
Abstract

Congenital scoliosis associated with split cord malformation raises the issue on how to best manage these patients to avoid neurologic injury while achieving satisfactory correction. We present the case of a 12-year-old girl who first presented when she was 11-year old with such combination but without much physical handicap or neurological deficit. The corrective surgery offered at that time was refused by the family. She again presented after 1 year with documented severe aggravation of the curve resulting in unstable walking and psychological upset. Her imaging studies showed multiple malformations in lower cervical and thoracic spine and a split cord malformation type 2 (fibrous septum with diplomyelia) at the apex of the deformity. A one-stage correction was deemed neurologically too risky. We therefore performed during a first stage a thoracotomy with anterior release. This was followed by skeletal traction with skull tongs and bilateral femoral pins. After gradual increase in traction weights a reasonable correction was achieved without any neurological deficit, over the next 10 days. A second-stage operation was done on the 11th day and a posterior instrumented fusion was performed. Post-operative recovery was uneventful and there were no complications. She was discharged with a Boston Brace to be worn for 3 months. At 2-year follow-up the patient outcome is excellent with excellent balance and correction of the deformity. In this grand round case, we discuss all the different option of treatment of congenital scoliosis associated with split cord malformation. In a medical environment where spinal cord monitoring is lacking, we recommend an initial release followed by skull and bifemoral traction over several days to monitor the neurologic status of the patient. Once optimal correction is achieved with the traction, a posterior instrumentation can be safely done.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Case presentation
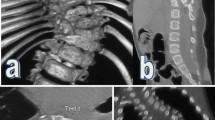
Our patient, an 11-year-old girl presented in our department in December 2004 with a rigid right thoracolumbar congenital scoliosis from T4 to L3, with an apex at T12. The curve measured 65° by the Cobb’s method (Fig. 1a). She had multiple congenitally anomalous vertebrae in lower cervical and whole of thoracic spine, with split cord malformation (SCM), without evidence of diastematomyelia. She had normal neurological examination without any sign of upper motor neuron lesion. She was Risser 1. There were no cardiac or renal anomalies on echocardiogram and ultrasound. She was offered surgery but parents refused the treatment at that time.

a Initial X-rays of the patient showing Rt thoracolumbar curve measuring 65° with an apex at T12. b, c One-year later AP and lateral X-rays showing increase in deformity to 84°. d Coronal MRI showing multiple anomalies involving almost whole of thoracic spine and consisting of congenital block hemivertebrae. e Axial MRI cut at T12 level showing split cord malformation without bony septum (malformation type II). f Standing posterior view of patient showing Rt thoracolumbar curve with tuft of hair at the upper thoracic level. g, h Cervical spine AP, Lat films showing Klippel–Feil malformation with left cervicothoracic congenital scoliosis
She presented again after 1 year in December 2005 with marked deterioration in deformity. She was unstable while walking and was disturbed mentally due to obvious deformity. On examination she had slightly brisk reflexes in lower limbs; rest of the neurological examination was unremarkable. On X-rays, curve was increased to 84°, and her sagittal balance was within normal limits (Fig. 1b, c). This time her parents agreed for surgery and an informed consent was taken for a two-stage procedure.
Diagnostic imaging section
Diagnosis and epidemiology
The exact incidence of congenital scoliosis is difficult to know with accuracy as a significant number may be asymptomatic. Some form of anomaly must be visible on X-rays, giving rise to a lateral curvature to call it a congenital scoliosis. Deformities visible on X-rays may be failures of formation or failure of segmentation or a combination of both. These deformities give an imbalanced growth and should be detected early in life so an appropriate surgical treatment can rebalance the growth of the spine. If left untreated, some of these cases end up as major rigid spinal deformities later in life. Correction of these large deformities may need either a lengthening procedure with its inherent neurologic risks or a shortening procedure, which is neurologically safer but technically more demanding. Mostly congenital scoliosis is thought to be caused by non-genetic, fetal environmental factors [17]. Rarely in association with various syndromes and multilevel deformities, there may be a genetic factor [10]. Natural history of congenital scoliosis shows that 25% cases will never progress, 25% will show mild progression and 50% will have severe progression, necessitating surgical intervention [14, 15, 17].
Congenital scoliosis can be diagnosed antenatally by ultrasound examination. After birth they may present with a deformity, skin manifestations, neurological symptoms, urinary problems or as an incidental finding on X-rays. Sometimes congenital anomalies are found on operations for what was thought to be an idiopathic scoliosis [1]. Clinical examination in these patients should be thorough, as there is a strong association with other anomalies in about 25% of cases [11–13]. At birth, VATER (vertebral, anal, tracheo-oesophageal and renal) anomalies should be sought and addressed to save patient’s life [7]. Urological anomalies are present in 18–25% of patients and congenital heart disease in 12% [8].
Other anomalies include: vertebral and spinal cord anomalies in up to 40% patients including; dysraphism (10%), diastematomyelia (5%), SCM, tethered cord, Arnold Chiari malformation, Sprengel’s deformity, Klippel-Feil syndrome, etc. [4, 5, 9]. There are many skin signs which may indicate the presence of a spinal dysraphism: hair patch, lipoma, dimple or scar in the back, foot anomaly such as club foot, atrophic calf muscles, leg length discrepancy and or any neurologic anomaly such as absent, increased or asymmetric reflexes and or urinary symptoms [13]. SCM is a closed neural tube defect where the spinal cord is split into two halves of variable size, each having its own (type 1) or combined (type 2) meningeal coverings [11]. The new nomenclature introduced by Pang was created to avoid the confusion between diastematomyelia and diplomyelia. In SCM type 1 (former diastematomyelia) there is a bony or fibrocartilaginous spur which separates the spinal cord into two halves with two different dural sleeves. In these patients, bony spur must be removed before undertaking corrective surgery at that level. This is the most frequent form of SCM. In SCM2 (former diplomyelia) there is no bony septum but a common dural sac and intradural fibrous band. The malformation of the spinal cord may not be addressed before corrective surgery.
Rationale for treatment and evidence-based literature
The treatment of congenital malformation of the spine associated with spinal dysraphisms is complex and must take into account the bony malformation and the spinal cord malformation. Little has been written for the surgical management of these complex deformities when the two co-exist.
For the deformity part of the spine, treatment modalities include either observation or surgery. Braces have a very limited role; however, they can be used in long flexible curves, secondary curves and as an adjunct to surgery to assist fusion. Observation is used for curves with little growth potential. Surgery is either prophylactic or corrective. Prophylactic surgery is indicated in younger children who have small and progressive curves. Fully segmented hemivertebra with a contralateral bar or other combinations of anomalies producing imbalance of growth on convex and concave sides of the spine may beneficiate from early prophylactic surgery. Prophylactic surgery is either in situ fusion or hemiepiphysiodesis. Posterior in situ fusion has potential for crankshaft phenomenon in younger children. Convex hemiepiphysiodesis is reserved for curves less than 50° and in children less than 5 years. It can be done either as a one-stage posterior transpedicular procedure or sequential or combined anterior and posterior procedure [16].
Corrective surgery takes the form of either a lengthening procedure or a shortening procedure; they should be done under strict neurologic surveillance. Lengthening procedure have a higher risk of neurological complications and if required can be done as staged procedure, with initial anterior or posterior release followed by skeletal traction gradually lengthening the spine over a short period of time and carefully monitoring the neurological status. The definitive posterior instrumented fusion is achieved in the best possible correction achieved with the skeletal traction. Halo femoral traction sliding rods have been described in the literature in the past for neurologically compromised patients [2]. If a lengthening procedure is performed in a single stage, risk for development of neurological deficit is ten times higher than idiopathic scoliosis [3]. In these cases spinal cord monitoring and wake-up test are mandatory. One of the advantages to perform an anterior release in these cases is that the removal of the discs anteriorly will allow some degree of shortening during the posterior correction and may avoid overstretching the spinal cord. Shortening procedures include hemivertebrectomy and vertebral column resection. Both procedures can be done either by a single-stage posterior approach or by a multistage approach. These cases are ideally done in the lumbar spine below the spinal cord; at the thoracic level they remain more challenging. Other treatment options include growing rods in flexible curves with apical fusion and or rib expanders for thoracic insufficiency syndromes associated with fused ribs and or chest hypoplasia [1].
The presence of the spinal cord dysraphism may change the strategy for the deformity correction. If there is a tethered cord syndrome with thick filum terminale, the section of the filum terminale is a simple neurosurgical procedure that can be performed before any attempt of deformity correction. In the presence of a SCM type 1, the bony septum must classically be removed before any corrective spinal surgery lengthening procedure. In the case of a SCM type 1 associated with a distal tethering with thick filum terminale, the section of the filum terminale may theoretically let the spinal cord snap at the level of the diastematomylelia as the spinal cord would migrate upward and this could therefore be dangerous for the neurologic status. Removing of the bony peg may therefore be the first step to start with in the rare association of a SCM type 1 with distal tethering and spinal deformity. This classic attitude has recently been challenged by Wang, who in a review of 11 cases of congenital scoliosis with type I SCM reported that the bone spur need not be excised before spinal correction if there is no clinical signs of spinal cord tethering and the bone spur is located in the middle of the split cord with much space around to accommodate it. The other logical option in SCM type 1 is to perform a shortening procedure such as a hemiresection or spinal column resection below the cord malformation to avoid any distraction of the spinal cord during correction. Other authors have reported a combined neurosurgical procedure where both the SCM and the deformity are addressed during the same anesthesia [6].
In the case of a SCM type 2 (without bony spur) it has been reported in the recent Chinese literature that a lengthening corrective procedure can be achieved safely without resection of the fibrous septum [12, 18].
In this grand round case the patient had no thick filum terminale and the spinal cord ended at a normal level. The SCM was a type 2 with no bony septum. It was therefore thought that no neurosurgical procedure was judged necessary before the correction of the deformity. A single-stage lengthening procedure was judged to be too risky neurologically as it could have potentially stretched the spinal cord and induced paralysis especially in our medical environment where we lack spinal cord monitoring. A vertebral column resection would have required a vertebrectomy at the thoracolumbar level where the SCM was located and was also thought to be too risky in our case. The patient also had an upper malformation of the cervicothoracic junction that compensated the thoracic malformation to some extent, so her head was in a straight position. Therefore, a hemiresection or spinal column resection at the thoracic spine level may have induced in a persisting head tilt or torticollis. With all these considerations in mind, it was therefore thought that the most appropriate treatment was to stage the surgery with skeletal traction between the two stages to monitor the patient neurologically and achieve satisfactory correction while keeping her balanced with her head aligned relative to her trunk. The advantage of the anterior release being also to provide increased flexibility of the spine, bringing the deformity to where it was several months or years before, and provide some relative shortening through the discs excisions at the time of the posterior correction.
Procedure
The first stage surgery took place on 6th of January 2006. Under general anesthesia, the patient was placed in a left lateral position. A right 8th rib thoracotomy was performed (Fig. 2b). A total rib head excision, release of anterior and posterior longitudinal ligaments with total discectomies, was performed at the apex of the curve from T9 to L1. Adequate mobilization was achieved at all these levels. Rib and locally harvested bone were morcellized and used as graft in the interspaces and pleura was closed over the graft (Fig. 2c). Patient recovered uneventfully and was placed on skull and bilateral femoral traction.

a AP X-rays showing correction of deformity under bipolar traction. b Stage one operation with right 8th rib thoracotomy and anterior release, after total discectomy, removal of the rib heads and release of the anterior and posterior longitudinal ligaments. c Closure of pleura after interbody grafting with morcellized bone graft. d Stage two operations, after correction of the deformity and bone grafting using pedicle screw system with four screws at the top and bottom of the construct and two screws to control the apex
Over next 10 days, gradual correction of deformity was achieved by increasing the weights daily. Patient was monitored neurologically and she did not develop any deficit. On the 10th day, chest X-rays revealed adequate correction of deformity (Fig. 2a).
Second-stage surgery was performed on 17th of January 2006. The patient was briefed about the Stagnara wake-up test before anesthesia. The patient was then placed prone on the operation table. A midline exposure was carried out from T4 to L3. At the upper part of the curve, pedicles landmarks were identified in the mass of the deformation and four screws were placed using the funnel technique. Two screws were placed in the pedicles at the apex of the curve at T11. Four screws were placed distally in the pedicles of L2 and L3. Contoured rods were applied and the deformity was further corrected using a rod derotation maneuver (CD Horizon M8, titanium, fixed and multi-axial pedicle screw system Memphis, TN, USA). At this stage, a wake-up test was performed and the patient moved both the feet normally. Extensive grafts were taken from both iliac crests and placed in the decorticated bed. The wound was closed over suction drains. Patient had an uneventful recovery.
Procedure imaging section
Outcome
Post-operative chest X-rays revealed a marked correction of the deformity. Drains were removed on second post-operative day and patient was allowed out of bed. The patient was advised to wear a Boston brace for 3 months. Patient was discharged from the hospital on 3rd post-operative day and was advised regular follow up. Stitches were removed at 2 weeks. Her wound was completely healed. After operation, the patient had very obvious posture and deformity correction even on simple inspection if compared with pre-operative picture (Fig. 4a). She was followed up at 6 weeks, 3 months, 6 months and then yearly. On last follow up at 2 years, she had only minimal loss of correction (Figs. 3c, d, 4b). She had solid fusion at that time, had no complaints and was fully satisfied with the procedure.

a, b Immediate post-operative AP and Lat X-rays. c, d AP and Lat X-rays 2 years after surgery

a One month after operation. b Two years after operation
References
Arlet V, Odent T, Aebi M (2003) Congenital scoliosis. Eur Spine J 12(5):456–463
Arlet V, Papin P, Marchesi D (1999) Halo femoral traction and sliding rods in the treatment of a neurologically compromised congenital scoliosis: technique. Eur Spine J 8(4):329–331
Banniza von Bazan UK, Rompe G, Krastel A, Martin K (1976) Diastematomyelia—its importance in the treatment of congenital scoliosis. Z Orthop Ihre Grenzgeb 114(6):881–889
Beals R, Robbins JR, Rolfe B (1993) Anomalies associated with vertebral malformations. Spine 18:1329–1332
Bradford DS, Heithoff KB, Cohen M (1991) Intraspinal abnormalities and congenital spine deformities: a radiographic and MRI study. J Pediatr Orthop 11:36–41
Hamzaoglu A, Ozturk C, Tezer M, Aydogan M, Sarier M, Talu U (2007) Simultaneous surgical treatment in congenital scoliosis and/or kyphosis associated with intraspinal abnormalities. Spine 32(25):2880–2884
Lawhon SM, MacEwen GD, Bunnell WP (1986) Orthopaedic aspects of the VATER association. J Bone Joint Surg Am 68(3):424–429
MacEwen GD, Winter RB, Hardy JH (1972) Evaluation of kidney anomalies in congenital scoliosis. J Bone Joint Surg 54A:1341
McMaster MJ (1998) Congenital scoliosis caused by a unilateral failure of vertebral segmentation with contralateral hemivertebrae. Spine 23:998–1005
McMaster MJ, Ohtsuka K (1982) The natural history of congenital scoliosis. A study of two hundred and fifty-one patients. J Bone Joint Surg Am 64:1128–1147
Pang D, Dias MS, Ahab-Barmada M (1992) Split cord malformation: Part I: a unified theory of embryogenesis for double spinal cord malformations. Neurosurgery 31(3):451–480
Wang T, Qiu GX, Shen JX, Zhang JG, Wang YP, Zhao H et al (2005) Evaluation and treatment of congenital scoliosis with split cord malformation. Zhonghua Wai Ke Za Zhi 43(12):770–773
Winter RB, Haven JJ, Moe JH et al (1974) Diastematomyelia and congenital spine deformities. J Bone Joint Surg A 56:27
Winter RB, Lonstein JE, Boachie-Adjei O (1996) Congenital spine deformity. J Bone Joint Surg Am 78:300–311
Winter RB, Moe JH, Lonstein JE (1983) A review of family histories in patients with congenital spine deformities. Orthop Trans 7:32
Winter RB, Moe JH, Lonstein JE (1984) Posterior arthrodesis for congenital scoliosis. An analysis of the cases of two hundred and ninety patients, five to nineteen years old. J Bone Joint Surg Am 66:1188–1197
Winter RB, Moe JH, Eilers VE (1968) Congenital scoliosis. A study of 234 patients treated and untreated. 1. Natural history. J Bone Joint Surg Am 50:1–15
Yu B, Wang YP, Qiu GX, Zhang JG, Li JY, Shen JX et al (2006) Corrective surgery of congenital scoliosis with type II split spinal cord malformation. Chin Med Sci J 21(1):48–52
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Qureshi, M.A., Asad, A., Pasha, I.F. et al. Staged corrective surgery for complex congenital scoliosis and split cord malformation. Eur Spine J 18, 1249–1254 (2009). https://doi.org/10.1007/s00586-009-1099-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00586-009-1099-1