2017).
Interestingly, loss-of-function mutations in one allele of the lysosomal enzyme β-glucocerebrosidase (GBA1) have been proven to be a significant risk factor in PD (Sidransky et al. 2009). Moreover, recent data indicate that the lysosomal link in PD extends beyond GBA1 with novel gene variants found in the autophagic-lysosomal pathways, including various lysosomal enzymes and lysosomal membrane proteins and transporters (Chang et al. 2017). Also, from a biochemical point of view, there is a strong relationship between lysosomal function and α-syn aggregation. Moreover, oligomeric α-syn conformers have been shown to negatively influence lysosomal pathways, for instance by blocking trafficking of lysosomal enzymes (e.g., GBA1), leading to a bidirectional pathogenic loop (Mazzulli et al. 2011). To fully understand underlying disease mechanisms in PD, molecular and structural details of α-syn aggregation have to be further deciphered. Typically, α-syn is a natively unfolded protein in aqueous solution, but is able to adopt oligomeric and fibrillar conformations under certain conditions [such as mutations in the α-syn gene (SNCA), lipid storage, oxidative stress and post-translational modifications]. Multiple studies have shown that glycosphingolipids metabolites, like the GBA1-substrate glucosylceramide (GluCer), can interact and induce α-syn aggregation. This is thought to potentially happen via a conversion of physiological α-syn oligomeric forms into neurotoxic oligomers, that, moreover, are able to seed amyloid fibril formation (Zunke et al. 2018) (Fig. 2).

The potential role of lysosomal function and metabolites on α-syn aggregation. Physiological α-syn can be found in monomeric as well as in oligomeric form in healthy human DA neurons. Lysosomal dysfunction and/or impairment in GBA1 activity leads to an increase in glucosylceramide (GluCer). Interaction of GluCer with α-syn oligomers leads to the formation of neurotoxic oligomers, which further accelerate fibril formation (Zunke et al. 2018)
Although genetic and mechanistic data strengthen the consequences of excess α-syn and its aggregation on a variety of cell functions in PD pathology, the full picture of molecular causes leading to the progressive loss of DA neurons is very complex and might be a reciprocal interplay of many different intra- and intercellular pathways.
Neuromelanin
Neuromelanin (NM) is an insoluble, granular pigment, chemically a complex organic polymer, which is responsible for giving certain parts of the brain a macroscopically dark appearance. It can be found in the brain in large concentrations principally in dopaminergic neurons of the SN and the ventral tegmental area, as well as in noradrenergic neurons of locus coeruleus, of the ventrolateral reticular formation and of the nucleus of the solitary tract in the medulla oblongata (Zecca et al. 2004a). Since the anatomical distribution of NM largely matches the pattern of neuronal degeneration in PD, it has long been hypothesized that it may be involved in the pathogenesis of PD. Surprisingly, the precise biochemical pathway leading to NM formation remains unknown. In dopamine (DA)-containing neurons, NM synthesis appears to result from iron-mediated oxidation of DA (Double et al. 2000; Jellinger and Paulus 1992; Gerlach et al. 2008), which leads to reaction with beta-sheet proteins (Zucca et al. 2017). It has been suggested that NM plays a role in the regulation of cytosolic dopamine by sequestering dopamine or its adducts in autophagic vacuoles/lysosomes (Fedorow et al. 2005).
NM has a strong affinity for transition metals such as iron (Zecca et al. 1994). Under physiological concentrations, iron may be cleaved in its redox-active form from the cytosol (Gotz et al. 2004; Gerlach et al. 2008). In the presence of iron overload, however, iron may be bound by a low-affinity site of NM where it could promote harmful redox reactions. Iron released from NM increases oxidative stress in mitochondria (Shamoto-Nagai et al. 2006). NM itself also has antioxidant properties and can bind to organic molecules, including those that have been linked with PD etiology (Karlsson and Lindquist 2016).
Retention of potentially toxic agents may, on the one hand, protect NM-containing neurons. On the other hand, such intracellular depots may slowly release accumulated compounds and cause cellular toxicity. Degeneration of DA neurons in PD is accompanied by the release of NM into the extracellular compartment. Subsequent phagocytosis of NM by microglia gives rise to neuroinflammation. Along with concomitant release of previously cleaved metals and toxic compounds from NM, neuroinflammation may trigger a vicious cycle, resulting in accelerated neurodegeneration.
The issues discussed significantly point to NM as a potential contributor to the Parkinsonian pathogenesis. An intriguing question is whether and how pathogenic pathways connected to NM converge with other cardinal features of Parkinsonian pathogenesis. As α-syn is regarded at the top of LB pathology as the histological hallmark in sporadic as well as in some monogenetic form of Parkinsonism, the question of whether and how NM is connected with accumulation of misfolded α-syn is of special interest.
The interaction of NM and α-syn in PD has recently been reviewed by Xu and Chan (Xu and Chan 2015). α-Syn is expressed in the SN and especially in neurons containing NM (Purisai et al. 2005). α-Syn increases with age (Xuan et al. 2011; Chu and Kordower 2007). This is associated with NM accumulation (Xuan et al. 2011) along with nigro-striatal dopamine depletion (Chu and Kordower 2007). Reasons for this might include cross-reactions of α-syn with NM (Fasano et al. 2003; Halliday et al. 2005). Here, NM is able to enhance the toxicity of α-syn (Li et al. 2012) as are early oxidation products of dopamine (Rekas et al. 2010; Bisaglia et al. 2007). Mechanisms of toxicity seem to include components of α-syn, NM, OS, iron and dopamine/dopamine oxidation products (Mandel et al. 2004).
Moreover, recent molecular genetic, biochemical and immunopathological studies in human specimens and animal models have implicated dysfunctional protein processing in the endosomal-lysosomal pathway as a unifying theme in the pathogenesis of PD (Perrett et al. 2015). Endosomes are critical hubs for the re-use, breakdown, trafficking of proteins and their extracellular release in exosomes. α-Syn overexpression and aggregation may disrupt endosomal and lysosomal function via multiple mechanisms (Perrett et al. 2015). NM granules are present in lysosomes of dopaminergic neurons (Tribl et al. 2006; Plum et al. 2016), and NM deposition is associated with α-syn accumulation in aging neurons (Xuan et al. 2011). According to one hypothesis, NM deposition in dopaminergic neurons may lead to α-syn accumulation by enhancing their susceptibility to stochastic molecular defects in the endosomal–lysosomal pathway (Perrett et al. 2015). Lysosomal dysfunction may subsequently lead to deficits in mitophagy and the buildup of dysfunctional mitochondria (Gegg and Schapira 2016). Hence, NM may mediate neuronal vulnerability in PD via induction of α-syn expression and aggregation (Xu and Chan 2015). Conversely, α-syn also facilitates biosynthesis of NM, presumably by increasing the levels of cytosolic dopamine (Pan et al. 2012). Dopaminergic neurons derived from PD patients displayed mitochondrial oxidant stress that led to oxidized dopamine accumulation and ultimately resulted in lysosomal dysfunction and α-syn accumulation (Burbulla et al. 2017). According to this scenario, another vicious cycle between NM formation, α-syn aggregation, endo-lysosomal dysfunction and deficient mitophagy may develop. Higher concentrations of dopamine in human SN neurons, compared to other species, may explain these neurons’ enhanced vulnerability as a consequence of enhanced NM formation (Zecca et al. 2008; Carballo-Carbajal et al. 2019; Aimi and McGeer 1996).
NM is composed of brown/black eumelanin and yellow/reddish pheomelanin that are arranged in a pheomelanin core and a eumelanin surface (Bush et al. 2006). Despite different biochemical synthesis, peripheral melanin contains the same two different melanin species as NM. The relationship of the two species appears to determine an individual’s pigmentation trait of skin and hair, a known modulator of the risk of developing PD. Ultrasound sonography of the midbrain region in healthy individuals revealed that a lighter skin phototype was associated with a larger echogenic SN area and increased prevalence of an abnormally enlarged echogenic SN area (Rumpf et al. 2015). Since the iron-binding capacity of pheomelanin exceeds that of eumelanin, vulnerability of dopaminergic SN neurons toward oxidative stress may be enhanced with higher concentrations of pheomelanin in individuals with a lighter phototype. Although the substrate of an expanded SN echogenic area is not known, it is regarded as a biomarker of PD. Therefore, finding enhanced prevalence of enhanced SN echogenicity in light hair phototypes may provide evidence linking pigmentation to the pathogenesis of PD. However, the nature of this link and its relationship to other elements of the molecular pathogenesis of PD remains speculative (Rumpf et al. 2015).
Since NM is a scavenger of paramagnetic metals, regions rich in NM appear hyperintense in MR T1-fast spin echo weighted imaging. Using NM-sensitive sequences, topographically specific reductions of the NM-signal have been demonstrated in patients with Parkinsonian disorders (Pavese and Tai 2018). Interestingly, the NM MR signal seems to be distinct from that of iron in T2* relaxometry of SN pars compacta, in that the former, but not the latter, is correlated to striatal dopamine function (Reimao et al. 2015). This finding suggests that both pathological pathways may be distinct at least at the time when the disease is clinically apparent.
Neuroinflammation
Another feature of PD pathology is neuroinflammation, which might also play a pivotal role in disease pathogenesis, since chronic microglial activation, potentially mediated by α-syn might contribute to the death of DA neurons (Sanchez-Guajardo et al. 2013).
In PD degeneration of dopaminergic neurons can be found, which is expedited by the risk factor of age and can be triggered by as yet unidentified factors. Until the first publication by McGeer et al. (1988) neurodegenerative disorders had rarely been associated with inflammatory processes in the brain (Eikelenboom and Stam 1982; Fischer 1907). In these seminal publications, an increase in the number of signs of neuroinflammation was described in the microglia of post-mortem brains. Neuroinflammatory processes contributing to the loss of dopaminergic neurons include activated microglia, reactive astrocytes and the release of cytokines and chemokines, component activation and reactive oxygen species. A number of authors were able to demonstrate that levels of pro-inflammatory mediators such as TNF-α, ILß, IL6, iNOS and COX2 in the SN are increased (Tansey et al. 2007; Mogi et al. 1994, 1995, 1996). It has been speculated that due to these neuroinflammatory processes the blood–brain barrier becomes permeable. Recently, substantial contribution has been added to our knowledge of neuroinflammatory changes in Parkinson patients (Hirsch et al. 1998, 2012). The relevance of T17 cells has gained special attention (Sommer et al. 2018).
In the meantime, we have sufficient evidence that there is a neuroinflammatory component in PD (Hirsch et al. 2012). However, neuroinflammation might be of low specificity since it has also been demonstrated in various psychiatric and neurological diseases such as for example Alzheimer’s disease, depression, schizophrenia and, again, in PD. There is, however, still considerable debate on the relevance of these neuroinflammatory changes for necrosis in the dopaminergic neurons of the SN. Zecca et al. demonstrated that the injection of neuromelanin induced acute microglia activation (Zecca et al. 2008). But in spite of the many indications for neuroinflammatory changes in the brains of Parkinson’s patients, the situation is similar to that of the status of α-syn in that it is still unresolved whether we have pathogenetic causation relevant for disease progression or rather a secondary alteration. One major concern here is that most of the data devolve from toxin-based animal models and not from humans. Thus, there is debate on whether α-syn protein fragments induce T cell-mediated immune reactions or, vice versa, whether the development of α-syn is triggered by such processes. α-Syn can induce the formation of LB, which are assumed to have pro-inflammatory effects. Overexpression of α-syn in cell cultures leads to an increase of microglia-induced release of pro-inflammatory molecules (Su et al. 2008).
Genetic background
Two parallel developments led to the discovery that the protein α-syn was a major component of LB. The non-amyloid component of AD plaques (NACP) was identified and cloned, revealing a precursor of 140 amino acid residue protein in the brains of patients with dementia (Ueda et al. 1993). Second, Polymeropoulos and colleagues identified in 1997 the alanine to threonine change at residue 53 (A53T) of the α-syn gene (SNCA) in a large Italian kindred and two seemingly unrelated Greek families with an early-onset autosomal dominant PD (Golbe et al. 1996; Polymeropoulos et al. 1997; Puschmann 2013). This A53T mutation, the first mutation in α-syn to be linked to a monogenic PD, has been detected in about 70 cases and is thus the most frequent of the α-syn mutations.
In the following years, α-syn mutations (A30P, E46K, Q51D and A53E) were discovered as additional PD-linked disease-causing mutations (Moore et al. 2005). More importantly, LB in brains of sporadic PD patients consisted of α-syn as their major component (Spillantini et al. 1997; Gai et al. 1998). Taken together, these neuropathological and genetic findings form the solid basis for the notion that neural LB are relevant for a number of diseases consequently named as synucleinopathies, such as sporadic PD, diffuse LB diseases and MSA. It should be noted that gene multiplications have also been linked to PD, indicating that duplications/triplications of SNCA are implicated in an apparent gene-phenotype association (Singleton et al. 2003; Ibanez et al. 2004; Kalia and Lang 2015; Lee and Trojanowski 2006).
Structure and function of α-syn
Monomeric α-syn is a soluble, presynaptic, 140 amino acid long protein which is involved in normal mitochondrial and lysosomal function, synaptic transmission and neurotransmitter release (Bendor et al. 2013; Lee and Trojanowski 2006).
The SNCA gene is located on the long arm of chromosome 4 and comprises 6 exons out of which 5 are coding. α-Syn belongs to a highly conserved protein family and its “brothers” are called beta and gamma synuclein (Bungeroth et al. 2014). The hydrophobic NAC domain (aa 61–95) is considered as the most important prerequisite for pathological α-syn assembly into fibrils involving a beta-sheet structure. α-Syn has multiple physiological functions in the CNS, which are not yet fully understood. A regular role in striatal dopamine release has been suggested using α-syn knock-out mice showing faster recovery from repetitive stimulation and partial striatal dopamine depletion (Abeliovich et al. 2000). Mostly observed within the presynaptic compartment, α-syn itself shows a SNARE-complex chaperone activity proposed to maintain neuronal synaptic homeostasis during aging. In general, α-syn is not restricted to the central nervous system, but interestingly had also been found to play an important role in membrane curvature and vesicular budding (Burre et al. 2010). In principle, α-syn is considered a natively unfolded protein with the potential to adopt alpha-helical but also beta-sheet structure, both tightly linked to its physiological or pathological properties. Finally, α-syn may occur physiologically as a helically folded tetramer, however, the aforementioned conformational flexibility of α-syn may be the underlying cause for its multi-functional properties (Bartels et al. 2010; Dettmer et al. 2015).
Although α-syn is predominantly expressed in the pre-synapse, it is released into the extracellular space facilitating the formation of LB and neuron-to-neuron transmission. Transsynaptic transmission of α-syn is believed to follow a stereotyped pattern of spread (Braak et al. 2003a). In consequence, α-syn can be found in the CSF (Shi et al. 2011). CSF α-syn levels have been investigated as potential biomarkers in patients and at-risk populations of PD (Sierks et al. 2011). In another clinical study, a panel of nine CSF biomarkers, including α-syn, was able to differentiate atypical Parkinson syndromes from patients with PD and dementia (Magdalinou et al. 2015). In addition, α-syn as a CSF biomarker may be useful to diagnose sporadic Creutzfeld–Jakob-Disease (Llorens et al. 2018). However, differences in analytical procedures yielded heterogeneous results with lower specificity.
While the causative role of pathologic SNCA mutations seems to be unquestionable in monogenic α-synopathies, the role of abnormal SNCA and α-syn aggregation in the pathogenesis of sporadic PD or other genetic variants of PD (e.g., those linked to Parkin, PINK1 or LRRK2 mutations) is less obvious. Parkin mutations cause autosomal recessive PD and heterozygous gene carriers have an increased risk for developing PD. The protein encoded in the Parkin gene is an ubiquitin ligase and its dysfunction might be associated with decreased autophagic degradation of α-syn aggregates (Tan et al. 2008). The PINK1 gene encodes a kinase that is believed to protect against stress-induced mitochondrial dysfunction and apoptosis.
PINK1 (PARK 6)-induced autosomal recessive, early PD shows neuronal loss in SN but no LB pathology (Schneider and Alcalay 2017). LRRK2 (PARK 8)—most frequent late-onset PD—shows neuronal loss in SN and locus coeruleus but inconsistent LB pathology (Pont-Sunyer et al. 2017). The LRRK2 gene is also associated with mitochondrial function and autophagy (Gomez-Suaga et al. 2012). While there is evidence for an association between these mutations and α-syn accumulation, the causal link is yet unclear. Here the notion is of importance, that there is no evidence for LB in postencephalitic parkinsonism (Jellinger 2009), MPTP-induced parkinsonism (Langston et al. 1999) and PD without nigral degeneration-SWEDD (Ling et al. 2016). In addition, lower brainstem pathology is not an obligatory trigger site of PD and in about 7–16% of PD the dorsal nucleus of the vagus is preserved as reviewed by Jellinger (2019). All this is indication to suggest that α-syn-related LB pathology is not necessarily a prerequisite of PD.
Dopaminergic terminal loss and its relationship to clinical symptoms depend on the type of monogenic disorder. Loss of dopaminergic terminals in relation to disease severity is much greater in Parkin carriers than in sporadic PD (Varrone et al. 2004), while loss of striatal dopamine neurons as indexed with Dopamine Transporter SPECT is similar in sporadic PD and LRRK2-associated PD (Sierra et al. 2017; Wile et al. 2017). Asymmetry of dopaminergic terminal loss was greater in LRRK2 carriers than in PD patients with SNCA, PINK1 or Parkin mutations (McNeill et al. 2013). Taken together, the clinical consequences of the underlying pathology in LRRK2-associated PD resemble those in sporadic PD more than those in SNCA, PINK1 or Parkin-associated forms.
Despite notable differences in dopamine transporter binding, the functional changes at a cortical network level in Parkin-associated PD and sporadic PD do not differ much (van Eimeren et al. 2010), and preclinical compensatory changes were found to be similar in PINK1 and Parkin mutation carriers (van Nuenen et al. 2009). This evidence implies that functional endophenotypes in terms of cortical network function might be similar in monogenic and sporadic forms of PD, while the molecular synaptic endophenotype might be different.
Abnormalities of α-syn-related genes have rarely been detected in sporadic PD. While homozygous abnormalities in α-syn genes (SNCA = PARK1 or PARK4) cause monogenic PD as discussed, heterozygous mutations in α-syn-related genes are infrequently found in sporadic PD, even in patients with positive family history. The conception is that different levels of α-syn involvement or genetic abnormality could define different clinical subtypes of Parkinsonian syndromes, but data supporting this hypothesis are lacking. In general, earlier twin studies showed that the degree of heritability in PD is moderate (Tanner 2003).
It is currently consensus that a minor (non-pathologic) abnormality in SNCA alone is not sufficient to cause PD. A second abnormality (“dual hit hypothesis”) is necessary for the defect in SNCA to become causative. Genome-wide association studies (GWAS) have revealed a number of candidate genes which could qualify as additional risk factors (Nalls et al. 2014). More recent meta-analysis of target genes of brain micro-RNA showed significant association of genetic variants in nine loci (Schulz et al. 2019). Post-translational modifications and other epigenetic factors are likely to play a significant role in this context (Lardenoije et al. 2015). Production and clearance of α-syn underlies an armada of enhancing, repressing and silencing gene-regulation mechanisms. The A53T SNCA mutation and hypomethylation of the SNCA gene can both cause increased transcription of SNCA mRNA and increase α-syn levels in the brain. One of six reported SNCA mutations, His50 Glu, was consistently identified in large population databases, but no enrichment was evident in PD cases compared to controls, thus showing insufficient evidence for pathogenicity (Blauwendraat et al. 2018). Other deregulations of SNCA gene transcription may shift the balance between α-syn production and clearance towards increased production (Miller et al. 2004). PD epigenetics is an evolving field which, however, has so far struggled to clearly identify disease-relevant epigenetic factors.
Despite this gain of knowledge, a number of controversies about α-syn genetics and function remain to be resolved. It has been difficult to clearly distinguish PD-related pathological processes from normal aging in the human brain. The initial trigger and sequence of pathological events which eventually result in the clinical manifestation of PD have been hard to identify. For example, the interaction of gut microbiota and α-syn dysfunction is a major focus of current research (Johnson et al. 2019). The roles of deleterious mitochondrial energy metabolism, cytosolic homeostasis, lysosomal dysfunction, oxidative stress and inflammation for aggregation of α-syn and manifestation of PD symptoms remain to be elucidated. The factors which lead to the clinically and pathologically distinct manifestations in PD, DLB or MSA are also unknown.
Αlpha-synuclein physiology and the involvement of SNCA regulation in Parkinson’s disease
Even though the exact function of α-syn is still unknown, the initial observation of α-syn in presynaptic terminals of the neurons suggested a physiological function of α-syn in neurotransmission. α-Syn was shown to modulate synaptic functions by facilitating vesicle clustering, recycling and docking to the cell membrane (Lashuel et al. 2013). As mentioned already, the unique character of α-syn in neurotransmission occurs via its chaperone activity, promoting SNARE-complex assembly and the rapid fusion of synaptic vesicles. However, as α-syn is (1) not present in all presynaptic terminal buttons and (2) presents a weak membrane-interaction pattern to synaptic vesicles, α-syn was suggested to have a more global function (Huang et al. 2019).
Accordingly, α-syn interplays with multiple members of Rab GTPase family, a family of proteins involved in the regulation of intracellular trafficking processes (Miraglia et al. 2018). These interactions suggest that α-syn also plays a role in intracellular protein trafficking by promoting vesicular transport. Moreover, α-syn also contributes to axonal transport by regulating the nucleation and the growth velocity of microtubules in the assembly and in the remodeling of growth cone (Carnwath et al. 2018). Therefore, α-syn behaves as a soluble-interacting protein binding to diverse organelles and dispersing from membranes for the fine tuning of cytoskeleton plasticity.
Mechanisms involved in SNCA expression
Molecular mechanisms regulating gene expression are highly complex and interconnected. A simplistic approach to review and examine the relevance of these mechanisms concerning the expression of SNCA in PD is to find an association between mutations in SNCA and gene expression. Genome-wide association studies (GWAS) had already demonstrated that SNCA is one of the most common and consistent susceptible genes for the sporadic form of PD (Krüger et al. 1998).
Most significant associated variants are located in the 5′, 3′ and intronic regions, respectively, suggesting a critical role of non-coding regions of SNCA on its own expression. One important example is the presence of the binding sites of two microRNAs (miR-7 and miR-153) in the SNCA 3′ untranslated region (UTR). As miR-7 was shown to decrease the expression of α-syn, variants in the binding site region affecting the affinity of binding may increase the expression of SNCA (Doxakis 2010). GWAS have highlighted an association of the promoter region of SNCA with PD. The microsatellite D4S3481, also termed REP1, is located about 10 kb upstream of the translational start of SNCA. GWAS had also contributed in the discovery of methylation-dependent putative promoter in SNCA, such as in the intron 1 in which the variant rs3756063 was associated with SNCA hypomethylation. Promoter hypomethylation is generally associated with increased expression, but the relevance of SNCA methylation on its expression has not been validated yet (Miranda-Morales et al. 2017) (see Fig. 3).

Human SNCA regions in which main SNPs reviewed in the present study are located. Promoter (REP1 repeats), exon2 and exon 3 (PM: point mutation A30P, E46K, G51D, A53T), intron 4 (CpGs) and 3′ UTR (miRNA-binding sites). Exons are represented in the black, untranslated region in the gray and introns in the white colors. En enhancer, PM point mutation
Multiple binding/enhancers/repressors were also identified in the genomic region surrounding SNCA but few were characterized in detail (Piper et al. 2018). For example, two enhancers located in intron 4 of the SNCA gene were identified using the Assay for Transposase-accessible Chromatin Sequencing (ATAC-seq) and functionally evaluated using reporter essay and SNPs was pinpointed within these regions and correlated with PD. Similarly, multiple transcription factors were also reported to play a critical role in the expression of SNCA, such as PARP-1 which was shown to bind to the Rep-1 repeat by NACP-Rep1 and regulate activity of an artificial REP1/SNCA reporter construct. Another example is the ability of α-syn to bind to DNA, such as to the mitochondrial transcriptional co-activator gene PPARGC1A, and to RNA as α-syn was reported to binds its own mRNA (Surguchev and Surguchov 2017).
More recently, mosaicism and somatic mutations were also suspected to play an important role in neurodegeneration (Leija-Salazar et al. 2018). Mosaicism arising from early development or aging has been well characterized as a pathological mechanism associated with cancer due to the availability of the numerous sample biopsies. The limited availability of brain tissues is making the study of mosaicism in neurodegenerative diseases more challenging. Despite this limitation, aneuploidy and more precisely increased copies of SNCA were observed in the human brain as well. This phenomenon was reported to occur more frequently in PD dopaminergic neurons (Mokretar et al. 2018), nevertheless, the cause and consequence of this mechanism remain unknown.
In conclusion, large efforts were performed over the last 20 years to determine the exact function of α-syn. Diverse functions were highlighted over multiple and complex research fields demonstrating the need of a unified community to decipher the mechanism linking SNCA and PD.
α-Synuclein conformation: a crucial target for overexpression, fibrillation and spreading
As reviewed above, the major hypothesis for the pathogenesis of PD currently focuses on an overexpression of α-syn. However, there are other additional metabolic disturbances initiating or at least accompanying α-syn overexpression. Increased concentrations of α-syn might be due to a loss of metabolic capacity to degrade α-syn via a dysfunctional proteasomal and/or lysosomal-autophagy system. Loss of metabolic capacity may be due to changes in the proteins conformational state. Here, the role of oxidative stress (OS), nitration, phosphorylation of α-syn, composition of cytosol, excitotoxicity, calcium metabolism and energy metabolism comes into play. In addition, the solubility of proteins, e.g., in the cytosol, is dependent on the size of the molecule, the molecular structure, size and order of aminoacid (AA) side chains, pH-value, content of electrolytes as well as on the concentration of the protein of interest, e.g., α-syn. Beside these aspects of general protein chemistry, more recent publications on molecular biological aspects are in line with the suggestion that components of the cytosol interact with proteins at conditions when cells fluid deranges (Porcari et al. 2015; Paleologou and El-Agnaf 2012; Narayanan and Scarlata 2001; Mattson 2011; Ugalde et al. 2019; Bernstein et al. 2004; Iofrida et al. 2017; Kim et al. 2011; Batelli et al. 2008; Jungermann and Möhler 1980; Löffler et al. 1979). The time lag between initiation of fibril formation and aggregation is dependent on such parameters.
α-Syn oxidation
α-Syn is a 140 amino acid intrinsically disordered protein (Ruf et al. 2008) (see Fig. 4). Importantly, this protein contains four tyrosine (TYR) residues. As described by Olteanu and Pielak (2004), peroxidative aggregation of α-syn requires TYRs (Olteanu and Pielak 2004). The combination of cytochrome c with hydrogen peroxide causes TYR-dependent peroxidative aggregation of proteins. TYR-39 is essential for wild type-like covalent aggregation, when α-syn adopts a collapsed conformation (Ruf et al. 2008). The importance of TYRs is also demonstrated by experiments showing that fibril formation is absent if the three C-terminal TYR-residues (TYR 125, 133,136) are replaced by alanine (Ulrih et al. 2008) (Fig. 4).

(modified from Ruf et al. 2008)
Location of tyrosine; this aminoacid is regarded as potential site of oxidation
The chemical reactivities of the TYRs in α-syn to form dityrosines have been studied by Ruf et al. (2008) using the reaction between cytochrome c and hydrogen peroxide. According to these studies by Ruf et al. (2008), TYR 133 and 136 are the most reactive ones, while TYR 125 is less able to accept a radical from cytochrome c. Lack of α-dimer and large amounts of degradation indicate that TYR 39 is the least reactive (Ruf et al. 2008). Upon aggregation, α-syn adopts a beta-sheet structure (Vamvaca et al. 2009).
Therefore, it is not farfetched to assume that OS plays a role in α-syn pathology, especially as α-syn has been shown to be a “ferrireductase”, thus catalyzing ROS and exacerbating neurodegeneration (McDowall and Brown 2016) (Table 1).
Additionally, α-syn interacts with membrane lipids like polyunsaturated fatty acids (PUFA) to stabilize its three-dimensional structure. Peroxidation of PUFAs may modify α-syn and may reduce their affinity to α-syn (Shamoto-Nagai et al. 2006). 4-Hydroxy-2-nonenal, a degradation product of Hn = 6 PUFA, has been detected in post-mortem brain tissue from PD and has been shown to induce oligomerization and modification of α-syn (Shamoto-Nagai et al. 2006). Moreover, peroxidation products, the role of ROS and iron-induced generation of toxic hydroxyl radicals with and without participation of mitochondrial pathology including reduction of respiratory chain activity have been frequently reviewed in the past (Shamoto-Nagai et al. 2006; Sian-Hulsmann et al. 2011).
At present, however, it remains to be determined whether and which of these oxidative-driven post-translational modifications (PTMs) are disease causative or rather reflect a bystanding role during disease progression. The precise pathways by which oxidative stress-associated α-syn modification mediate toxicity are also poorly understood at present. Moreover, PTMs of α-syn may not only be present within cells but may also occur in the extracellular space, leading to additional cellular damage.
This, however, is not a one-way pathology, as multifactorial players interact and finally contribute to disease onset and disease progression (Dickson 2007; Jellinger 2010, 2011; Sian-Hulsmann et al. 2011; Berg et al. 2001; Zecca et al. 2004b).
For example, pigmented neurons of the SN pars compacta (SNpc) and locus coeruleus (LC) are the most vulnerable ones in the pathology of PD (Hirsch et al. 1997).
This vulnerability (Surmeier et al. 2017; Surmeier 2018) has been associated with the regionally characteristic pigment NM. The earliest intra-cellular changes have been attributed to an increase of NM density in A9 neurons of normal morphological appearance and no characteristic pathology in PD and has been associated with oxidation and iron load (Halliday et al. 2005). Interestingly, it has been found that even in this early stage, α-syn can concentrate in the lipid component of the pigment (Halliday et al. 2005).
Other reactions of α-syn also may be of importance for changes in α-syn conformation including nitration (Uversky et al. 2005), glycation (Munch et al. 2000; Vicente Miranda et al. 2017), polyamine complexes (Fernandez et al. 2004) and phosphorylation (Pinho et al. 2019; Mbefo et al. 2010; Paleologou et al. 2010, 2008). Phosphorylation of α-syn along with copper also affects the protein aggregation process as described by Castillo-Gonzalez et al. (2017). It is, therefore, not surprising that there is an interaction of α-syn with divalent metal ions as reviewed recently (Binolfi et al. 2006, 2008, 2011; Miotto et al. 2014a, b, 2017; Gentile et al. 2018; Rasia et al. 2005). Metals such as iron and copper have been associated with oxidative stress and generation of ROS (Sian-Hulsmann et al. 2011; Zecca et al. 2004a; Gotz et al. 2004; Double et al. 2003).
While most of these interactions of α-syn have been described in models of α-syn conformation and fibrillation, verification in PD is lacking. The reasons for this are manifold: first, there is limited availability of post-mortem (p.m.) brain tissue. Second, isolation and purification of α-syn from such tissue requires pooling of p.m. tissue due to the low concentration of the various expected α-syn species. Third, highly sensitive analytical methods are required to detect changes in α-syn molecular structure. The methodological armamentarium includes NMR studies (Li et al. 2009; Salmon et al. 2010; Cho et al. 2009; Schwalbe et al. 2014) and highly sophisticated methodology like N–H spin–spin couplings (Xiang et al. 2013), a six-dimensional alpha-proton detection-based automated projection spectroscopy (APSY) (Yao et al. 2014) or neutron reflectometry and fluorescence spectroscopy (Jiang et al. 2015). For the determination of protein-bound 3,4-dihydroxy-phenylalanine as a marker for post-translational protein hydroxylation of tyrosine in human tissues ex vivo, a HPLC-method equipped with an electrochemical detector resp. fluorescence detector has been described by Harth et al. (Harth et al. 2001a, b).
An intermediate conclusion from these facts is that the pathology of α-syn is multifactorial and based on both genetic and non-genetic disturbances/dysregulations.
Hierarchical spreading of Lewy body pathology
An important feature of α-syn pathology is its propensity to form aggregates upon missense mutations causing misfolded α-syn or (relative) overexpression of the wild-type protein, the latter being represented by either multiplications of the α-syn gene locus (SNCA; duplications, triplications) or by common genetic variants (SNPs) in the SNCA gene that alter the expression levels of physiological α-syn in vitro and in vivo (Fuchs et al. 2008; Cronin et al. 2009). This explains how mutations in the SNCA gene contribute to either monogenic forms of PD with nearly 100% penetrance of the autosomal dominantly inherited mutations, or to the common sporadic form of the disease with SNPs acting as a genetic modifier of PD susceptibility.
The observation that regional differences of α-syn aggregation, in form of LB and Lewy neurites, occur in the brain of patients with PD led to the hypothesis of an intercellular spread of α-syn pathology along neuroanatomical structures (Braak et al. 2003a). As the toxic α-syn deposition in human brains occurs primarily in distal axons and synapses, this confers to the distal accentuation of α-syn pathology with primary loss of function reflected by impaired synaptic contacts before neurodegeneration occurs. In fact, axonopathy in presymptomatic PD is followed by neuronal degeneration (Longhena et al. 2017; Bridi and Hirth 2018). α-Syn is a naturally unfolded soluble protein that can randomly adopt a beta-sheet structure and subsequently polymerize into larger units of protofibrillar structures which finally form insoluble fibrils that coalesce to form the pathognomonic intraneuronal protein inclusions in PD. This process seems to be stochastically favored by increased concentrations of physiological α-syn protein or misfolded, more aggregation prone, mutant α-syn. Indeed, it was shown that synthetic preformed fibrillary α-syn precursors are able to seed the aggregation of α-syn by recruiting physiologically unfolded α-syn into amyloid-like protein inclusions, reviewed in Uchihara and Giasson (2016).
Here, it has been postulated that the conformational change of α-syn acts as a template for itself to form aggregates which leads to a spread of pathology as in prion diseases. In line with this hypothesis of a prion-like mechanism of α-syn spreading and neurodegeneration, a post-mortem study of patients with implantation of fetal human midbrain neurons revealed the development of LB pathology in the transplanted cells (Kordower et al. 2008). Using animal models, the injection of α-syn aggregates from brains of patients with PD or dementia with LB resulted in the formation of inclusions in wild-type mice and monkeys even in distant brain regions, further supporting the concept of prion-like spreading of α-syn pathology (reviewed in (Goedert et al. 2017). As described in detail by Niu et al. (2018), the spread of misfolded α-syn starts in the olfactory bulb (OB) and the gut with a progressive invasion of the posterior part of the brain. Indeed human mutant α-syn applied to the OB induces pathological changes in sensitive brain areas (Niu et al. 2018). However, caveats have been published by Gelpi and Colom-Cadena as well as by Masaracchia et al. indicating that the prion hypothesis of human synucleinopathies has to be reconsidered (Gelpi and Colom-Cadena 2019; Masaracchia et al. 2018).
As pathological α-syn aggregates were not only observed in the central nervous system, but may occur early in the enteric nervous system, a retrograde spreading of the pathology from the gut to the brain via the vagal nerve was considered. Indeed, the dorsal motor nucleus of the vagal nerve is, together with the olfactory bulb, among the first anatomical structures in the brain related to LB formation in PD (Braak et al. 2003a).
Interaction of α-synuclein and gut microbioma
Although there is still some discussion, experimental and epidemiological data provide evidence that vagotomy might be protective against the development of PD. Scandinavian retrospective registry studies show that truncal but not selective vagotomy reduces the risk of later developing PD, an effect which seems to be time dependent and only observed after a long follow-up period of > 5–20 years (Liu et al. 2017; Svensson et al. 2015). Similar evidence is provided by Pan-Montojo and colleagues in a mouse model of PD, by showing that the spread of α-syn pathology from the gut into the brain is inhibited by vagotomy (Pan-Montojo et al. 2010, 2012). Moreover, vagotomy completely blocks the dopaminergic degeneration within the SN after enteral treatment with the PD-inducing compound rotenone in this PD mouse model. Additionally, recent epidemiological data showed an association between appendectomy and PD risk, with a lower risk for the development of PD after appendectomy (Killinger et al. 2018). This might be of great interest since the appendix is known to harbor particularly strong α-syn immunoreactivity in conjunction with neural structures and receives substantial input from the vagal nerve. These data suggest the vagal nerve as a highway facilitating the spread of α-syn aggregates from the gut to the brain and supports the hypothesis that pathological α-syn aggregation could be initiated in the gut. However, other factors might be involved in the peripheral and central distribution patterns of α-syn and its pathological aggregates.
What are the triggers of α-syn pathology in the gastro-intestinal tract as a potential initiation event of subsequent central PD pathology? A first potential trigger is the presence of infection or inflammatory disease in the gastro-intestinal tract, since α-syn expression in this region might be a mechanism of immune response against infections and other inflammatory events (Stolzenberg et al. 2017). Indeed, a vicious cycle with mutual potentiation of inflammation and α-syn deposition was described as a mechanism contributing to the chronic progressive neurodegeneration in a mouse model of PD (Gao et al. 2011). Another potential factor contributing to the initiation of α-syn pathology in the gut and subsequently in the brain might be dysbiosis of the gut microbiota. Indeed, constipation as one clinical presentation of dysbiota is an early premotor symptom of PD, and the gut microbiome that was found to be altered in PD patients compared to healthy controls showed a characteristic over- and underrepresentation of distinct bacterial species (Hill-Burns et al. 2017; Petrov et al. 2017; Scheperjans et al. 2015). Interestingly, some of the dysregulated gut microbes were found to interfere with the intestinal mucosal barrier and, therefore, might elicit neuroinflammatory responses (Hill-Burns et al. 2017; Petrov et al. 2017) including OS (Sian-Hulsmann et al. 2015). In an elegant set of experiments, Sampson and colleagues were able to show that signals from microbes are able to promote α-syn-mediated motor dysfunction and brain pathology in a mouse model of PD, most likely via microglia activation (Sampson et al. 2016). Interestingly, even rotenone toxicity in PD mouse models might be mediated through gastro-intestinal dysbiota (Yang et al. 2017) Indeed, human gut microbiota from PD patients induced enhanced motor dysfunction in this PD mouse model with overexpression of human α-syn (Sampson et al. 2016). Other experimental/toxic PD models affect mitochondrial function and ROS production (Jiang and Dickson 2018; Bringmann et al. 1995).
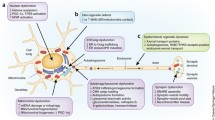
While most researchers are in favor of the gut-brain axis pathology in PD, recent evidence from time-dependent analyses following 6-hydroxydopamine-induced degeneration of the nigro-striatal tract in animal models suggest peripheral nervous system denervation, including peripheral inflammation, respectively, immunity (Armentero et al. 2006; Engler et al. 2009; Ambrosi et al. 2017), respiratory disturbances (Oliveira et al. 2019), cardiac sympathetic denervation (Jones et al. 2014) and gastrointestinal disturbances (Blandini et al. 2009). These data suggest that degenerative processes leading to SN degeneration and PD might start from both poles, e.g., gut, respectively, other peripheral sites as well as from the SN, respectively, brain stem nuclei depending on the various triggers of PD and the patients individual genetic vulnerability (Fig. 5).

Schematic presentation of pathways underlying PD pathology: gut-brain axis (Braak et al. 2003a, b; Pan-Montojo et al. 2010, 2012; Killinger et al. 2018; Machado et al. 2011), brain-periphery axis (Armentero et al. 2006; Engler et al. 2009; Blandini et al. 2009; Ambrosi et al. 2017; Oliveira et al. 2019)
α-Synuclein and its interactions with proteins involved in neuronal pathology
There are many more questions remaining to be solved, all of which could not be discussed in this review. For example, (1) why is there frequently a generation of more than one LB in a SN dopaminergic neuron and (2) is α-syn the key dominator of LB generation? If so it should be detected in the core of LBs rather than in the halo (Fig. 1). This, however, seems not to be the case. According to Kurt Jellinger (pers. communication), LB have a distinct central Parkin- and ubiquitin-positive domain while α-syn is localized in the halo of the LB. This should suggest a primary pathological process focusing on the proteasomal pathway of protein degradation and would be in line with the fact that α-syn interacts with other key players of neurodegenerative diseases, such as Aβ, Tau and TDP 43.
These interactions may result in a synergism of toxic events underlying synucleinopathies such as PD, MSA and LBD, depending on their appearance in specific brain regions and relative proteins with pathological contributions, thus contributing to the discrimination potential between these disorders (Tables 2 and 3).
α-Synuclein and proteasomal degradation
The fidelity of cellular machineries implicated in the removal of damaged and dysfunctional proteins, such as the ubiquitin–proteasome system (UPS) or the autophagy–lysosome pathway (ALP), plays a crucial role in maintaining protein homeostasis (proteostasis). A decrease in the efficiency of these systems paralleled by an increase in the abundance of misfolded proteins occurs during aging and is a common denominator of neurodegenerative diseases (reviewed in Vilchez et al. 2014; Hipp et al. 2019).
α-Syn protein levels have been reported to increase in the SN with aging (Li et al. 2004; Chu and Kordower 2007), suggesting an inefficient clearance of α-syn. This may particularly pertain to misfolded, aggregated and oxidatively modified α-syn species. Both the UPS and the ALP have been implicated in the degradation of α-syn, and there is an ongoing discussion on the preferences of these degrading systems regarding the type of α-syn species.
The 26S proteasome holoenzyme is composed of the 20S barrel-shaped core complex and the 19S regulatory particle that is attached to one or both ends of the 20S core (rev. in Finley and Prado 2019). The 20S core complex harbors caspase-like, trypsin-like, and chymotrypsin-like activities inside the barrel, whereas the 19S cap recognizes, unfolds, deubiquitinates and translocates substrates into the proteasomal core. In fact, substrate ubiquitination is usually a prerequisite for degradation via the proteasome.
LB found in synucleinopathies stain positive for ubiquitin and co-localize with proteasomal subunits. Although the proteasome is probably not able to deal with large aggregates, it can handle monomeric or oligomeric α-syn extracted from LB with the help of the segregase VCP (valosin-containing protein)/p97. Several recent publications add evidence for a role of the UPS in clearing α-syn. Based on overexpression approaches in cellular models, RER1 (retention in endoplasmic reticulum 1) was reported to promote α-syn degradation by the proteasome (Park et al. 2017). RER1 localizes to early compartments of the secretory pathway and cycles between the cis-Golgi and the ER. It may promote retrieval of membrane-bound α-syn to the ER and subsequent proteasomal degradation, although an effect of endogenous RER1 has not yet been demonstrated in this context.
An interesting link between the transcriptional regulation of α-syn and proteasomal degradation was provided in a study that identified ZSCAN21 (zink finger and SCAN domain containing 21) as a transcription factor driving the expression of α-syn (Lassot et al. 2018; Clough et al. 2009; Dermentzaki et al. 2016). Under normal conditions, ZSCAN21 levels are controlled by TRIM41-mediated ubiquitination and subsequent proteasomal degradation. Cellular stress, for example induced by MPTP treatment, induces up-regulation of TRIM17 that inhibits TRIM41 and thus causes stabilization of ZSCAN21 and enhanced α-syn expression. In support of a disease-relevant role of this pathway, genetic variants of TRIM41 and ZSCAN21 co-segregate with PD and result in stabilization of ZSCAN21 protein.
A recent study indicated that the proteasome has an ATP-dependent, ubiquitin- and proteolysis-independent fragmentation function in vitro that disassembles large tau and α-syn fibrils into smaller aggregates (Cliffe et al. 2019). These smaller aggregates were more toxic than large fibrils and caused cell lysis when added exogenously to HEK293 cells. Whether this mechanism plays a role in vivo remains to be determined. For the proteasomal degradation, the N-terminal part of α-syn is essential. Experimental work demonstrates that aggregated α-syn is also degraded by the proteasome, although with a reduced rate due to Met 1/5 oxidation (Alvarez-Castelao et al. 2014). These authors also report that even mild oxidation inhibits the proteasome, especially when hydrogen peroxide is used as source of oxidation. The result is an accumulation of oxidatively damaged α-syn and initiation of PD (Alvarez-Castelao et al. 2014). Vice versa, Melo et al. (2018) and Pinho et al. (2019) demonstrate that α-syn dysfunction as a cellular stressor impairs mitochondria, endoplasmic reticulum, autophagy and cellular dynamics causing dopamine depletion, LB formation and PD.
In fact, a controversial issue has been the question whether misfolded α-syn species can impair proteasomal function. Conflicting results have been reported depending on the approach (in vitro versus in vivo), cellular models (neuronal versus non-neuronal) and type of α-syn species used (Lindersson et al. 2004; Snyder et al. 2003; Petrucelli et al. 2002; Bence et al. 2001; Tanaka et al. 2001; Zhang et al. 2008; Tofaris et al. 2003). It appears that cell type- and context-specific features account for these discrepancies and that an inhibitory effect of α-syn on proteasomal activities can contribute to the selective vulnerability of dopaminergic neurons in PD (Zondler et al. 2017). In conclusion, α-syn is not only degraded by the proteasome, pathogenic α-syn species obviously have the propensity to compromise the UPS, indicating a pathologically relevant reciprocal interaction.
Mitochondrial functioning in Parkinson’s disease
Reactive oxygen species (ROS) are permanently produced during metabolic intraneural processes and contribute in particular to important cellular maintenance functions. The imbalance in the level of ROS and their degradation by antioxidative mechanisms is referred to as “oxidative stress”. To maintain ROS levels, mitochondrial function is the crucial intracellular organelle, in particular in neurons. The fact that neurons have high energy requirements and functionally depend on fine-tuned levels of calcium helps to explain the role of mitochondria for the functionality of the nervous system, in particular the tight link of mitochondrial impairment to neurodegenerative diseases. Genetic evidence links important genes such as Parkin and the mitochondrial kinase PINK1 to genetic forms of PD, in particular regulating the elimination of damaged mitochondria (≙ mitophagy) by Parkin signaling downstream to PINK1 (Trempe and Fon 2013; Baker et al. 2011). However, the first evidence connecting mitochondria to PD was derived from the observation of severe Parkinsonism and cognitive dysfunction in individuals using “synthetic heroin”, primarily consisting of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), in a region of Northern California (Langston et al. 1983). Consequently, it became evident that in particular the MPTP’ metabolite MPP+ produced by oxidation via the monoamine oxidase (MAO-B) was toxic. MPP+ is selectively taken up by dopaminergic neurons within the SN, leading to severe dopamine depletion. Further evidence supporting the important role of mitochondrial damage is that specifically decreased catalytic activity of complex 1 has been described in the SN and frontal cortex of PD patients. In addition, exposure to specific pesticides such as rotenone with its potential to inhibit complex 1 catalytic activity results in an increased susceptibility for these exposed individuals to develop PD later in life. Furthermore, the accumulation of α-syn may impair mitochondrial homeostasis, i.e., by decreasing the activity of mitochondrial complex 1 (Devi et al. 2008). In general, the kidney bean-shaped mitochondria form a highly dynamic reticular network controlled by a fusion and fission machinery enabling mitochondria to constantly fuse and to divide. There is a plethora of mitochondrial quality pathways, within a given cell governing molecular quality control by antioxidative chaperones and protease activity, but additionally an organelle quality control by removing dysfunctional mitochondria via mitophagy (Baker et al. 2011).
Interaction of ubiquitin proteasome and lysosomal functioning in Parkinson’s disease
Protein degradation via the ubiquitin-proteasome system and the autophagy-lysosome pathway regulates both the intracellularly produced and ingested protein cargo. To maintain protein homeostasis, the proteasomal function within the SN is crucial, since evidence suggests that in particular α-syn is ubiquitinated in LB (Furukawa et al. 2002). In general, the autophagosom–lysosomal pathway (ALP) involves three major modes, namely microautophagy, chaperone-mediated autophagy and macroautophagy (Deter and De Duve 1967). By definition, microautophagy is the non-selective engulfment of small cytoplasmic portions by lysosomes (Mijaljica et al. 2011). In contrast, chaperone-mediated autophagy is a very selective process by which proteins possessing a distinct motif (KFERQ) like α-syn are recognized and guided to the lysosomal membrane transporter LAMP2A thereby entering the lysosome (Cuervo and Wong 2014).
Autophagy
More recent work demonstrates that autophagy is eventually controlled by GTP-ase–p38 MAPK signaling (Obergasteiger et al. 2018), a pathway which might be disturbed in PD. It has been shown that modulators of autophagy such as FOXO 1, SESN 3 and TSC 2 are present in LB and that TSC 2 increases after α-syn overexpression (Miki et al. 2018). On the other hand, impaired formation auf autophagosomes, which under physiological conditions reduce the intracellular burden of α-syn, increases the exosomal formation of α-syn (Fussi et al. 2018).
Macroautophagy refers to the process of degradation of large portions of cytoplasm, including protein aggregates and organelles (i.e., mitochondria), via inclusion into double-membrane lipid structures called “autophagosomes” and finally fusing into lysosomes (Takeshige et al. 1992; Tsukada and Ohsumi 1993). Currently, in the light of recent ultra-structural evidence showing that LB are composed of proteins, lipids and increased mitochondria, autophagosomes and lysosomes suggest that the degradation machinery is severely impaired and thereby providing a direct link between ALP-impairment and LB formation (Shahmoradian et al. 2019).
Therapy
The heterogeneity of PD symptoms calls for an individually adapted therapeutic regime. It aims to improve these individual combinations of motoric and non-motoric symptoms (Lee and Koh 2015). Their onset and severity are related to one another to a certain extent, based on the considerable impact of applied therapies (Muller et al. 2017). A diverse drug portfolio is employed for amelioration of PD symptoms with individually balanced combination of various drugs being applied. Careful and slow drug titration with continuous attention to the tolerability, safety and the needs of patients and their caregivers is essential for a successful treatment of PD in the long term. An optimized therapeutic regime can prevent an adjustment of the human body to features of PD, as unconscious learning processes may aggravate certain typical symptoms such as bound posture or walking with small steps. Currently, the best pharmacological treatment options exist for balancing the dopamine deficit in PD (Muller 2012). Generally, the focus of early PD treatment is on alleviating mild motoric symptoms. As PD progresses, L-dopa is used to mitigate more severe motoric symptoms. Subsequently, L-dopa is employed alongside other therapies, such as monoamine oxidase-B (MAO-B) inhibitors, dopamine agonists, NMDA antagonists or catechol-O-methyltransferase inhibitors (COMT-I). These substances aim to minimize L-dopa-associated complications of motoric response. Deep-brain stimulation (DBS) or pump systems with application of apomorphine or L-dopa intestinal gel (LCIG) are mostly used as a final therapeutic approach in more advanced PD patients.
A definite cure for PD is still not available. Cumbersome transplantation trials, gene therapies or disease course-modifying treatments have not provided a significant promising breakthrough in the past years. The resurgence of complex stem cell implantation in the basal ganglia as a dopamine substitution method still has to prove its superiority over simple drug intake (Cyranoski 2018). Effects of drugs on specific non-motoric symptoms or balance problems are still somehow neglected.
Future therapeutic concepts
New treatment concepts are underway, most of them focusing on α-syn pathology. A better understanding of α-syn pathophysiology will be key for developing new therapeutic strategies. Such strategies could include a reduction of α-syn production by modification of SNCA gene transcription or RNA interference, inhibition of α-syn aggregation by insertion of “intrabodies” or viral-vector therapies, and clearance of α-syn aggregates by targeted immunotherapy or promoting autophagy of α-syn oligomers (Lardenoije et al. 2015; Brundin et al. 2017). Vaccination or application of antibodies has also been tested (Wang et al. 2019; Vaikath et al. 2019; Shen et al. 2019; Wood 2014). It is far from clear whether neuropathological findings, such as LB accumulation or misfolded protein enrichment, definitely play an active role in the ongoing chronic disease process itself. They could also merely reflect well-wrapped protein waste produced by disease-affected neurons. In general, chronic neurodegenerative processes result from several different metabolic cascades. It is well known that they ultimately result in cell death via well-described mechanisms. These processes induce individually different clinical signs and symptoms. Driven by experimental research, many hypotheses support the concepts of protein misfolding. However, occurrence of protein misfolding is a first-line defence process. It involves protein refolding mediated by chaperone proteins. A failure of these processes can cause protein degradation and/or accumulation. If this refolding/degradation machinery cannot prevent misfolded proteins, a stress response will be activated involving up-regulation of refolding and degradation processes. If stress levels induced by protein misfolding become too severe, cell death programs are activated. Accordingly, potential therapeutic strategies currently being tested in experimental settings include reduction of protein misfolding, repair of misfolded proteins and facilitation of degradation of proteins, particularly when they are damaged beyond repair. There is a certain capacity of the human brain to compensate these initial events for considerable intervals before the clinical onset of initially mild and unspecific symptoms of neurodegenerative disease. Thus, initiation and rate of disease progression varies individually. Preclinical and experimental researchers still primarily focus on these processes, although animal models can only partially reflect the variability of PD symptoms, disease progress and severity as observed in clinical practice. A similar development is known from Alzheimer’s disease, with all β-amyloid trials having failed to date. Future trials on disease modification will also test the usefulness of wearable, non-invasive body worn companion devices that collect movement-related measurements to assess patients’ daily activities in relation to disease progression and symptomatic drug effects (Schlachetzki et al. 2017; Klucken et al. 2018). They will also further estimate the close relationship between the presence of motoric and non-motoric features in PD, i.e., the close association between motion, apathy and mood (Muller et al. 2017).
Based on this outlook, there are potential therapeutic approaches which could develop into viable treatment options and influence disease progression. Very new strategies are also being applied to influence the effects of micro-RNA molecules which, as non-coding RNA, are decisive in regulating gene expression. The objective here is (1) to decrease both pro-inflammatory reactions in microglia and MHC II-expression or (2) to modulate the inflammasome, for example with tyrosine kinase-inhibitors. Corresponding studies have been and are at present being conducted (Lawana et al. 2017; Thome et al. 2016; Brahmachari et al. 2017).
Altogether these approaches in the area of microglia are at present highly promising, due to the fact that medications already in use can be “repurposed” and because a re-orientation from a purely anti-inflammatory strategy to active immunomodulation can be pursued (Abushouk et al. 2018; Pagan et al. 2016; Pena-Altamira et al. 2016).
What we have learned about α-syn knock-out in mice, however, is a somewhat discomforting factor because these mice showed an increase in microglia-induced inflammation (Austin et al. 2011). This could entail considerable risk for patients if modern therapeutic strategies, which are used to reduce abnormal α-syn, could conversely induce a reactive inflammation by completely depleting α-syn. Further studies are needed to reveal whether such a therapeutic approach is feasible.
Generally, one has to accept that treatment of chronic neurodegenerative disorders such as PD necessitates a combination of therapeutic options which are individually adapted to disease process and progression. The individual patient, the caregiver and the treating physician must determine the value of a patient tailored therapy in the long run. The interaction between the physician, the caregiver and the patient decide and determine the feasibility of a treatment approach in PD. PD asks for a personalized medicine concept, which adds or switches treatments to best manage the symptoms of PD.
References
Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25(1):239–252
Abushouk AI, Negida A, Elshenawy RA, Zein H, Hammad AM, Menshawy A, Mohamed WMY (2018) C-Abl inhibition; a novel therapeutic target for Parkinson’s disease. CNS Neurol Disord Drug Targets 17(1):14–21. https://doi.org/10.2174/1871527316666170602101538
Ahmed H, Abushouk AI, Gabr M, Negida A, Abdel-Daim MM (2017) Parkinson’s disease and pesticides: a meta-analysis of disease connection and genetic alterations. Biomed Pharmacother 90:638–649. https://doi.org/10.1016/j.biopha.2017.03.100
Aimi Y, McGeer PL (1996) Lack of toxicity of human neuromelanin to rat brain dopaminergic neurons. Parkinsonism Relat Disord 2(2):69–74. https://doi.org/10.1016/1353-8020(96)00004-1
Alonso-Navarro H, Jimenez-Jimenez FJ, Garcia-Martin E, Agundez JA (2014) Genomic and pharmacogenomic biomarkers of Parkinson’s disease. Curr Drug Metab 15(2):129–181
Alvarez-Castelao B, Goethals M, Vandekerckhove J, Castano JG (2014) Mechanism of cleavage of alpha-synuclein by the 20S proteasome and modulation of its degradation by the RedOx state of the N-terminal methionines. Biochim Biophys Acta 1843(2):352–365. https://doi.org/10.1016/j.bbamcr.2013.11.018
Ambrosi G, Kustrimovic N, Siani F, Rasini E, Cerri S, Ghezzi C, Dicorato G, Caputo S, Marino F, Cosentino M, Blandini F (2017) Complex changes in the innate and adaptive immunity accompany progressive degeneration of the nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine in the rat. Neurotox Res 32(1):71–81. https://doi.org/10.1007/s12640-017-9712-2
Armentero MT, Levandis G, Nappi G, Bazzini E, Blandini F (2006) Peripheral inflammation and neuroprotection: systemic pretreatment with complete Freund’s adjuvant reduces 6-hydroxydopamine toxicity in a rodent model of Parkinson’s disease. Neurobiol Dis 24(3):492–505. https://doi.org/10.1016/j.nbd.2006.08.016
Austin SA, Rojanathammanee L, Golovko MY, Murphy EJ, Combs CK (2011) Lack of alpha-synuclein modulates microglial phenotype in vitro. Neurochem Res 36(6):994–1004. https://doi.org/10.1007/s11064-011-0439-9
Baker MJ, Tatsuta T, Langer T (2011) Quality control of mitochondrial proteostasis. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a007559
Bartels T, Ahlstrom LS, Leftin A, Kamp F, Haass C, Brown MF, Beyer K (2010) The N-terminus of the intrinsically disordered protein alpha-synuclein triggers membrane binding and helix folding. Biophys J 99(7):2116–2124. https://doi.org/10.1016/j.bpj.2010.06.035
Batelli S, Albani D, Rametta R, Polito L, Prato F, Pesaresi M, Negro A, Forloni G (2008) DJ-1 modulates alpha-synuclein aggregation state in a cellular model of oxidative stress: relevance for Parkinson’s disease and involvement of HSP70. PLoS One 3(4):e1884. https://doi.org/10.1371/journal.pone.0001884
Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292(5521):1552–1555
Bendor JT, Logan TP, Edwards RH (2013) The function of alpha-synuclein. Neuron 79(6):1044–1066. https://doi.org/10.1016/j.neuron.2013.09.004
Berg D, Gerlach M, Youdim MB, Double KL, Zecca L, Riederer P, Becker G (2001) Brain iron pathways and their relevance to Parkinson’s disease. J Neurochem 79(2):225–236
Berg D, Godau J, Seppi K, Behnke S, Liepelt-Scarfone I, Lerche S, Stockner H, Gaenslen A, Mahlknecht P, Huber H, Srulijes K, Klenk J, Fassbender K, Maetzler W, Poewe W (2013) The PRIPS study: screening battery for subjects at risk for Parkinson’s disease. Eur J Neurol 20(1):102–108. https://doi.org/10.1111/j.1468-1331.2012.03798.x
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F (1973) Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci 20(4):415–455
Bernstein SL, Liu D, Wyttenbach T, Bowers MT, Lee JC, Gray HB, Winkler JR (2004) Alpha-synuclein: stable compact and extended monomeric structures and pH dependence of dimer formation. J Am Soc Mass Spectrom 15(10):1435–1443. https://doi.org/10.1016/j.jasms.2004.08.003
Binolfi A, Rasia RM, Bertoncini CW, Ceolin M, Zweckstetter M, Griesinger C, Jovin TM, Fernandez CO (2006) Interaction of alpha-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement. J Am Chem Soc 128(30):9893–9901. https://doi.org/10.1021/ja0618649
Binolfi A, Lamberto GR, Duran R, Quintanar L, Bertoncini CW, Souza JM, Cervenansky C, Zweckstetter M, Griesinger C, Fernandez CO (2008) Site-specific interactions of Cu(II) with alpha and beta-synuclein: bridging the molecular gap between metal binding and aggregation. J Am Chem Soc 130(35):11801–11812. https://doi.org/10.1021/ja803494v
Binolfi A, Valiente-Gabioud AA, Duran R, Zweckstetter M, Griesinger C, Fernandez CO (2011) Exploring the structural details of Cu(I) binding to alpha-synuclein by NMR spectroscopy. J Am Chem Soc 133(2):194–196. https://doi.org/10.1021/ja107842f
Bisaglia M, Mammi S, Bubacco L (2007) Kinetic and structural analysis of the early oxidation products of dopamine: analysis of the interactions with alpha-synuclein. J Biol Chem 282(21):15597–15605. https://doi.org/10.1074/jbc.M610893200
Blandini F, Balestra B, Levandis G, Cervio M, Greco R, Tassorelli C, Colucci M, Faniglione M, Bazzini E, Nappi G, Clavenzani P, Vigneri S, De Giorgio R, Tonini M (2009) Functional and neurochemical changes of the gastrointestinal tract in a rodent model of Parkinson’s disease. Neurosci Lett 467(3):203–207. https://doi.org/10.1016/j.neulet.2009.10.035
Blauwendraat C, Kia DA, Pihlstrom L, Gan-Or Z, Lesage S, Gibbs JR, Ding J, Alcalay RN, Hassin-Baer S, Pittman AM, Brooks J, Edsall C, Chung SJ, Goldwurm S, Toft M, Schulte C, Hernandez D, Singleton AB, Nalls MA, Brice A, Scholz SW, Wood NW (2018) Insufficient evidence for pathogenicity of SNCA His50Gln (H50Q) in Parkinson’s disease. Neurobiol Aging 64:159 e155–159 e158. https://doi.org/10.1016/j.neurobiolaging.2017.12.012
Braak H, Del Tredici K (2017) Neuropathological staging of brain pathology in sporadic parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis 7(s1):S71–S85. https://doi.org/10.3233/JPD-179001
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003a) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211
Braak H, Rub U, Gai WP, Del Tredici K (2003b) Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna) 110(5):517–536. https://doi.org/10.1007/s00702-002-0808-2
Brahmachari S, Karuppagounder SS, Ge P, Lee S, Dawson VL, Dawson TM, Ko HS (2017) c-Abl and Parkinson’s disease: mechanisms and therapeutic potential. J Parkinsons Dis 7(4):589–601. https://doi.org/10.3233/JPD-171191
Bridi JC, Hirth F (2018) Mechanisms of alpha-Synuclein induced synaptopathy in Parkinson’s disease. Front Neurosci 12:80. https://doi.org/10.3389/fnins.2018.00080
Bringmann G, God R, Feineis D, Wesemann W, Riederer P, Rausch WD, Reichmann H, Sontag KH (1995) The TaClo concept: 1-trichloromethyl-1,2,3,4-tetrahydro-beta-carboline (TaClo), a new toxin for dopaminergic neurons. J Neural Transm Suppl 46:235–244
Brooks DJ (2010) Examining Braak’s hypothesis by imaging Parkinson’s disease. Mov Disord 25(Suppl 1):S83–S88. https://doi.org/10.1002/mds.22720
Brundin P, Dave KD, Kordower JH (2017) Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol 298(Pt B):225–235. https://doi.org/10.1016/j.expneurol.2017.10.003
Bungeroth M, Appenzeller S, Regulin A, Volker W, Lorenzen I, Grotzinger J, Pendziwiat M, Kuhlenbaumer G (2014) Differential aggregation properties of alpha-synuclein isoforms. Neurobiol Aging 35(8):1913–1919. https://doi.org/10.1016/j.neurobiolaging.2014.02.009
Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, Savas JN, Kiskinis E, Zhuang X, Kruger R, Surmeier DJ, Krainc D (2017) Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357(6357):1255–1261. https://doi.org/10.1126/science.aam9080
Burke WJ, Kumar VB, Pandey N, Panneton WM, Gan Q, Franko MW, O’Dell M, Li SW, Pan Y, Chung HD, Galvin JE (2008) Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol 115(2):193–203. https://doi.org/10.1007/s00401-007-0303-9
Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329(5999):1663–1667. https://doi.org/10.1126/science.1195227
Bush WD, Garguilo J, Zucca FA, Albertini A, Zecca L, Edwards GS, Nemanich RJ, Simon JD (2006) The surface oxidation potential of human neuromelanin reveals a spherical architecture with a pheomelanin core and a eumelanin surface. Proc Natl Acad Sci USA 103(40):14785–14789. https://doi.org/10.1073/pnas.0604010103
Carballo-Carbajal I, Laguna A, Romero-Gimenez J, Cuadros T, Bove J, Martinez-Vicente M, Parent A, Gonzalez-Sepulveda M, Penuelas N, Torra A, Rodriguez-Galvan B, Ballabio A, Hasegawa T, Bortolozzi A, Gelpi E, Vila M (2019) Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson's disease pathogenesis. Nat Commun 10(1):973. https://doi.org/10.1038/s41467-019-08858-y
Carnwath T, Mohammed R, Tsiang D (2018) The direct and indirect effects of alpha-synuclein on microtubule stability in the pathogenesis of Parkinson’s disease. Neuropsychiatr Dis Treat 14:1685–1695. https://doi.org/10.2147/NDT.S166322
Castillo-Gonzalez JA, Loera-Arias MJ, Saucedo-Cardenas O, Montes-de-Oca-Luna R, Garcia-Garcia A, Rodriguez-Rocha H (2017) Phosphorylated alpha-Synuclein-copper complex formation in the pathogenesis of Parkinson’s disease. Parkinsons Dis 2017:9164754. https://doi.org/10.1155/2017/9164754
Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49(10):1511–1516. https://doi.org/10.1038/ng.3955
Cheng HC, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67(6):715–725. https://doi.org/10.1002/ana.21995
Cho MK, Nodet G, Kim HY, Jensen MR, Bernado P, Fernandez CO, Becker S, Blackledge M, Zweckstetter M (2009) Structural characterization of alpha-synuclein in an aggregation prone state. Protein Sci 18(9):1840–1846. https://doi.org/10.1002/pro.194
Chu Y, Kordower JH (2007) Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson’s disease? Neurobiol Dis 25(1):134–149. https://doi.org/10.1016/j.nbd.2006.08.021
Cliffe R, Sang JC, Kundel F, Finley D, Klenerman D, Ye Y (2019) Filamentous aggregates are fragmented by the proteasome holoenzyme. Cell Rep 26(8):2140 e2143–2149 e2143. https://doi.org/10.1016/j.celrep.2019.01.096
Clough RL, Dermentzaki G, Stefanis L (2009) Functional dissection of the alpha-synuclein promoter: transcriptional regulation by ZSCAN21 and ZNF219. J Neurochem 110(5):1479–1490. https://doi.org/10.1111/j.1471-4159.2009.06250.x
Cronin KD, Ge D, Manninger P, Linnertz C, Rossoshek A, Orrison BM, Bernard DJ, El-Agnaf OM, Schlossmacher MG, Nussbaum RL, Chiba-Falek O (2009) Expansion of the Parkinson disease-associated SNCA-Rep1 allele upregulates human alpha-synuclein in transgenic mouse brain. Hum Mol Genet 18(17):3274–3285. https://doi.org/10.1093/hmg/ddp265
Cuervo AM, Wong E (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24(1):92–104. https://doi.org/10.1038/cr.2013.153
Cyranoski D (2018) ‘Reprogrammed’ stem cells approved to mend human hearts for the first time. Nature 557(7707):619–620. https://doi.org/10.1038/d41586-018-05278-8
Del Rey NL, Quiroga-Varela A, Garbayo E, Carballo-Carbajal I, Fernandez-Santiago R, Monje MHG, Trigo-Damas I, Blanco-Prieto MJ, Blesa J (2018) Advances in Parkinson’s disease: 200 years later. Front Neuroanat 12:113. https://doi.org/10.3389/fnana.2018.00113
Dermentzaki G, Paschalidis N, Politis PK, Stefanis L (2016) Complex effects of the ZSCAN21 transcription factor on transcriptional regulation of alpha-Synuclein in primary neuronal cultures and in vivo. J Biol Chem 291(16):8756–8772. https://doi.org/10.1074/jbc.M115.704973
Deter RL, De Duve C (1967) Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol 33(2):437–449
Dettmer U, Newman AJ, von Saucken VE, Bartels T, Selkoe D (2015) KTKEGV repeat motifs are key mediators of normal alpha-synuclein tetramerization: their mutation causes excess monomers and neurotoxicity. Proc Natl Acad Sci USA 112(31):9596–9601. https://doi.org/10.1073/pnas.1505953112
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK (2008) Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 283(14):9089–9100. https://doi.org/10.1074/jbc.M710012200
Dickson DW (2007) Linking selective vulnerability to cell death mechanisms in Parkinson’s disease. Am J Pathol 170(1):16–19. https://doi.org/10.2353/ajpath.2007.061011
Dickson DW (2012) Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a009258
Dorsey ER, Sherer T, Okun MS, Bloem BR (2018) The emerging evidence of the Parkinson pandemic. J Parkinsons Dis 8(s1):S3–S8. https://doi.org/10.3233/JPD-181474
Double KL, Zecca L, Costi P, Mauer M, Griesinger C, Ito S, Ben-Shachar D, Bringmann G, Fariello RG, Riederer P, Gerlach M (2000) Structural characteristics of human substantia nigra neuromelanin and synthetic dopamine melanins. J Neurochem 75(6):2583–2589
Double KL, Gerlach M, Schunemann V, Trautwein AX, Zecca L, Gallorini M, Youdim MB, Riederer P, Ben-Shachar D (2003) Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem Pharmacol 66(3):489–494
Doxakis E (2010) Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem 285(17):12726–12734. https://doi.org/10.1074/jbc.M109.086827
Eikelenboom P, Stam FC (1982) Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol 57(2–3):239–242
Engelhardt E (2017) Lafora and Tretiakoff: the naming of the inclusion bodies discovered by Lewy. Arq Neuropsiquiatr 75(10):751–753. https://doi.org/10.1590/0004-282X20170116
Engler H, Doenlen R, Riether C, Engler A, Niemi MB, Besedovsky HO, del Rey A, Pacheco-Lopez G, Feldon J, Schedlowski M (2009) Time-dependent alterations of peripheral immune parameters after nigrostriatal dopamine depletion in a rat model of Parkinson’s disease. Brain Behav Immun 23(4):518–526. https://doi.org/10.1016/j.bbi.2009.01.018
Fahn S (2003) Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci 991:1–14
Fasano M, Giraudo S, Coha S, Bergamasco B, Lopiano L (2003) Residual substantia nigra neuromelanin in Parkinson’s disease is cross-linked to alpha-synuclein. Neurochem Int 42(7):603–606
Fedorow H, Tribl F, Halliday G, Gerlach M, Riederer P, Double KL (2005) Neuromelanin in human dopamine neurons: comparison with peripheral melanins and relevance to Parkinson’s disease. Prog Neurobiol 75(2):109–124. https://doi.org/10.1016/j.pneurobio.2005.02.001
Fernandez CO, Hoyer W, Zweckstetter M, Jares-Erijman EA, Subramaniam V, Griesinger C, Jovin TM (2004) NMR of alpha-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J 23(10):2039–2046. https://doi.org/10.1038/sj.emboj.7600211
Finley D, Prado MA (2019) The proteasome and its network: engineering for adaptability. Cold Spring Harbor Perspect Biol. https://doi.org/10.1101/cshperspect.a033985
Fischer O (1907) Miliare Nekrosen mit drusigen Wucherungen der Neurofibrillen, eine regelmässige Veränderung der Hirnrinde bei seniler Demenz. Monatsschr Psychiatr Neurol. https://doi.org/10.1159/000211873
Foley P, Riederer P (1999) Pathogenesis and preclinical course of Parkinson’s disease. J Neural Transm Suppl 56:31–74
Foley P, Riederer P (2000) Influence of neurotoxins and oxidative stress on the onset and progression of Parkinson’s disease. J Neurol 247(Suppl 2):II82–II94
Fuchs J, Tichopad A, Golub Y, Munz M, Schweitzer KJ, Wolf B, Berg D, Mueller JC, Gasser T (2008) Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB J 22(5):1327–1334. https://doi.org/10.1096/fj.07-9348com
Furukawa Y, Vigouroux S, Wong H, Guttman M, Rajput AH, Ang L, Briand M, Kish SJ, Briand Y (2002) Brain proteasomal function in sporadic Parkinson’s disease and related disorders. Ann Neurol 51(6):779–782. https://doi.org/10.1002/ana.10207
Fussi N, Hollerhage M, Chakroun T, Nykanen NP, Rosler TW, Koeglsperger T, Wurst W, Behrends C, Hoglinger GU (2018) Exosomal secretion of alpha-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis 9(7):757. https://doi.org/10.1038/s41419-018-0816-2
Gai WP, Power JH, Blumbergs PC, Blessing WW (1998) Multiple-system atrophy: a new alpha-synuclein disease? Lancet 352(9127):547–548
Gao HM, Zhang F, Zhou H, Kam W, Wilson B, Hong JS (2011) Neuroinflammation and alpha-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ Health Perspect 119(6):807–814. https://doi.org/10.1289/ehp.1003013
Gegg ME, Schapira AH (2016) Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiol Dis 90:43–50. https://doi.org/10.1016/j.nbd.2015.09.006
Gelpi E, Colom-Cadena M (2019) Oligomers: a hot topic for neurodegeneration and a note of caution for experimental models. Brain 142(2):228–230. https://doi.org/10.1093/brain/awy342
Gentile I, Garro HA, Delgado Ocana S, Gonzalez N, Strohaker T, Schibich D, Quintanar L, Sambrotta L, Zweckstetter M, Griesinger C, Menacho Marquez M, Fernandez CO (2018) Interaction of Cu(i) with the Met-X3-Met motif of alpha-synuclein: binding ligands, affinity and structural features. Metallomics 10(10):1383–1389. https://doi.org/10.1039/c8mt00232k
Gerlach M, Riederer P, Double KL (2008) Neuromelanin-bound ferric iron as an experimental model of dopaminergic neurodegeneration in Parkinson’s disease. Parkinsonism Relat Disord 14(Suppl 2):S185–S188. https://doi.org/10.1016/j.parkreldis.2008.04.028
Goedert M, Masuda-Suzukake M, Falcon B (2017) Like prions: the propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain 140(2):266–278. https://doi.org/10.1093/brain/aww230
Golbe LI, Di Iorio G, Sanges G, Lazzarini AM, La Sala S, Bonavita V, Duvoisin RC (1996) Clinical genetic analysis of Parkinson’s disease in the Contursi kindred. Ann Neurol 40(5):767–775. https://doi.org/10.1002/ana.410400513
Gomez-Suaga P, Fdez E, Blanca Ramirez M, Hilfiker S (2012) A link between autophagy and the pathophysiology of LRRK2 in Parkinson’s disease. Parkinsons Dis 2012:324521. https://doi.org/10.1155/2012/324521
Gotz ME, Double K, Gerlach M, Youdim MB, Riederer P (2004) The relevance of iron in the pathogenesis of Parkinson’s disease. Ann N Y Acad Sci 1012:193–208
Halliday GM, Ophof A, Broe M, Jensen PH, Kettle E, Fedorow H, Cartwright MI, Griffiths FM, Shepherd CE, Double KL (2005) Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson’s disease. Brain 128(Pt 11):2654–2664. https://doi.org/10.1093/brain/awh584
Harth R, Gerlach M, Riederer P, Gotz ME (2001a) A highly sensitive method for the determination of protein bound 3,4-dihydroxyphenylalanine as a marker for post-translational protein hydroxylation in human tissues ex vivo. Free Radic Res 35(2):167–174
Harth R, Gerlach M, Riederer P, Gotz ME (2001b) A sensitive procedure for the determination of protein bound 3,4-dihydroxyphenyl-alanine as a marker for posttranslational protein hydroxylation in human frontal cortex, liver, and red blood cells. Adv Exp Med Biol 500:517–519
Hill-Burns EM, Debelius JW, Morton JT, Wissemann WT, Lewis MR, Wallen ZD, Peddada SD, Factor SA, Molho E, Zabetian CP, Knight R, Payami H (2017) Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov Disord 32(5):739–749. https://doi.org/10.1002/mds.26942
Hipp MS, Kasturi P, Hartl FU (2019) The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. https://doi.org/10.1038/s41580-019-0101-y
Hirsch EC, Faucheux B, Damier P, Mouatt-Prigent A, Agid Y (1997) Neuronal vulnerability in Parkinson’s disease. J Neural Transm Suppl 50:79–88
Hirsch EC, Hunot S, Damier P, Faucheux B (1998) Glial cells and inflammation in Parkinson’s disease: a role in neurodegeneration? Ann Neurol 44(3 Suppl 1):S115–S120
Hirsch EC, Vyas S, Hunot S (2012) Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord 18(Suppl 1):S210–S212. https://doi.org/10.1016/S1353-8020(11)70065-7
Holdorff B, Rodrigues e Silva AM, Dodel R (2013) Centenary of Lewy bodies (1912–2012). J Neural Transm (Vienna) 120(4):509–516. https://doi.org/10.1007/s00702-013-0984-2
Huang M, Wang B, Li X, Fu C, Wang C, Kang X (2019) alpha-Synuclein: a multifunctional player in exocytosis, endocytosis, and vesicle recycling. Front Neurosci 13:28. https://doi.org/10.3389/fnins.2019.00028
Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364(9440):1169–1171. https://doi.org/10.1016/S0140-6736(04)17104-3
Iofrida C, Daniele S, Pietrobono D, Fusi J, Galetta F, Trincavelli ML, Bonuccelli U, Franzoni F, Martini C (2017) Influence of physical exercise on beta-amyloid, alpha-synuclein and tau accumulation: an in vitro model of oxidative stress in human red blood cells. Arch Ital Biol 155(1–2):33–42. https://doi.org/10.12871/000398292017124
Jellinger KA (2009) Absence of alpha-synuclein pathology in postencephalitic parkinsonism. Acta Neuropathol 118(3):371–379. https://doi.org/10.1007/s00401-009-0537-9
Jellinger KA (2010) Neurochemical biomarkers in the differential diagnosis of movement disorders. Mov Disord 25(4):500. https://doi.org/10.1002/mds.22853
Jellinger KA (2011) Synuclein deposition and non-motor symptoms in Parkinson disease. J Neurol Sci 310(1–2):107–111. https://doi.org/10.1016/j.jns.2011.04.012
Jellinger KA (2019) Is Braak staging valid for all types of Parkinson's disease? J Neural Transm (Vienna) 126(4):423–431. https://doi.org/10.1007/s00702-018-1898-9
Jellinger KA, Paulus W (1992) Clinico-pathological correlations in Parkinson’s disease. Clin Neurol Neurosurg 94(Suppl):S86–S88
Jiang P, Dickson DW (2018) Parkinson’s disease: experimental models and reality. Acta Neuropathol 135(1):13–32. https://doi.org/10.1007/s00401-017-1788-5
Jiang Z, Hess SK, Heinrich F, Lee JC (2015) Molecular details of alpha-synuclein membrane association revealed by neutrons and photons. J Phys Chem B 119(14):4812–4823. https://doi.org/10.1021/jp512499r
Johnson ME, Stecher B, Labrie V, Brundin L, Brundin P (2019) Triggers, facilitators, and aggravators: redefining Parkinson’s disease pathogenesis. Trends Neurosci 42(1):4–13. https://doi.org/10.1016/j.tins.2018.09.007
Jones DR, Moussaud S, McLean P (2014) Targeting heat shock proteins to modulate alpha-synuclein toxicity. Ther Adv Neurol Disord 7(1):33–51. https://doi.org/10.1177/1756285613493469
Jungermann K, Möhler H (1980) Biochemie. Ein Lehrbuch für Studierende der Medizin, Biologie und Pharmazie. Springer, Berlin
Kalia LV, Lang AE (2015) Parkinson’s disease. Lancet 386(9996):896–912. https://doi.org/10.1016/S0140-6736(14)61393-3
Karlsson O, Lindquist NG (2016) Melanin and neuromelanin binding of drugs and chemicals: toxicological implications. Arch Toxicol 90(8):1883–1891. https://doi.org/10.1007/s00204-016-1757-0
Killinger BA, Madaj Z, Sikora JW, Rey N, Haas AJ, Vepa Y, Lindqvist D, Chen H, Thomas PM, Brundin P, Brundin L, Labrie V (2018) The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aar5280
Kim YH, Lussier S, Rane A, Choi SW, Andersen JK (2011) Inducible dopaminergic glutathione depletion in an alpha-synuclein transgenic mouse model results in age-related olfactory dysfunction. Neuroscience 172:379–386. https://doi.org/10.1016/j.neuroscience.2010.10.072
Klucken J, Kruger R, Schmidt P, Bloem BR (2018) Management of Parkinson’s disease 20 years from now: towards digital health pathways. J Parkinsons Dis 8(s1):S85–S94. https://doi.org/10.3233/JPD-181519
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14(5):504–506. https://doi.org/10.1038/nm1747
Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet 18(2):106–108. https://doi.org/10.1038/ng0298-106
Langston JW, Ballard P, Tetrud JW, Irwin I (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219(4587):979–980
Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D (1999) Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol 46(4):598–605
Lardenoije R, Iatrou A, Kenis G, Kompotis K, Steinbusch HW, Mastroeni D, Coleman P, Lemere CA, Hof PR, van den Hove DL, Rutten BP (2015) The epigenetics of aging and neurodegeneration. Prog Neurobiol 131:21–64. https://doi.org/10.1016/j.pneurobio.2015.05.002
Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14(1):38–48. https://doi.org/10.1038/nrn3406
Lassot I, Mora S, Lesage S, Zieba BA, Coque E, Condroyer C, Bossowski JP, Mojsa B, Marelli C, Soulet C, Tesson C, Carballo-Carbajal I, Laguna A, Mangone G, Vila M, Brice A, Desagher S (2018) The E3 ubiquitin ligases TRIM17 and TRIM41 modulate alpha-synuclein expression by regulating ZSCAN21. Cell Rep. 25(9):2484 e2489–2496 e2489. https://doi.org/10.1016/j.celrep.2018.11.002
Lawana V, Singh N, Sarkar S, Charli A, Jin H, Anantharam V, Kanthasamy AG, Kanthasamy A (2017) Involvement of c-Abl kinase in microglial activation of NLRP3 inflammasome and impairment in autolysosomal system. J Neuroimmune Pharmacol 12(4):624–660. https://doi.org/10.1007/s11481-017-9746-5
Lee HM, Koh SB (2015) Many faces of Parkinson’s disease: non-motor symptoms of Parkinson’s disease. J Mov Disord 8(2):92–97. https://doi.org/10.14802/jmd.15003
Lee VM, Trojanowski JQ (2006) Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 52(1):33–38. https://doi.org/10.1016/j.neuron.2006.09.026
Leija-Salazar M, Piette C, Proukakis C (2018) Review: somatic mutations in neurodegeneration. Neuropathol Appl Neurobiol 44(3):267–285. https://doi.org/10.1111/nan.12465
Li W, Lesuisse C, Xu Y, Troncoso JC, Price DL, Lee MK (2004) Stabilization of alpha-synuclein protein with aging and familial parkinson’s disease-linked A53T mutation. J Neurosci 24(33):7400–7409. https://doi.org/10.1523/JNEUROSCI.1370-04.2004
Li C, Lutz EA, Slade KM, Ruf RA, Wang GF, Pielak GJ (2009) 19F NMR studies of alpha-synuclein conformation and fibrillation. Biochemistry 48(36):8578–8584. https://doi.org/10.1021/bi900872p
Li J, Yang J, Zhao P, Li S, Zhang R, Zhang X, Liu D, Zhang B (2012) Neuromelanin enhances the toxicity of alpha-synuclein in SK-N-SH cells. J Neural Transm (Vienna) 119(6):685–691. https://doi.org/10.1007/s00702-011-0753-z
Lindersson E, Beedholm R, Hojrup P, Moos T, Gai W, Hendil KB, Jensen PH (2004) Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem 279(13):12924–12934. https://doi.org/10.1074/jbc.M306390200
Ling H, Kearney S, Yip HL, Silveira-Moriyama L, Revesz T, Holton JL, Strand C, Davey K, Mok KY, Polke JM, Lees AJ (2016) Parkinson's disease without nigral degeneration: a pathological correlate of scans without evidence of dopaminergic deficit (SWEDD)? J Neurol Neurosurg Psychiatry 87(6):633–641. https://doi.org/10.1136/jnnp-2015-310756
Liu B, Fang F, Pedersen NL, Tillander A, Ludvigsson JF, Ekbom A, Svenningsson P, Chen H, Wirdefeldt K (2017) Vagotomy and Parkinson disease: a Swedish register-based matched-cohort study. Neurology 88(21):1996–2002. https://doi.org/10.1212/WNL.0000000000003961
Llorens F, Kruse N, Karch A, Schmitz M, Zafar S, Gotzmann N, Sun T, Kochy S, Knipper T, Cramm M, Golanska E, Sikorska B, Liberski PP, Sanchez-Valle R, Fischer A, Mollenhauer B, Zerr I (2018) Validation of alpha-Synuclein as a CSF biomarker for sporadic Creutzfeldt-Jakob disease. Mol Neurobiol 55(3):2249–2257. https://doi.org/10.1007/s12035-017-0479-5
Löffler G, Petrides PE, Weiss L, Harper HA (1979) Physiologische Chemie. Lehrbuch der medizinischen Biochemie und Pathobiochemie für Studierende der Medizin und Ärzte. Springer, Berlin
Longhena F, Faustini G, Missale C, Pizzi M, Spano P, Bellucci A (2017) The contribution of alpha-Synuclein spreading to Parkinson’s disease synaptopathy. Neural Plast 2017:5012129. https://doi.org/10.1155/2017/5012129
Lunati A, Lesage S, Brice A (2018) The genetic landscape of Parkinson’s disease. Rev Neurol (Paris) 174(9):628–643. https://doi.org/10.1016/j.neurol.2018.08.004
Magdalinou NK, Paterson RW, Schott JM, Fox NC, Mummery C, Blennow K, Bhatia K, Morris HR, Giunti P, Warner TT, de Silva R, Lees AJ, Zetterberg H (2015) A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry 86(11):1240–1247. https://doi.org/10.1136/jnnp-2014-309562
Mandel S, Maor G, Youdim MB (2004) Iron and alpha-synuclein in the substantia nigra of MPTP-treated mice: effect of neuroprotective drugs R-apomorphine and green tea polyphenol (−)-epigallocatechin-3-gallate. J Mol Neurosci 24(3):401–416. https://doi.org/10.1385/JMN:24:3:401
Masaracchia C, Hnida M, Gerhardt E, Lopes da Fonseca T, Villar-Pique A, Branco T, Stahlberg MA, Dean C, Fernandez CO, Milosevic I, Outeiro TF (2018) Membrane binding, internalization, and sorting of alpha-synuclein in the cell. Acta Neuropathol Commun 6(1):79. https://doi.org/10.1186/s40478-018-0578-1
Mattson MP (2011) Commentary: proteooxidotoxic process of aggregation. Neuromolecular Med 13(2):91–92. https://doi.org/10.1007/s12017-011-8146-x
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146(1):37–52. https://doi.org/10.1016/j.cell.2011.06.001
Mbefo MK, Paleologou KE, Boucharaba A, Oueslati A, Schell H, Fournier M, Olschewski D, Yin G, Zweckstetter M, Masliah E, Kahle PJ, Hirling H, Lashuel HA (2010) Phosphorylation of synucleins by members of the Polo-like kinase family. J Biol Chem 285(4):2807–2822. https://doi.org/10.1074/jbc.M109.081950
McDowall JS, Brown DR (2016) Alpha-synuclein: relating metals to structure, function and inhibition. Metallomics 8(4):385–397. https://doi.org/10.1039/c6mt00026f
McGeer PL, Itagaki S, Akiyama H, McGeer EG (1988) Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol 24(4):574–576. https://doi.org/10.1002/ana.410240415
McNeill A, Wu RM, Tzen KY, Aguiar PC, Arbelo JM, Barone P, Bhatia K, Barsottini O, Bonifati V, Bostantjopoulou S, Bressan R, Cossu G, Cortelli P, Felicio A, Ferraz HB, Herrera J, Houlden H, Hoexter M, Isla C, Lees A, Lorenzo-Betancor O, Mencacci NE, Pastor P, Pappata S, Pellecchia MT, Silveria-Moriyama L, Varrone A, Foltynie T, Schapira AH (2013) Dopaminergic neuronal imaging in genetic Parkinson’s disease: insights into pathogenesis. PLoS One 8(7):e69190. https://doi.org/10.1371/journal.pone.0069190
Melo TQ, Copray S, Ferrari MFR (2018) Alpha-Synuclein toxicity on protein quality control, mitochondria and endoplasmic reticulum. Neurochem Res 43(12):2212–2223. https://doi.org/10.1007/s11064-018-2673-x
Mijaljica D, Prescott M, Devenish RJ (2011) Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7(7):673–682
Miki Y, Tanji K, Mori F, Utsumi J, Sasaki H, Kakita A, Takahashi H, Wakabayashi K (2018) Autophagy mediators (FOXO1, SESN3 and TSC2) in Lewy body disease and aging. Neurosci Lett 684:35–41. https://doi.org/10.1016/j.neulet.2018.06.052
Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, Singleton AB (2004) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62(10):1835–1838. https://doi.org/10.1212/01.wnl.0000127517.33208.f4
Miotto MC, Binolfi A, Zweckstetter M, Griesinger C, Fernandez CO (2014a) Bioinorganic chemistry of synucleinopathies: deciphering the binding features of Met motifs and His-50 in AS-Cu(I) interactions. J Inorg Biochem 141:208–211. https://doi.org/10.1016/j.jinorgbio.2014.08.012
Miotto MC, Rodriguez EE, Valiente-Gabioud AA, Torres-Monserrat V, Binolfi A, Quintanar L, Zweckstetter M, Griesinger C, Fernandez CO (2014b) Site-specific copper-catalyzed oxidation of alpha-synuclein: tightening the link between metal binding and protein oxidative damage in Parkinson’s disease. Inorg Chem 53(9):4350–4358. https://doi.org/10.1021/ic4031377
Miotto MC, Pavese MD, Quintanar L, Zweckstetter M, Griesinger C, Fernandez CO (2017) Bioinorganic chemistry of Parkinson’s disease: affinity and structural features of Cu(I) binding to the full-length beta-Synuclein protein. Inorg Chem 56(17):10387–10395. https://doi.org/10.1021/acs.inorgchem.7b01292
Miraglia F, Ricci A, Rota L, Colla E (2018) Subcellular localization of alpha-synuclein aggregates and their interaction with membranes. Neural Regen Res 13(7):1136–1144. https://doi.org/10.4103/1673-5374.235013
Miranda-Morales E, Meier K, Sandoval-Carrillo A, Salas-Pacheco J, Vazquez-Cardenas P, Arias-Carrion O (2017) Implications of DNA methylation in Parkinson’s disease. Front Mol Neurosci 10:225. https://doi.org/10.3389/fnmol.2017.00225
Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T (1994) Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett 180(2):147–150
Mogi M, Harada M, Kondo T, Riederer P, Nagatsu T (1995) Brain beta 2-microglobulin levels are elevated in the striatum in Parkinson’s disease. J Neural Transm Parkinson’s Dis Dement Sect 9(1):87–92
Mogi M, Harada M, Kondo T, Riederer P, Nagatsu T (1996) Interleukin-2 but not basic fibroblast growth factor is elevated in parkinsonian brain. Short communication. J Neural Transm (Vienna) 103(8–9):1077–1081. https://doi.org/10.1007/BF01291792
Mokretar K, Pease D, Taanman JW, Soenmez A, Ejaz A, Lashley T, Ling H, Gentleman S, Houlden H, Holton JL, Schapira AHV, Nacheva E, Proukakis C (2018) Somatic copy number gains of alpha-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain 141(8):2419–2431. https://doi.org/10.1093/brain/awy157
Moore DJ, West AB, Dawson VL, Dawson TM (2005) Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci 28:57–87. https://doi.org/10.1146/annurev.neuro.28.061604.135718
Muller T (2012) Drug therapy in patients with Parkinson’s disease. Transl Neurodegener 1(1):10. https://doi.org/10.1186/2047-9158-1-10
Muller T, Ohm G, Eilert K, Mohr K, Rotter S, Haas T, Kuchler M, Lutge S, Marg M, Rothe H (2017) Benefit on motor and non-motor behavior in a specialized unit for Parkinson’s disease. J Neural Transm (Vienna) 124(6):715–720. https://doi.org/10.1007/s00702-017-1701-3
Munch G, Luth HJ, Wong A, Arendt T, Hirsch E, Ravid R, Riederer P (2000) Crosslinking of alpha-synuclein by advanced glycation endproducts—an early pathophysiological step in Lewy body formation? J Chem Neuroanat 20(3–4):253–257
Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46(9):989–993. https://doi.org/10.1038/ng.3043
Narayanan V, Scarlata S (2001) Membrane binding and self-association of alpha-synucleins. Biochemistry 40(33):9927–9934
Ndayisaba A, Kaindlstorfer C, Wenning GK (2019) Iron in neurodegeneration—cause or consequence? Front Neurosci 13:180. https://doi.org/10.3389/fnins.2019.00180
Niu H, Shen L, Li T, Ren C, Ding S, Wang L, Zhang Z, Liu X, Zhang Q, Geng D, Wu X, Li H (2018) Alpha-synuclein overexpression in the olfactory bulb initiates prodromal symptoms and pathology of Parkinson’s disease. Transl Neurodegener 7:25. https://doi.org/10.1186/s40035-018-0128-6
Obergasteiger J, Frapporti G, Pramstaller PP, Hicks AA, Volta M (2018) A new hypothesis for Parkinson’s disease pathogenesis: GTPase-p38 MAPK signaling and autophagy as convergence points of etiology and genomics. Mol Neurodegener 13(1):40. https://doi.org/10.1186/s13024-018-0273-5
Oliveira LM, Oliveira MA, Moriya HT, Moreira TS, Takakura AC (2019) Respiratory disturbances in a mouse model of Parkinson’s disease. Exp Physiol 104(5):729–739. https://doi.org/10.1113/EP087507
Olteanu A, Pielak GJ (2004) Peroxidative aggregation of alpha-synuclein requires tyrosines. Protein Sci 13(11):2852–2856. https://doi.org/10.1110/ps.04947204
Pagan F, Hebron M, Valadez EH, Torres-Yaghi Y, Huang X, Mills RR, Wilmarth BM, Howard H, Dunn C, Carlson A, Lawler A, Rogers SL, Falconer RA, Ahn J, Li Z, Moussa C (2016) Nilotinib effects in Parkinson’s disease and dementia with Lewy bodies. J Parkinsons Dis 6(3):503–517. https://doi.org/10.3233/JPD-160867
Paleologou KE, El-Agnaf OM (2012) alpha-Synuclein aggregation and modulating factors. Subcell Biochem 65:109–164. https://doi.org/10.1007/978-94-007-5416-4_6
Paleologou KE, Schmid AW, Rospigliosi CC, Kim HY, Lamberto GR, Fredenburg RA, Lansbury PT Jr, Fernandez CO, Eliezer D, Zweckstetter M, Lashuel HA (2008) Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J Biol Chem 283(24):16895–16905. https://doi.org/10.1074/jbc.M800747200
Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim HY, Lamberto GR, Fernandez CO, Schmid A, Chegini F, Gai WP, Chiappe D, Moniatte M, Schneider BL, Aebischer P, Eliezer D, Zweckstetter M, Masliah E, Lashuel HA (2010) Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein–membrane interactions. J Neurosci 30(9):3184–3198. https://doi.org/10.1523/JNEUROSCI.5922-09.2010
Pan T, Zhu J, Hwu WJ, Jankovic J (2012) The role of alpha-synuclein in melanin synthesis in melanoma and dopaminergic neuronal cells. PLoS One 7(9):e45183. https://doi.org/10.1371/journal.pone.0045183
Pan-Montojo F, Anichtchik O, Dening Y, Knels L, Pursche S, Jung R, Jackson S, Gille G, Spillantini MG, Reichmann H, Funk RH (2010) Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One 5(1):e8762. https://doi.org/10.1371/journal.pone.0008762
Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O’Sullivan GA, Pal A, Said J, Marsico G, Verbavatz JM, Rodrigo-Angulo M, Gille G, Funk RH, Reichmann H (2012) Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep 2:898. https://doi.org/10.1038/srep00898
Park HJ, Ryu D, Parmar M, Giasson BI, McFarland NR (2017) The ER retention protein RER1 promotes alpha-synuclein degradation via the proteasome. PLoS One 12(9):e0184262. https://doi.org/10.1371/journal.pone.0184262
Pavese N, Brooks DJ (2009) Imaging neurodegeneration in Parkinson’s disease. Biochim Biophys Acta 1792(7):722–729. https://doi.org/10.1016/j.bbadis.2008.10.003
Pavese N, Tai YF (2018) Nigrosome imaging and neuromelanin sensitive MRI in diagnostic evaluation of Parkinsonism. Mov Disord Clin Pract 5(2):131–140. https://doi.org/10.1002/mdc3.12590
Pena-Altamira E, Prati F, Massenzio F, Virgili M, Contestabile A, Bolognesi ML, Monti B (2016) Changing paradigm to target microglia in neurodegenerative diseases: from anti-inflammatory strategy to active immunomodulation. Expert Opin Ther Targets 20(5):627–640. https://doi.org/10.1517/14728222.2016.1121237
Perrett RM, Alexopoulou Z, Tofaris GK (2015) The endosomal pathway in Parkinson’s disease. Mol Cell Neurosci 66(Pt A):21–28. https://doi.org/10.1016/j.mcn.2015.02.009
Petrov VA, Saltykova IV, Zhukova IA, Alifirova VM, Zhukova NG, Dorofeeva YB, Tyakht AV, Kovarsky BA, Alekseev DG, Kostryukova ES, Mironova YS, Izhboldina OP, Nikitina MA, Perevozchikova TV, Fait EA, Babenko VV, Vakhitova MT, Govorun VM, Sazonov AE (2017) Analysis of gut microbiota in patients with Parkinson’s disease. Bull Exp Biol Med 162(6):734–737. https://doi.org/10.1007/s10517-017-3700-7
Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR (2002) Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36(6):1007–1019
Pinho R, Paiva I, Jercic KG, Fonseca-Ornelas L, Gerhardt E, Fahlbusch C, Garcia-Esparcia P, Kerimoglu C, Pavlou MAS, Villar-Pique A, Szego E, Lopes da Fonseca T, Odoardi F, Soeroes S, Rego AC, Fischle W, Schwamborn JC, Meyer T, Kugler S, Ferrer I, Attems J, Fischer A, Becker S, Zweckstetter M, Borovecki F, Outeiro TF (2019) Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum Mol Genet 28(1):31–50. https://doi.org/10.1093/hmg/ddy326
Piper DA, Sastre D, Schule B (2018) Advancing stem cell models of alpha-synuclein gene regulation in neurodegenerative disease. Front Neurosci 12:199. https://doi.org/10.3389/fnins.2018.00199
Plum S, Steinbach S, Attems J, Keers S, Riederer P, Gerlach M, May C, Marcus K (2016) Proteomic characterization of neuromelanin granules isolated from human substantia nigra by laser-microdissection. Sci Rep 6:37139. https://doi.org/10.1038/srep37139
Poewe W, Karamat E, Kemmler GW, Gerstenbrand F (1990) The premorbid personality of patients with Parkinson’s disease: a comparative study with healthy controls and patients with essential tremor. Adv Neurol 53:339–342
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276(5321):2045–2047
Pont-Sunyer C, Tolosa E, Caspell-Garcia C, Coffey C, Alcalay RN, Chan P, Duda JE, Facheris M, Fernandez-Santiago R, Marek K, Lomena F, Marras C, Mondragon E, Saunders-Pullman R, Waro B (2017) The prodromal phase of leucine-rich repeat kinase 2-associated Parkinson disease: clinical and imaging studies. Mov Disord 32(5):726–738. https://doi.org/10.1002/mds.26964
Porcari R, Proukakis C, Waudby CA, Bolognesi B, Mangione PP, Paton JF, Mullin S, Cabrita LD, Penco A, Relini A, Verona G, Vendruscolo M, Stoppini M, Tartaglia GG, Camilloni C, Christodoulou J, Schapira AH, Bellotti V (2015) The H50Q mutation induces a 10-fold decrease in the solubility of alpha-synuclein. J Biol Chem 290(4):2395–2404. https://doi.org/10.1074/jbc.M114.610527
Przuntek H, Muller T, Riederer P (2004) Diagnostic staging of Parkinson’s disease: conceptual aspects. J Neural Transm (Vienna) 111(2):201–216. https://doi.org/10.1007/s00702-003-0102-y
Purisai MG, McCormack AL, Langston WJ, Johnston LC, Di Monte DA (2005) Alpha-synuclein expression in the substantia nigra of MPTP-lesioned non-human primates. Neurobiol Dis 20(3):898–906. https://doi.org/10.1016/j.nbd.2005.05.028
Puschmann A (2013) Monogenic Parkinson’s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord 19(4):407–415. https://doi.org/10.1016/j.parkreldis.2013.01.020
Rasia RM, Bertoncini CW, Marsh D, Hoyer W, Cherny D, Zweckstetter M, Griesinger C, Jovin TM, Fernandez CO (2005) Structural characterization of copper(II) binding to alpha-synuclein: insights into the bioinorganic chemistry of Parkinson’s disease. Proc Natl Acad Sci USA 102(12):4294–4299. https://doi.org/10.1073/pnas.0407881102
Reimao S, Pita Lobo P, Neutel D, Correia Guedes L, Coelho M, Rosa MM, Ferreira J, Abreu D, Goncalves N, Morgado C, Nunes RG, Campos J, Ferreira JJ (2015) Substantia nigra neuromelanin magnetic resonance imaging in de novo Parkinson’s disease patients. Eur J Neurol 22(3):540–546. https://doi.org/10.1111/ene.12613
Rekas A, Knott RB, Sokolova A, Barnham KJ, Perez KA, Masters CL, Drew SC, Cappai R, Curtain CC, Pham CL (2010) The structure of dopamine induced alpha-synuclein oligomers. Eur Biophys J 39(10):1407–1419. https://doi.org/10.1007/s00249-010-0595-x
Rietdijk CD, Perez-Pardo P, Garssen J, van Wezel RJ, Kraneveld AD (2017) Exploring Braak’s hypothesis of Parkinson’s disease. Front Neurol 8:37. https://doi.org/10.3389/fneur.2017.00037
Ruf RA, Lutz EA, Zigoneanu IG, Pielak GJ (2008) Alpha-Synuclein conformation affects its tyrosine-dependent oxidative aggregation. Biochemistry 47(51):13604–13609. https://doi.org/10.1021/bi801884z
Rumpf JJ, Schirmer M, Fricke C, Weise D, Wagner JA, Simon J, Classen J (2015) Light pigmentation phenotype is correlated with increased substantia nigra echogenicity. Mov Disord 30(13):1848–1852. https://doi.org/10.1002/mds.26427
Salmon L, Nodet G, Ozenne V, Yin G, Jensen MR, Zweckstetter M, Blackledge M (2010) NMR characterization of long-range order in intrinsically disordered proteins. J Am Chem Soc 132(24):8407–8418. https://doi.org/10.1021/ja101645g
Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, Chesselet MF, Keshavarzian A, Shannon KM, Krajmalnik-Brown R, Wittung-Stafshede P, Knight R, Mazmanian SK (2016) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167(6):1469–1480. https://doi.org/10.1016/j.cell.2016.11.018
Sanchez-Guajardo V, Barnum CJ, Tansey MG, Romero-Ramos M (2013) Neuroimmunological processes in Parkinson’s disease and their relation to alpha-synuclein: microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro 5(2):113–139. https://doi.org/10.1042/AN20120066
Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, Haapaniemi E, Kaakkola S, Eerola-Rautio J, Pohja M, Kinnunen E, Murros K, Auvinen P (2015) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30(3):350–358. https://doi.org/10.1002/mds.26069
Schlachetzki JCM, Barth J, Marxreiter F, Gossler J, Kohl Z, Reinfelder S, Gassner H, Aminian K, Eskofier BM, Winkler J, Klucken J (2017) Wearable sensors objectively measure gait parameters in Parkinson’s disease. PLoS One 12(10):e0183989. https://doi.org/10.1371/journal.pone.0183989
Schneider SA, Alcalay RN (2017) Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov Disord 32(11):1504–1523. https://doi.org/10.1002/mds.27193
Schulz J, Takousis P, Wohlers I, Itua IOG, Dobricic V, Rucker G, Binder H, Middleton L, Ioannidis JPA, Perneczky R, Bertram L, Lill CM (2019) Meta-analyses identify differentially expressed micrornas in Parkinson’s disease. Ann Neurol 85(6):835–851. https://doi.org/10.1002/ana.25490
Schwalbe M, Ozenne V, Bibow S, Jaremko M, Jaremko L, Gajda M, Jensen MR, Biernat J, Becker S, Mandelkow E, Zweckstetter M, Blackledge M (2014) Predictive atomic resolution descriptions of intrinsically disordered hTau40 and alpha-synuclein in solution from NMR and small angle scattering. Structure 22(2):238–249. https://doi.org/10.1016/j.str.2013.10.020
Shahmoradian SH, Lewis AJ, Genoud C, Graff-Meyer A, Hench J, Moors T, Schweighauser G, Wang J, Goldie KN, Suetterlin R, Castano-Diez D, Perez-Navarro P, Huisman E, Ipsen S, Ingrassia A, de Gier Y, Rozemuller AJM, Da Paepe A, Erny J, Staempfli A, Hoernschemeyer J, Grosserueschkamp F, Niedieker D, El-Mashtoly SF, Quadri M, van IJcken WFJ, Bonifati V, Gerwert K, Bohrmann B, Frank S, Britschgi M, Stahlberg H, van de Berg W, Lauer ME (2019) Lewy pathology in Parkinson’s disease consists of a crowded organellar, membranous medley. bioRxiv. https://doi.org/10.1101/137976
Shamoto-Nagai M, Maruyama W, Yi H, Akao Y, Tribl F, Gerlach M, Osawa T, Riederer P, Naoi M (2006) Neuromelanin induces oxidative stress in mitochondria through release of iron: mechanism behind the inhibition of 26S proteasome. J Neural Transm (Vienna) 113(5):633–644. https://doi.org/10.1007/s00702-005-0410-5
Shen N, Song G, Yang H, Lin X, Brown B, Hong Y, Cai J, Cao C (2019) Identifying the pathological domain of Alpha-Synuclein as a therapeutic for Parkinson’s disease. Int J Mol Sci. https://doi.org/10.3390/ijms20092338
Shi M, Bradner J, Hancock AM, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Kim HM, Leverenz JB, Montine TJ, Ginghina C, Kang UJ, Cain KC, Wang Y, Aasly J, Goldstein D, Zhang J (2011) Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol 69(3):570–580. https://doi.org/10.1002/ana.22311
Sian-Hulsmann J, Mandel S, Youdim MB, Riederer P (2011) The relevance of iron in the pathogenesis of Parkinson’s disease. J Neurochem 118(6):939–957. https://doi.org/10.1111/j.1471-4159.2010.07132.x
Sian-Hulsmann J, Monoranu C, Strobel S, Riederer P (2015) Lewy bodies: a spectator or salient killer? CNS Neurol Disord Drug Targets 14(7):947–955
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Durr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361(17):1651–1661. https://doi.org/10.1056/NEJMoa0901281
Sierks MR, Chatterjee G, McGraw C, Kasturirangan S, Schulz P, Prasad S (2011) CSF levels of oligomeric alpha-synuclein and beta-amyloid as biomarkers for neurodegenerative disease. Integr Biol (Camb) 3(12):1188–1196. https://doi.org/10.1039/c1ib00018g
Sierra M, Martinez-Rodriguez I, Sanchez-Juan P, Gonzalez-Aramburu I, Jimenez-Alonso M, Sanchez-Rodriguez A, Berciano J, Banzo I, Infante J (2017) Prospective clinical and DaT-SPECT imaging in premotor LRRK2 G2019S-associated Parkinson disease. Neurology 89(5):439–444. https://doi.org/10.1212/WNL.0000000000004185
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302(5646):841. https://doi.org/10.1126/science.1090278
Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B (2003) Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem 278(14):11753–11759 Epub 12003 Jan 11724
Sommer A, Maxreiter F, Krach F, Fadler T, Grosch J, Maroni M, Graef D, Eberhardt E, Riemenschneider MJ, Yeo GW, Kohl Z, Xiang W, Gage FH, Winkler J, Prots I, Winner B (2018) Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell 23(1):123 e126–131 e126. https://doi.org/10.1016/j.stem.2018.06.015
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388(6645):839–840. https://doi.org/10.1038/42166
Stolzenberg E, Berry D, Yang Lee EY, Kroemer A, Kaufman S, Wong GCL, Oppenheim JJ, Sen S, Fishbein T, Bax A, Harris B, Barbut D, Zasloff MA (2017) A role for neuronal Alpha-Synuclein in gastrointestinal immunity. J Innate Immun 9(5):456–463. https://doi.org/10.1159/000477990
Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ (2008) Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging 29(11):1690–1701. https://doi.org/10.1016/j.neurobiolaging.2007.04.006
Surguchev AA, Surguchov A (2017) Synucleins and gene expression: ramblers in a crowd or cops regulating traffic? Front Mol Neurosci 10:224. https://doi.org/10.3389/fnmol.2017.00224
Surmeier DJ (2018) Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J 285(19):3657–3668. https://doi.org/10.1111/febs.14607
Surmeier DJ, Obeso JA, Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18(2):101–113. https://doi.org/10.1038/nrn.2016.178
Svensson E, Horvath-Puho E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, Sorensen HT (2015) Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 78(4):522–529. https://doi.org/10.1002/ana.24448
Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y (1992) Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol 119(2):301–311
Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, Dawson VL, Dawson TM, Lim KL (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet 17(3):431–439. https://doi.org/10.1093/hmg/ddm320
Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, Dawson VL, Dawson TM, Ross CA (2001) Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet 10(9):919–926
Tanner CM (2003) Is the cause of Parkinson’s disease environmental or hereditary? Evidence from twin studies. Adv Neurol 91:133–142
Tansey MG, McCoy MK, Frank-Cannon TC (2007) Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 208(1):1–25. https://doi.org/10.1016/j.expneurol.2007.07.004
Thome AD, Harms AS, Volpicelli-Daley LA, Standaert DG (2016) microRNA-155 regulates Alpha-Synuclein-induced inflammatory responses in models of Parkinson disease. J Neurosci 36(8):2383–2390. https://doi.org/10.1523/JNEUROSCI.3900-15.2016
Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG (2003) Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem 278(45):44405–44411 (Epub 42003 Aug 44415)
Trempe JF, Fon EA (2013) Structure and function of parkin, PINK1, and DJ-1, the three musketeers of neuroprotection. Front Neurol 4:38. https://doi.org/10.3389/fneur.2013.00038
Tribl F, Marcus K, Meyer HE, Bringmann G, Gerlach M, Riederer P (2006) Subcellular proteomics reveals neuromelanin granules to be a lysosome-related organelle. J Neural Transm (Vienna) 113(6):741–749. https://doi.org/10.1007/s00702-006-0452-3
Trigo-Damas I, Del Rey NL, Blesa J (2018) Novel models for Parkinson’s disease and their impact on future drug discovery. Expert Opin Drug Discov 13(3):229–239. https://doi.org/10.1080/17460441.2018.1428556
Tsukada M, Ohsumi Y (1993) Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 333(1–2):169–174
Uchihara T, Giasson BI (2016) Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol 131(1):49–73. https://doi.org/10.1007/s00401-015-1485-1
Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T (1993) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA 90(23):11282–11286
Uhl GR (1998) Hypothesis: the role of dopaminergic transporters in selective vulnerability of cells in Parkinson’s disease. Ann Neurol 43(5):555–560. https://doi.org/10.1002/ana.410430503
Ulrih NP, Barry CH, Fink AL (2008) Impact of Tyr to Ala mutations on alpha-synuclein fibrillation and structural properties. Biochim Biophys Acta 1782(10):581–585. https://doi.org/10.1016/j.bbadis.2008.07.004
Uversky VN, Yamin G, Munishkina LA, Karymov MA, Millett IS, Doniach S, Lyubchenko YL, Fink AL (2005) Effects of nitration on the structure and aggregation of alpha-synuclein. Brain Res Mol Brain Res 134(1):84–102. https://doi.org/10.1016/j.molbrainres.2004.11.014
Vaikath NN, Hmila I, Gupta V, Erskine D, Ingelsson M, El-Agnaf OMA (2019) Antibodies against alpha-synuclein: tools and therapies. J Neurochem. https://doi.org/10.1111/jnc.14713
Vamvaca K, Volles MJ, Lansbury PT Jr (2009) The first N-terminal amino acids of alpha-synuclein are essential for alpha-helical structure formation in vitro and membrane binding in yeast. J Mol Biol 389(2):413–424. https://doi.org/10.1016/j.jmb.2009.03.021
van Eimeren T, Binkofski F, Buhmann C, Hagenah J, Strafella AP, Pramstaller PP, Siebner HR, Klein C (2010) Imaging movement-related activity in medicated Parkin-associated and sporadic Parkinson’s disease. Parkinsonism Relat Disord 16(6):384–387. https://doi.org/10.1016/j.parkreldis.2010.04.003
van Nuenen BF, van Eimeren T, van der Vegt JP, Buhmann C, Klein C, Bloem BR, Siebner HR (2009) Mapping preclinical compensation in Parkinson’s disease: an imaging genomics approach. Mov Disord 24(Suppl 2):S703–S710. https://doi.org/10.1002/mds.22635
Varrone A, Pellecchia MT, Amboni M, Sansone V, Salvatore E, Ghezzi D, Garavaglia B, Brice A, Brunetti A, Bonavita V, De Michele G, Salvatore M, Pappata S, Barone P (2004) Imaging of dopaminergic dysfunction with [123I]FP-CIT SPECT in early-onset parkin disease. Neurology 63(11):2097–2103
Vicente Miranda H, Szego EM, Oliveira LMA, Breda C, Darendelioglu E, de Oliveira RM, Ferreira DG, Gomes MA, Rott R, Oliveira M, Munari F, Enguita FJ, Simoes T, Rodrigues EF, Heinrich M, Martins IC, Zamolo I, Riess O, Cordeiro C, Ponces-Freire A, Lashuel HA, Santos NC, Lopes LV, Xiang W, Jovin TM, Penque D, Engelender S, Zweckstetter M, Klucken J, Giorgini F, Quintas A, Outeiro TF (2017) Glycation potentiates alpha-synuclein-associated neurodegeneration in synucleinopathies. Brain 140(5):1399–1419. https://doi.org/10.1093/brain/awx056
Vilchez D, Saez I, Dillin A (2014) The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun 5:5659. https://doi.org/10.1038/ncomms6659
Wang Z, Gao G, Duan C, Yang H (2019) Progress of immunotherapy of anti-alpha-synuclein in Parkinson’s disease. Biomed Pharmacother 115:108843. https://doi.org/10.1016/j.biopha.2019.108843
Weiner WJ (2008) There is no Parkinson disease. Arch Neurol 65(6):705–708. https://doi.org/10.1001/archneur.65.6.705
Wile DJ, Agarwal PA, Schulzer M, Mak E, Dinelle K, Shahinfard E, Vafai N, Hasegawa K, Zhang J, McKenzie J, Neilson N, Strongosky A, Uitti RJ, Guttman M, Zabetian CP, Ding YS, Adam M, Aasly J, Wszolek ZK, Farrer M, Sossi V, Stoessl AJ (2017) Serotonin and dopamine transporter PET changes in the premotor phase of LRRK2 parkinsonism: cross-sectional studies. Lancet Neurol 16(5):351–359. https://doi.org/10.1016/S1474-4422(17)30056-X
Wong YC, Krainc D (2017) alpha-Synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 23(2):1–13. https://doi.org/10.1038/nm.4269
Wood H (2014) Parkinson disease: a monoclonal antibody targeting misfolded alpha-synuclein has therapeutic potential in Parkinson disease. Nat Rev Neurol 10(8):426. https://doi.org/10.1038/nrneurol.2014.119
Wypijewska A, Galazka-Friedman J, Bauminger ER, Wszolek ZK, Schweitzer KJ, Dickson DW, Jaklewicz A, Elbaum D, Friedman A (2010) Iron and reactive oxygen species activity in parkinsonian substantia nigra. Parkinsonism Relat Disord 16(5):329–333. https://doi.org/10.1016/j.parkreldis.2010.02.007
Xiang SQ, Narayanan RL, Becker S, Zweckstetter M (2013) N-H spin-spin couplings: probing hydrogen bonds in proteins. Angew Chem Int Ed Engl 52(12):3525–3528. https://doi.org/10.1002/anie.201209641
Xu S, Chan P (2015) Interaction between neuromelanin and Alpha-Synuclein in Parkinson’s disease. Biomolecules 5(2):1122–1142. https://doi.org/10.3390/biom5021122
Xuan Q, Xu SL, Lu DH, Yu S, Zhou M, Ueda K, Cui YQ, Zhang BY, Chan P (2011) Increased expression of alpha-synuclein in aged human brain associated with neuromelanin accumulation. J Neural Transm (Vienna) 118(11):1575–1583. https://doi.org/10.1007/s00702-011-0636-3
Ugalde CL, Lawson VA, Finkelstein DI, Hill AF (2019) The role of lipids in alpha-synuclein misfolding and neurotoxicity. J Biol Chem. https://doi.org/10.1074/jbc.rev119.007500
Yang X, Qian Y, Xu S, Song Y, Xiao Q (2017) Longitudinal analysis of fecal microbiome and pathologic processes in a rotenone induced mice model of Parkinson’s disease. Front Aging Neurosci 9:441. https://doi.org/10.3389/fnagi.2017.00441
Yao X, Becker S, Zweckstetter M (2014) A six-dimensional alpha proton detection-based APSY experiment for backbone assignment of intrinsically disordered proteins. J Biomol NMR 60(4):231–240. https://doi.org/10.1007/s10858-014-9872-9
Zecca L, Pietra R, Goj C, Mecacci C, Radice D, Sabbioni E (1994) Iron and other metals in neuromelanin, substantia nigra, and putamen of human brain. J Neurochem 62(3):1097–1101
Zecca L, Stroppolo A, Gatti A, Tampellini D, Toscani M, Gallorini M, Giaveri G, Arosio P, Santambrogio P, Fariello RG, Karatekin E, Kleinman MH, Turro N, Hornykiewicz O, Zucca FA (2004a) The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci USA 101(26):9843–9848. https://doi.org/10.1073/pnas.0403495101
Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR (2004b) Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 5(11):863–873. https://doi.org/10.1038/nrn1537
Zecca L, Wilms H, Geick S, Claasen JH, Brandenburg LO, Holzknecht C, Panizza ML, Zucca FA, Deuschl G, Sievers J, Lucius R (2008) Human neuromelanin induces neuroinflammation and neurodegeneration in the rat substantia nigra: implications for Parkinson’s disease. Acta Neuropathol 116(1):47–55. https://doi.org/10.1007/s00401-008-0361-7
Zhang NY, Tang Z, Liu CW (2008) alpha-Synuclein protofibrils inhibit 26 S proteasome-mediated protein degradation: understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J Biol Chem 283(29):20288–20298. https://doi.org/10.1074/jbc.M710560200
Zondler L, Kostka M, Garidel P, Heinzelmann U, Hengerer B, Mayer B, Weishaupt JH, Gillardon F, Danzer KM (2017) Proteasome impairment by alpha-synuclein. PLoS One 12(9):e0184040. https://doi.org/10.1371/journal.pone.0184040
Zucca FA, Segura-Aguilar J, Ferrari E, Munoz P, Paris I, Sulzer D, Sarna T, Casella L, Zecca L (2017) Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog Neurobiol 155:96–119. https://doi.org/10.1016/j.pneurobio.2015.09.012
Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, Toker NJ, Jeon S, Fredriksen K, Mazzulli JR (2018) Reversible conformational conversion of alpha-Synuclein into toxic assemblies by glucosylceramide. Neuron 97(1):92 e110–107 e110. https://doi.org/10.1016/j.neuron.2017.12.012
Acknowledgements
PR is thankful to the “Verein zur Durchführung neurowissenschaftlicher Tagungen e. V.” for supporting this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Riederer, P., Berg, D., Casadei, N. et al. α-Synuclein in Parkinson’s disease: causal or bystander?. J Neural Transm 126, 815–840 (2019). https://doi.org/10.1007/s00702-019-02025-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-019-02025-9