
Abstract
Background
Prognosis of esophageal adenocarcinoma (EAC) is still poor. Therefore, the development of novel therapeutic modalities is necessary to improve therapeutic outcomes in EAC. Here, we report a novel promoter-controlled oncolytic adenovirus targeting CDX2 (Ad5/3-pCDX2) and its specific anticancer effect for EAC.
Methods
We used OE19, OE33, HT29, MKN28, RH30, and HEL299 cell lines. To establish CDX2 overexpressing OE19 cells, pCMV-GLI1 plasmid was transfected to OE19 (OE19 + GLI1). The virus replication and cytocidal effect of replication competent Ad5/3-pCDX2 were analyzed in vitro. Antitumor effect of Ad5/3-pCDX2 was assessed in xenograft mouse models by intratumoral injection of the viruses. Finally, efficacy of combination therapy with Ad5/3-pCDX2 and 5FU was evaluated.
Results
EAC cells and HT29 showed high mRNA levels of CDX2, but not MKN28, RH30, and HEL299. We confirmed that deoxycholic acid (DCA) exposure enhanced CDX2 expression in EAC cells and OE19 + GLI1 had persistent CDX2 overexpression without DCA. Ad5/3-pCDX2 showed stronger cytocidal effect in OE19 + GLI1 than OE19, whereas Ad5/3-pCDX2 did not kill CDX2-negative cells. Ad5/3-pCDX2 was significantly replicated in EAC cells and the virus replication was higher in OE19 + GLI1 and OE19 with DCA compared to OE19 without DCA exposure. In vivo, Ad5/3-pCDX2 significantly suppressed OE19 tumor growth and the antitumor effect was enhanced in OE19 + GLI1 tumor. In contrast, Ad5/3-pCDX2 did not show significant antitumor effect in MKN28 tumor. Moreover, Ad5/3-pCDX2 significantly increased the efficacy of 5FU in vitro and in vivo.
Conclusions
Ad5/3-pCDX2 showed specific anticancer effect for EAC, which was enhanced by bile acid exposure. Ad5/3-pCDX2 has promising potential for EAC therapy in the clinical setting.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Esophageal cancer is the sixth leading cause of cancer-associated death worldwide [1] and esophageal adenocarcinoma (EAC) is the most common subtype of esophagus cancer in Western countries [2]. EAC mostly emerges from Barrett’s esophagus (BE) that is characterized by replacement of squamous epithelium in the distal esophagus in response to gastroesophageal reflux disease (GERD) [3]. The incidence of EAC has dramatically risen in the last few decades; the burden was estimated to be 20,640 new cases and 16,410 deaths in 2022 in the United States [4]. Although EAC patients with earlier stage have a favorable prognosis, most EAC patients are diagnosed with an advanced stage and the 5-year survival rate of these patients is less than 20% [5]. For locally advanced EAC, the treatment has evolved from single to multimodal therapy, namely surgery in combination with perioperative chemotherapy. Surgical resection remains the mainstay treatment option and surgery combined with neoadjuvant therapy recently plays an important role for the curative treatment of resectable advanced EAC [6, 7]. However, radical esophagectomy combine with neoadjuvant therapies is a very invasive procedure with a high rate of surgical complications, and thus is not feasible for all patients due to their comorbidities and general conditions [6]. In particular, neoadjuvant chemoradiotherapy (nCRT) causes higher rates of postoperative complications and in-hospital mortality than neoadjuvant chemotherapy (nCT) [8]. In addition, in local EACs, the therapeutic response to neoadjuvant radiotherapy may be weaker than that in esophagus squamous cell carcinoma (ESCC) [7, 8]. Therefore, innovative options that can be combined with nCT are needed to control the local tumor efficiently to improve therapeutic outcomes and prognosis in a wide range of EAC patients.
Oncolytic virotherapy employs cytocidal function of viruses to kill cancer cells and is a very promising approach to treat cancers. Among them, oncolytic adenovirus (OAd) is an attractive modality in cancer virotherapy [9]. To restrict cytocidal effect to cancer cells, OAds can be engineered for selective replication within cancer cells. One approach to target adenovirus replication to cancer cells is controlling the expression of adenovirus E1-gene, which is essential for virus replication, with tumor specific promoters [10]. Tumor-specific promoter-controlled OAds can replicate within cancer cells where the controlling promoter is active and selectively attack cancers. We have already developed several OAds targeting gastrointestinal cancers including EAC [11,12,13]. Considering the fact that high intratumor heterogeneity is a significant feature in EAC, especially in advanced stage [14], development of novel OAds controlled by alternative promoters is essential to overcome the limited therapeutic efficacy due to the tumor heterogeneity of EAC. Moreover, local injection of OAds showing “normal tissue off” effect is a reasonable strategy to control primary EAC tumor with less damage of the normal esophagus tissue in neoadjuvant setting. Thus, we constructed a novel promoter-controlled OAd aiming for the clinical application for locally advanced EAC treatment.
Caudal-related homologue 2 (CDX2) is an important homeodomain transcription factor in the maintenance of adult intestinal epithelium and regulating target genes involved in various processes including cell differentiation and proliferation [15]. CDX2 expression in the esophagus increases with BE progression [16] and remains elevated in EAC [18]. The chronic bile acids reflux into the distal esophagus by GERD plays an important role in the progression from BE to EAC [18] and stimulates invasion and metastasis of EAC [19,20,21]. Although squamous epithelium of the esophagus under normal conditions shows no CDX2 expression, bile acids exposure increases CDX2 expression in human EAC and BE cells [22, 23]. In patients, bile acids induce CDX2 promoter activity in esophageal squamous cells from GERD patients with BE, but not in those without BE [24]. These data suggest that the CDX2 promoter is a promising target for promoter-controlled OAds to selectively eliminate EAC under GERD with minimal damage in normal esophagus. We, therefore, hypothesized that a CDX2 promoter-controlled OAd will show selective antitumor effect for EAC via specific replication and cytolysis in EAC cells, where CDX2 expression is turned on by bile acid exposure. While efficient transduction of the target cells is crucial for realization of the expected efficacy of OAds, EAC cells cannot be easily transduced with wild type adenovirus fiber due to scarce expression of its receptor, coxsackie adenovirus receptor [12]. To overcome this hurdle, we employed Ad5/3-fiber chimera virus, which has a replacement of the Ad5 fiber knob with Ad3 fiber knob. This virus has shown much better transduction and virus-cell binding in EAC cells [12]. In the present study, we generated a novel CDX2 promoter-controlled OAd with adenovirus 5/3 chimeric fiber (Ad5/3-pCDX2) and investigated specific replication and killing in EAC cells in vitro and the in vivo effects for EAC tumor growth in mouse models after its intratumoral injection.
Materials and methods
Cell lines
Two human EAC cell lines (OE19 and OE33) from the European Collection of Authenticated Cell Cultures (#96071721 and #96070808. ECACC. Salisbury, UK) and two human colorectal cancers (HT29 and Caco2), gastric cancer (MKN28), rhabdomyosarcoma (RH30), and human normal fibroblast (HEL299) cell lines were purchased from ATCC (Manassas, VA, USA). Gastrointestinal stromal tumor (GIST-T1) cell line was kindly provided by Dr. Yujiro Hayashi, Ph.D. (Mayo Clinic, Rochester, MN). These cell lines were grown in RPMI1640 (OE19, OE33, and RH30), Dulbecco’s modified Eagle’s (Caco2, MKN28, GIST-T1, and HEL299), or McCOY’S 5A (HT29) medium. All cell lines were cultured with medium supplemented with 10% (V/V) fetal bovine serum and 1% penicillin–streptomycin mixture (100 and 100 µg/mL, respectively), and maintained as adherent monolayers at 37 °C in a humidified incubator with 5% CO2. To establish CDX2 overexpressing OE19 cells (OE19 + GLI1), OE19 cells were plated in 60-mm dishes and then transfected with the pCMV-GLI1 cDNA plasmid (#37113. Addgene. Watertown, MA, USA) using Lipofectamine 3000 Reagent (Invitrogen. Carlsbad, CA, USA). Stable transfectants were isolated in the presence of 500 μg/ml G418 (Roche. Indianapolis, IN, USA).
Reagents
Deoxycholic acid (DCA) was purchased from Sigma-Aldrich (D2510-10G. St. Louis, MO, USA). Drugs were diluted according to the manufacture’s protocol. EAC cells were exposed with DCA to induce CDX2 overexpression. DCA was added into growth medium at 50 to 300 µM and cultured for 24 h.
Adenoviral vectors
We generated adenoviruses with 5/3 chimeric fiber (Ad5/3) containing CDX2 promoter-driven luciferase (Luc) expression cassette (Ad5/3-pCDX2-GL3B) as replication deficient viruses and CDX2 promoter-driven E1 gene (Ad5/3-pCDX2) as replication competent virus. Short (1516 bp) and long (2687 bp) lengths of CDX2 promoter region [pCDX2(S) and pCDX2(L)] were isolated from the OE33 genome by PCR. Two lengths of pCDX2 were cloned using Zero Blant TOPO PCR Cloning Kit (Invitrogen) and the sequences were confirmed. For generating Luc reporter-expressing plasmid with pCDX2 (pGL3B-pCDX2), we adopted the In-Fusion cloning system. pGL3-Basic (4818 bp; #E1751. Promega. Madison, WI, USA) was digested with XhoI and BglII, and then insert PCR products (pCDX(S) and (L)) were inserted into the linearized pGL3-Basic using the In-Fusion HD Cloning Kit (Takara Bio. San Jose, CA, USA) according to the manufacturer's instructions.
To generate adenovirus shuttle plasmid with pCDX2 and luciferase (Luc) (pShuttle-GL3B-pCDX2) for replication deficient adenovirus, pGL3B-pCDX2 and pShuttle-GL3B (8533 bp: Supplementary Fig. 1) was digested with KpnI and HindIII. The inserts with pCDX2 from pGL3B-pCDX2 and the linear pShuttle-GL3B were ligated using the Fast-Link DNA Ligation Kit (Lucigen. Madison, WI, USA) according to the manufacturer’s instructions. For pShuttle of replication competent adenovirus, pCDX2(S) from pShuttle-GL3B-pCDX2(S) and E1-gene plus protein IX (pIX) region from pShuttle-Cox2LH-E1-XpIXF (11,692 bp: Supplementary Fig. 2) were amplified by PCR. In parallel, pShuttle-Cox2LH-E1-XpIXF was digested with SalI to remove the Cox2-E1-pIX cassette. The insert PCR products (pCDX2 and E1-pIX) were inserted into the linear plasmid backbone using the In-Fusion HD Cloning Kit. The pShuttle-GL3B-pCDX2 or pShuttle-pCDX2-E1-XpIXF were amplified in E. coli. The resulting plasmids of interest were extracted by EndoFree Plasmid Maxi Kit (Qiagen. Hilden, Germany). The shuttle plasmids were linearized with PmeI and subsequently co-transformed into E. Coli BJ5183 cells (Agilent Technology. Santa Clara, CA, USA) with an adenoviral backbone pAdEasy-5/3F (replication deficient) or pMG553 (replication competent). All adenovirus backbones were based on human adenovirus type 5. Finally, the linearized recombinant plasmids were transfected into 293 cells using SuperFect Transfection Reagent (Qiagen). Recombinant adenoviruses were generated around 7 days post-transfection and purified by double cesium chloride density gradient ultracentrifugation. The titer of the viruses was determined by optical absorbance at 260 nm and plaque forming unit assays [11]. The primers for adenovirus construction are listed in Supplementary Table 1.
RNA isolation and quantitative reverse-transcription polymerase chain reaction (RT-PCR)
Total RNA was isolated using the RNeasy Mini Kit (74,106. Qiagen) from samples and reverse-transcribed with PrimeScript RT Master Mix including oligo dT primer (RR036A. Takara Bio) using the manufacturers’ protocols. Quantitative RT-PCR was performed using a Roche LightCycler 480 II and PowerUp SYBR Green Master Mix (A25742. Thermo Fisher, Waltham, MA, USA). Gene expression was calculated by LightCycler Software (Roche). Primer sequences are described in Supplementary Table 2.
Luciferase reporter assay by plasmid transfection or virus infection
Cells (3 × 104) were plated in a 96-well plate and transfected with pGL3B-pCDX2(S and L) or pShuttle-pCMV-Luc (control) using Lipofectamine 3000 Reagent (Invitrogen). The same number of cells were infected with Ad5/3-pCDX2-GL3B at 0, 10, and 100 vp/cell. Ad5/3-pCMV-Luc was used as a control to standardize the infectivity of the viruses between the cell lines. For the assessment of promoter activity in combination therapy with 5-fluorouracil (5FU), Ad5/3-pCDX2(S)-GL3B (100 vp/cell) was infected in OE19. Two days after transfection or infection, Luc activity was determined with a Luc Assay System (Promega) according to the manufacturers’ protocols.
In vitro analysis of cytocidal effect by crystal violet staining
Crystal violet staining was performed as described previously [25]. Briefly, a total of 5 × 104 cells were plated in 12-well plates and infected with virus at 0.1–10 vp/cell. As a positive control, we used Ad5/3 with a normal promoter (Ad5/3). The cells were fixed and stained with crystal violet when Ad5/3 completely killed cells at 0.1 vp/cell.
Quantitative in vitro cytotoxicity assay
Cells (3000/well) cultured in a 96-well plate were infected with virus at 10 vp/cell in 100 µl of medium. The cells were incubated for 12 days and cell viability was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay. A detailed protocol is provided in a previous report [12]. To assess the efficacy of combination therapy with Ad5/3-pCDX2 and 5FU, OE19 cell suspensions (5000 cells/100 μl) were added to each well in a 96-well plate and incubated at 37 °C for 24 h. Cells were infected with Ad5/3-pCDX2 at 100 or 10 vp/cell, and 5FU (0, 5, 10, 20 μM) was subsequently added to each well after 4 h post-infection. The cells were incubated for 7 (100 vp/cell) or 10 (10 vp/cell) days. The results were expressed as the percentage viable with respect to the untreated control. Calculation of the combination index (CI) in combination therapy with Ad5/3-pCDX2 and 5FU was performed using Compusyn software.
Analysis of viral replication
Cells (1 × 105) in 12-well plates were infected with virus (10 vp/cell), and the growth medium was collected at day 2 and 5 after virus infection. The DNA was isolated from the medium using QIAamp DNA Mini Kit (Qiagen) and the total viral copy number of the E4-gene was analyzed by qPCR. For a detailed protocol, see our previous report [25]. To assess the influence of DCA for virus replication in OE19, DCA (50 µM) was added into the growth medium at day 3 after virus infection.
Binding assay
One day after seeding cells (5 × 104 cells/24 well plate), cells were infected with Ad5 or Ad5/3 at 100 and 1000 vp/cell. The assays were performed as previously described [12].
In vivo antitumor effect in xenograft mouse models
All experimental procedures using animals were approved by the University of Minnesota Institutional Animal Care and Use Committee (IACUC, #1412-32188A). Male nude mice (6–8 weeks of age, Charles River Laboratories, Wilmington, MA, USA) were used to established xenografts. First, OE19 or OE19 + GLI1 cells (3 × 106 per inject site) were inoculated into the flanks of mice. Once subcutaneous tumors developed, we harvested and cut the tumors and then implanted a chunk of tumor (2 × 2 × 2 mm) subcutaneously into the flank of another nude mice. When nodules reached a size of 8–10 mm in maximum diameter, a single virus dose (1010 vp in 100 µl PBS) of replication competent adenovirus [Ad5/3-pCDX2 and Ad5/3 with normal promoter (control)] or PBS were injected intratumorally (i.t.). The condition of the mice was monitored daily, and the tumor diameter was measured twice a week. The tumor volume was calculated as width2 × length/2. The mice were euthanized 20 days after virus injection and tumors were harvested. Half of each tumor specimen was quickly frozen, and the second half was fixed with formalin for hematoxylin eosin (HE) stain and immunostaining. In a separate experiment under the same conditions, mice were sacrificed at day 7 to assess the virus replication in the tumors. The expression of hexon protein in the tumor was analyzed by immunostaining using the FITC-labeled anti-hexon polyclonal antibody (#AB1056F. Millipore. Burlington, MA, USA) and counterstained with DAPI. The DNA was purified from frozen tumor tissue (Day 20), and the adenovirus DNA copy number of the E4-gene was quantified by qPCR as described [25]. To establish the MKN28 and HT29 subcutaneous tumors, we directly injected MKN28 (5 × 106 cells) and HT29 (3 × 106 cells) into the flanks of nude mice. The schedule of virus treatment and assay methods using tumor samples were same as mentioned before in the experiments for EAC xenograft models.
5FU and Ad5/3-pCDX2 combination therapy in xenograft mouse models
After OE19 subcutaneous tumor formation in nude mice by implanting tumor chunks, mice were divided two groups; 5FU and 5FU plus Ad5/3-pCDX2 groups. The 5FU plus Ad5/3-pCDX2 group underwent intratumoral injection of Ad5/3-pCDX2 (101o vp in 100 µl PBS) at day 0. In both groups, 5FU (10 mg/kg) were intraperitoneally (i.p.) injected every 2 days from day 1 to 5. The tumor diameter was measured three times a week, and tumor volume was calculated using the formula mentioned before. The mice were euthanized 20 days after virus injection, and tumor samples were harvested.
Statistical analysis
Data are shown as mean ± standard deviation (SD). Statistical analysis was performed using student’s t-tests or a non-parametric Wilcoxon test. For all tests, differences with P < 0.05 were considered significant. All statistical analyses were performed using JMP 9.0 software (SAS Institute, Cary, NC, USA).
Results
CDX2 level is higher in EAC cells and further elevated by bile acids
CDX2 mRNA levels were analyzed in several cell lines by RT-PCR. EAC (OE19 and OE33) and CRC (HT29 and Caco2) cell lines showed significantly high levels of CDX2, compared to GIST-T1, MKN28, RH30 and HEL299 cell lines (Fig. 1a). In addition, DCA exposure (100 µM, 24 h) significantly increased CDX2 levels in EAC cells (Fig. 1b). To resemble persistent CDX2 overexpressing EAC cells without DCA exposure, we overexpressed a hedgehog pathway target gene GLI1, which has been reported to be involved in CDX2 overexpression in BE under bile acids exposure. GLI1 expression also induces CDX2 overexpression in BE cells [23]. We confirmed that DCA exposure (100 or 300 µM, 24 h) increased not only CDX2 but also GLI1 mRNA levels in a dose dependent manner in OE19 (Fig. 1c and d). CDX2 overexpressing OE19 cells (OE19 + GLI1), which was made by transfecting cells with pCMV-GLI1, showed significantly increased CDX2 level compared to OE19 transfected with empty plasmid (pcDNA3.1) as a negative control (Fig. 1e). There was no significant difference in the cell migration and growth capability between OE19 and OE19 + GLI1 (Supplementary Fig. 3a and b).

CDX2 levels in cancer cells a CDX2 mRNA levels in OE19, OE33 (esophageal adenocarcinoma), HT29, Caco2 (colorectal cancer), GIST-T1 (gastrointestinal stromal tumor), MKN28 (gastric cancer), RH30 (rhabdomyosarcoma), and HEL299 (normal fibroblast) cells by RT-PCR. P value is compared to GIST-T1, MKN28, RH30, and HEL299. b CDX2 mRNA levels in esophageal adenocarcinoma cells after deoxycholic acid (DCA) exposure (100 µM, 24 h). c CDX2 and d GLI1 mRNA levels in OE19 after DCA exposure at 100 and 300 µM (24 h). e CDX2 mRNA levels in OE19 transfected with empty vector (control) and pCMV-GLI1 cDNA plasmid (OE19 + GLI1). Results are presented as mean ± SD (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001 (paired t-test; two-tailed)
The short CDX2 promoter in replication deficient adenovirus vector demonstrates higher promoter activity and selectivity in vitro
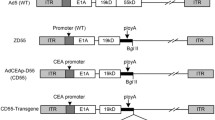
Before constructing replication competent adenovirus, replication deficient adenoviruses with short (S) or long (L) CDX2 promoter regions (Fig. 2a and b) were used to assess the promoter activity in cell lines. We first compared the promoter activities using Luc-expressing plasmid with short or long CDX2 promoter regions (pGL3B-pCDX2 (S)) and pGL3B-pCDX2 (L)). Both plasmids showed higher promoter activity in EAC and CRC cell lines than CDX2 negative cells (Supplementary Fig. 3c). In Luc assays, using replication deficient viruses (Ad5/3-pCDX2-GL3B, Fig. 2a), the promoter activity of Ad5/3-pCDX2(S)-GL3B in CDX2 positive cell lines (OE19 and Caco2) was significantly increased compared to Ad5/3-pCDX2(L)-GL3B (Fig. 2c). In CDX2 negative cell lines (RH30 and GIST-T1), both Ad5/3-pCDX2(L and S)-GL3B did not show significant increase of promoter activity. The contrast between CDX2 positive and negative cell lines was better with the short promoter pCDX2(S). The promoter activity of Ad5/3-pCDX2(S)-GL3B was also enhanced in OE19 + GLI1 or OE19 exposed by DCA (100 or 300 µM) (Fig. 2d). Based on these results, we decided to use the short length promoter region to establish replication competent virus for the following experiments.

Promoter activity of replication deficient CDX2 promoter-controlled adenovirus in vitro. a A schematic diagram of Ad5/3-pCDX2-GL3B (replication deficient). The vectors were constructed based on human adenovirus type 5. CDX2 promoter-driven E1 gene or firefly luciferase expression cassette was inserted in the corresponding position. The E3 region of Ad5/3-pCDX2-GL3B was removed. b Two lengths of CDX2 promoter region used for Ad5/3-pCDX2-GL3B vectors. c Promoter activity in adenovirus construct with Ad5/3-pCDX2-GL3B vectors with long and short CDX2 promoters measured by luciferase reporter assay (n = 5). Values are expressed as ratio to luciferase expression at 0 vp/cell. b Promoter activity of Ad5/3-pCDX2-GL3B with short CDX2 promoter normalized to luciferase expression of AdEasy-pCMV in RH30 (CDX2-negative), OE19 transfected with empty vector (control), OE19 + GLI1 (CDX2-overexpressing OE19), and OE19 exposed with deoxycholic acid (DCA, 48 h exposure) by luciferase reporter assay (n = 5)
CDX2 promoter-controlled OAd selectively replicates and kills CDX2-positive EAC cells
Next, we constructed replication competent adenovirus with the short CDX2 promoter (Ad5/3-pCDX2. Figure 3a). The cytolytic effect and virus replication of Ad5/3-pCDX2 were compared in EAC, CRC, and CDX2 negative cells. In crystal violet assay (Fig. 3b), Ad5/3-pCDX2 at a titer of 1 and 10 VP/cell killed OE19 and OE19 + GLI1, but not MKN28 (CDX2 negative), after 10 days post-infection. The cytolytic effect of Ad5/3-pCDX2 in OE19 + GLI1 was stronger than that in OE19. Ad5/3-pCDX2 also started to kill HT29 (CDX2-positive CRC) at a titer of 1 VP/cell after 14 days post-infection. Quantitative analyses of cell viability revealed that Ad5/3-pCDX2 caused significantly more cell death in OE19 and OE19 + GLI1 compared to RH30 (CDX2 negative), while the cytolysis of Ad5/3-pCDX2 was stronger in OE19 + GLI1 than OE19 (Fig. 3c), similar to results in the crystal violet assay. In both the crystal violet and quantitative cytotoxicity assays (Fig. 3b and c), Ad5/3-pCDX2 did not kill normal fibroblast HEL299 cells, suggesting that Ad5/3-pCDX2 does not attack CDX2-negative background tissues. The virus DNA copy number increased from day 2 to day 5 in OE19 and OE19 + GLI1 indicating steady virus replication, but not in RH30 and MKN28 (Fig. 3d). At day 5, the DNA copy number in OE19 + GLI1 and OE19 with DCA was significantly higher than that in OE19 without DCA. The replication of Ad5/3-pCDX2 in HT29 also increased significantly from day 2 to 14 (Supplementary Fig. 3d). These in vitro experiments revealed that the cytolytic effect and replication capability of Ad5/3-pCDX2 was regulated by CDX2 levels in the cancer cells. In the context of virus binding, 5/3 fiber showed significantly higher infectivity to OE19 compared to wild type Ad5 fiber (Fig. 3e).

Measurement of replication ability and cytolytic effect of CDX2 promoter-controlled adenovirus in vitro. a A schematic diagram of Ad5/3-pCDX2 (replication competent). The E3 region of Ad5/3-pCDX2 was maintained. b Cytolytic effect of Ad5/3-pCDX2 in HEL299 (CDX2-negative normal fibroblast), MKN28 (CDX2-negative), HT29, OE19, and OE19 + GLI1 cells by crystal violet assay. Cells were stained with crystal violet when Ad5/3 completely eliminated the cells at 0.1 vp/cell. c Quantitative cell viability analysis after 12 days of Ad5/3-pCDX2 treatment in HEL299, RH30, OE19, and OE19 + GLI1 in quantitative cytotoxicity assay (MTS assay, n = 5). Values are expressed as ratio to cell viability without virus. †P < 0.05 for vs. HEL299. **P < 0.01, ***P < 0.001 for vs. RH30 d E4 gene copy number 2 and 5 days after Ad5/3-pCDX2 (replication competent) infection to RH30, MKN28, OE19, OE19 with DCA (50 µM/L, 48 h from day 3), and OE19 + GLI1 by qPCR (n = 4). †P < 0.05 for OE19 without DCA vs. OE19 with DCA at day 5. **P < 0.01, ***P < 0.001 for vs. MKN28 and RH30 at day 5. e Infectivity of adenovirus with wild type Ad5 and 5/3 fiber to OE19 by binding assay (n = 4). Results are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 (paired t-test; two-tailed)
CDX2 promoter-controlled OAd suppresses EAC tumor growth in xenograft mouse models
After establishment of OE19 subcutaneous tumors in nude mice, 1010 vp of Ad5/3-pCDX2, Ad5/3 (positive control), or PBS (control) was intratumorally injected (Fig. 4a). Ad5/3-pCDX2 and Ad5/3 significantly suppressed OE19 tumor growth compared to PBS treatment (Fig. 4b–e). Ad5/3-pCDX2 showed comparable effect to those injected with positive control virus Ad5/3. Intratumoral spread of virus was evaluated in the tumor samples at day 7 and 20 after the treatment by staining of viral structural protein (hexon) (Fig. 4f). Cancer cells in the OE19 tumors expressed high levels of hexon protein, which were sustained over 20 days in both Ad5/3-pCDX2 and Ad5/3 groups. This indicated that the viruses successfully spread in the tumors.

Antitumor effect of CDX2 promoter-controlled replication competent adenovirus in OE19 xenograft model in mice. a Experimental schedule; intratumoral injection of Ad5/3-pCDX2 (1010 vp), Ad5/3 (positive control, 1010 vp), or PBS (control) into OE19 subcutaneous tumors at day 0 (n = 8 each group). b Chronological change of relative tumor volume in control, Ad5/3, and Ad5/3-pCDX2 groups. Individual tumor volume in control (c), Ad5/3 (d), and Ad5/3-pCDX2 (e). f Expression of viral structural protein (hexon: green) in the OE19 tumors and hematoxylin eosin stain (HE) at day 7 and 20 of control, Ad5/3 and Ad5/3-pCDX2 groups. The tumors at day7 were harvested in a separate experiment under the same conditions. Results are presented as mean ± SD. *P < 0.05, **P < 0.01 (paired t-test; two-tailed)
In mice with MKN28 (CDX2-negative) tumors treated with 1010 vp of Ad5/3-pCDX2, Ad5/3, or PBS (Fig. 5a), Ad5/3 inhibited tumor growth similar to the result of OE19 tumors. However, Ad5/3-pCDX2 did not suppress the tumor growth significantly (Fig. 5b–e). The virus replication of Ad5/3 was significantly increased in MKN28 tumors at day 20, but Ad5/3-pCDX did not show significant replication (Fig. 5f). These results indicated that Ad5/3-pCDX2 did not have replicative capability or antitumor effect in CDX2-negative tumors. Regarding the CDX2 expression in subcutaneous tumors, OE19 tumors showed significantly higher expression than MKN28 tumors (Fig. 5g). Interestingly, OE19 tumors showed nearly three hundred folds increased CDX2 expression, compared to OE19 cell lines in vitro. We also confirmed that CDX2 protein expression was significantly higher in OE19 than MKN28 tumors (data not shown).

Comparison of the effectiveness of CDX2 promoter-controlled replication competent adenovirus in CDX2-negative xenograft mouse models. a Experimental schedule; intratumorally injection of Ad5/3-pCDX2 (1010 vp), Ad5/3 (positive control, 1010 vp), or PBS (control) into MKN28 subcutaneous tumors at day 0 (Control n = 6, Ad5/3 n = 5, and Ad5/3-pCDX2 n = 5 mice). b Chronological change of relative tumor volume in control, Ad5/3, and Ad5/3-pCDX2 groups. Individual tumor volume in control (c), Ad5/3 (d), and Ad5/3-pCDX2 (e). f E4 gene copy number (virus replication) in MKN28 tumors at day 20 in control, Ad5/3, and Ad5/3-pCDX2 group by qPCR. g CDX2 mRNA level in OE19 and MKN28 cells in vitro, and MKN28 and OE19 subcutaneous tumor in vivo (n = 3). Results are presented as mean ± SD. *P < 0.05, **P < 0.01, n.s.; no statistical significance. (paired t-test; two-tailed)
CDX2 promoter-controlled OAd shows enhanced antitumor effect in OE19 + GLI1 tumors
We next assessed the antitumor effect of Ad5/3-pCDX2 in OE19 + GLI1 (CDX2 overexpressing cell) subcutaneous tumors (Fig. 6a). Ad5/3-pCDX2 (1010 vp) significantly suppressed OE19 + GLI1 tumor growth from day 10 to 20 after the treatments, compared to the control. (Fig. 6b–e). Most importantly, Ad5/3-pCDX2 group showed significantly stronger antitumor effect than the Ad5/3 group. The hexon expression surrounding necrotic regions were observed in the OE19 + GLI1 tumors at day 7 in the Ad5/3-pCDX2 group (Fig. 6f). As for virus replication, the virus E4-gene copy number of Ad5/3-pCDX2 was significantly higher than that of Ad5/3 in OE19 + GLI1 tumors, whereas there was no significant difference between Ad5/3-pCDX2 and Ad5/3 replication in OE19 tumors (Fig. 6g). When the relative tumor volumes were compared between OE19 and OE19 + GLI1 tumors at day 10 after treatment, the OE19 + GLI1 tumors were significantly smaller than OE19 tumors in the Ad5/3-pCDX2 but not Ad5/3 treatment (Fig. 6h), indicating the antitumor effect of Ad5/3-pCDX2 was enhanced in OE19 + GLI1 with the higher virus replication. In addition to EAC tumors, we also confirmed that Ad5/3-pCDX2 suppressed HT29 (CDX2-positive) subcutaneous tumor growth (Supplementary Fig. 3e).

Enhanced antitumor effect of CDX2 promoter-controlled replication competent adenovirus in CDX2 overexpressing EAC subcutaneous tumors. a Experimental schedule; intratumorally injection of Ad5/3-pCDX2 (1010 vp), Ad5/3 (positive control, 1010 vp), or PBS (control) into OE19 + GLI1 (CDX2-overexpressing OE19) subcutaneous tumors at day 0 (n = 4 mice each group). b Chronological change of relative tumor volume in control, Ad5/3, and Ad5/3-pCDX2 groups. Individual tumor volume in control (c), Ad5/3 (d), and Ad5/3-pCDX2 (e). f Expression of viral structural protein (hexon: green) in the tumors and hematoxylin eosin stain (HE) at day 7 of Ad5/3-pCDX2 groups. The tumors at day7 were harvested in a separate experiment under the same conditions. g E4 gene copy number (virus replication) of Ad5/3 and Ad5/3-pCDX2 at day 20 compared between OE19 and OE19 + GLI1 tumors by qPCR. h Relative volume at day 10 of OE19 and OE19 + GLI1 tumors compared between control, Ad5/3, and Ad5/3-pCDX2 groups. Results are presented as mean ± SD. *P < 0.05, **P < 0.01 (paired t-test; two-tailed)
Ad5/3-pCDX2 combining with 5FU chemotherapy inhibits EAC tumor growth
Because 5FU is one of the main drugs of nCT for locally advanced EAC, we evaluated whether CDX2 promoter-controlled OAd can affect the efficacy of 5FU in EAC. Using the OE19 subcutaneous tumor mouse model, combination therapy with Ad5/3-pCDX2 and 5FU was performed (Fig. 7a). The 5FU (10 mg/kg) plus Ad5/3-pCDX2 (1010 vp) combination treatment significantly suppressed tumor growth compared to the 5FU monotherapy (Fig. 7b and c). Both groups did not show body weight loss and deterioration of general condition during the experiment. In Luc assays for assessment of the promoter activity in vitro using Ad5/3-pCDX2-GL3B (non-replicative), 5FU did not affect the CDX2 promoter activity in OE19 cells, suggesting 5FU will not impact the replication of Ad5/3-pCDX2. (Fig. 7d). MTS assays revealed that Ad5/3-pCDX2 (replicative, 100 vp/cell) enhanced the killing effect of 5FU in OE19 at day 7 (Fig. 7e). Low titer of Ad5/3-pCDX2 (10 vp/cell) also increased the cell killing of 5FU at day 10 after the treatment (Supplementary Fig. 3f). Calculation of the CI to determine synergism (CI < 1), antagonism (CI > 1), or additive effect (CI = 1) revealed that the combination therapy with Ad5/3-pCDX2 and 5FU showed synergistic anticancer effect in OE19 (Fig. 7f).

Efficacy of combination therapy with 5-fluorouracil (5FU) and Ad5/3-pCDX2 in mouse models. a Efficacy of Ad5/3-pCDX2 (1010 vp intratumorally injection, day 0) in combination therapy with 5FU (10 mg/kg intraperitoneally injection, day 1, 3, and 5) in OE19 subcutaneous tumor mouse model (n = 3 mice each group). The method of tumor establishment was same as Fig. 4a. b Macroscopic findings of OE19 tumors at day 10 in 5FU only and 5FU plus Ad5/3-pCDX2 groups. c Chronological change of relative tumor volume in 5FU and 5FU plus Ad5/3-pCDX2 groups. d Assessment of promoter activity of Ad5/3-pCDX2(S)-GL3B (non-replicative) after 5FU treatment in OE19 by luciferase reporter assay (n = 5). Values are expressed as ratio to luciferase expression at 0 µM of 5FU. e The in vitro killing effect of 5FU combining with Ad5/3-pCDX2 (100 vp/cell) in OE19 at day 7 in quantitative cytotoxicity assay (MTS assay, n = 5). Values are expressed as ratio to cell viability without treatment. f Calculation of the combination index (CI) to determine synergism (CI < 1), antagonism (CI > 1), or additive effect (CI = 1) in 5FU combining with Ad5/3-pCDX2. Results are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 (paired t-test; two-tailed)
Discussion
Despite recent advances of EAC treatment, the 5-year survival rates of stage II and III tumors, which are defined as locally advanced EAC and have indication for neoadjuvant therapy, are 45.1% and 17.6%, respectively [26]. While neoadjuvant therapy improves survival in locally advanced EAC, there is no definitive standard regimen for neoadjuvant therapy and the rate of pathological complete response after nCT is still disappointing [8, 14, 27]. Combining with radiotherapy is one of the reasonable options to control the local EAC tumor in the neoadjuvant setting. A recent report showed that major histological reduction of viable carcinoma in primary EAC tumor by nCRT was associated with improved overall survival after radical esophagectomy [28]. However, the 5-year survival after nCRT in EAC patients is lower than that in ESCC patients, meaning the long-term prognostic benefit of radiotherapy is less in EAC compared to ESCC [7]. Thus, the development of novel therapeutic modalities, instead of radiotherapy, is essential to improve therapeutic response in the local tumor, and is expected to contribute to better prognosis of EAC patients after neoadjuvant therapy. Since we reported cyclooxygenase-2 (COX-2) promoter-controlled OAd showing antitumor effect for EAC [12, 29], novel OAds aimed at EAC treatment have not been established due to lack of an appropriate tumor-specific promoter. For the treatment of advanced EAC with high intratumoral heterogeneity, a novel OAd with an alternative promoter is needed to eliminate EAC that COX-2 promoter-controlled OAd cannot target. Furthermore, COX-2 expression is found at some level in normal squamous epithelium in the esophagus [30], and COX-2 mRNA levels in normal esophageal epithelium is elevated in EAC patients compared to healthy control [31]. This suggests that the COX-2 promoter-controlled OAd may also attack not only EAC but also the surrounding normal esophagus and thus may not be suitable for neoadjuvant therapy. In the present study, our novel CDX2 promoter-controlled OAd showed selective cytolysis corresponding with CDX2 expression in cancer cells as well as significant EAC tumor growth suppression in mouse models. In addition, the Ad5/3-pCDX2 combination therapy with 5FU demonstrated significant antitumor effect, suggesting that this OAd can be a promising therapeutic option for locally advanced EAC in the clinical setting.
To generate a novel promoter-controlled OAd for EAC treatment, we focused on the CDX2 promoter that possess “tumor on” and “normal tissue off” promoter selectivity in the esophagus. According to previous reports, CDX2 was not expressed in normal esophagus or stomach [17, 32], whereas its expression was increased in BE and EAC [16, 17, 33, 34]. The present study exhibited that EAC and CRC cell lines had higher CDX2 mRNA level, and DCA exposure upregulated CDX2 in EAC cells. We successfully generated the CDX2 promoter-controlled replication deficient and subsequently replication competent adenoviruses. In the experiments using the replication deficient virus, the virus with the short promoter region of CDX2 (Ad5/3-pCDX2(S)-GL3B) showed higher promoter activity than that with the long promoter region and the contrast between CDX2 positive and negative cell lines was more significant. Previously, we observed a promoter-controlled OAd for rhabdomyosarcoma also demonstrated strongest promoter activity when the shortest sequence of promoter region was inserted into the adenovirus genome [25]. The short CDX2 promoter region included some binding sites for essential factors such as NF-κB, STAT, and AP-1, that can be induced by bile acid exposure or inflammation [35, 36]. Therefore, Ad5/3-pCDX2(S)-GL3B could preserve the selectivity of promoter activity in CDX2-positive EAC cells and the minimal promoter region might be sensitive to activate the CDX2 promoter than the longer one. Moreover, the CDX2 promoter activity was significantly enhanced in OE19 cells by DCA exposure or GLI1 expression, corresponding with CDX2 upregulation. Based on the results of the replication deficient virus, we pursued to assess anticancer effects of the short CDX2 promoter-controlled OAd.
The replication competent virus, Ad5/3-pCDX2, showed cytolytic effect and virus replication in EAC but not CDX2-negative cancers both in vitro and in vivo. Although the in vitro cytolytic effect of Ad5/3-pCDX2 in OE19 was weaker than that of Ad5/3 wild type, in vivo OE19 tumor growth suppression by Ad5/3-pCDX2 was remarkable and comparable to Ad5/3. This could be caused by significantly increased CDX2 levels in OE19 tumors in vivo compared to that in OE19 cell line in culture. To assess the efficacy of Ad5/3-pCDX2 in EAC tumors under GERD condition, therapeutic experiments in mouse orthotopic EAC models are preferred but are not reliable because observation and maintenance of mice bearing tumor in the esophagus are very complicated. The subcutaneous tumor model is a practical alternative, but it is not realistic to inject DCA in the subcutaneous tumor. Instead of bile acid exposure for CDX2 upregulation, we therefore expressed GLI1 to persistently induce CDX2 expression in OE19 instead of DCA administration (OE19 + GLI1). OE19 + GLI1 showed significant overexpression of CDX2, at a level slightly lower than the OE19 exposed to DCA (100 µM, 24 h). Data from the literature shows that peak bile acid concentration in the distal esophagus was greater than 100 µM in GERD patients [37,38,39]. Thus, the induction of CDX2 expression in OE19 + GLI1 was not too strong compared to that of in vivo EAC with bile acid exposure in GERD patients. Corresponding with CDX2 levels, Ad5/3-pCDX2 showed higher replication and cytolytic effect in OE19 + GLI1 than that in original OE19. In OE19 + GLI1 subcutaneous tumors, the Ad5/3-pCDX2 significantly suppressed tumor growth, compared to not only PBS (negative control) but also Ad5/3 (positive control) treatment groups, indicating that the antitumor effect of Ad5/3-pCDX2 was strongly boosted by driving CDX2 expression in the OE19 + GLI1 tumors. Considering the lack of a good EAC animal model representing bile acid exposure of lower esophagus, establishment of subcutaneous OE19 + GLI1 tumors is a good model mimicking the DCA-exposed EAC in the distal esophagus to predict the antitumor effect of Ad5/3-pCDX2 in EAC patients with GERD.
To evaluate the translational value of Ad5/3-pCDX2 in EAC treatment, we assessed the efficacy of combination therapy with 5FU and Ad5/3-pCDX2. For locally advanced EAC patients, neoadjuvant therapy using multidrug modalities, including 5FU, is one of the common options to improve resectability and prognosis after surgery and 5FU is the key drug in both nCRT and nCT protocols [8, 27, 40]. In vivo, Ad5/3-pCDX2 plus 5FU therapy showed significantly stronger antitumor effect and tumor shrinkage in EAC tumors without body weight loss or deterioration of general condition. Of note, in vitro data showed that 5FU did not affect the CDX2 promoter activity in the replication deficient adenovirus construct, indicating that 5FU will not impact the replication of Ad5/3-pCDX2. The CI analysis showed that CI of the combination therapy of Ad5/3-pCDX2 with 5-FU was around 0.5, meaning synergistic effect (CI < 1). OAds were reported to sensitize the effect of chemotherapy and adenoviral E4 proteins can inhibit cellular DNA repair pathways [41,42,43]. Thus, Ad5/3-pCDX2 could show synergistic effect in 5FU combination therapy. Our results suggested that combining Ad5/3-pCDX2 with 5FU-based regimen is a promising strategy in neoadjuvant therapy to control the primary tumor and improve therapeutic outcomes of locally advanced EAC. Furthermore, the off-target effects of Ad5/3-pCDX2 in normal esophagus can reduce complications after neoadjuvant therapy. Neoadjuvant radiotherapy causes normal esophagus damage and radiation over 40 Gy to the mediastinum increases perioperative complications including anastomotic leak and respiratory infection [8]. Thus, Ad5/3-pCDX2 is an alternative modality when EAC patients have worse general conditions that cannot tolerate full dose radiation before surgery. A potential issue for clinical application is the heterogeneity of CDX2 expression in EAC primary tumors because reduced CDX2 protein expression in EAC compared to BE is detected in some patients by immunohistochemistry [44]. To overcome this issue, evaluation of CDX2 status by pretreatment endoscopic biopsy can help to determine the target patients and predict the efficacy of the Ad5/3-pCDX2 treatment.
In this study, we generated a CDX2 promoter-controlled OAd for EAC treatment and demonstrated the significant antitumor effects in EAC mouse models. The CDX2 promoter-controlled OAd is promising as a novel therapeutic modality for EAC with CDX2 expression induced by bile acid reflux. Combining with conventional chemotherapy, the CDX2 promoter-controlled OAd can provide a beneficial option to achieve the curative therapy and improve the prognosis of local advanced EAC patients.
Abbreviations
- BE:
-
Barrett’s esophagus
- CDX2:
-
Caudal-related homologue 2
- COX-2:
-
Cyclooxygenase-2
- EAC:
-
Esophageal adenocarcinoma
- ESCC:
-
Esophagus squamous cell carcinoma
- GERD:
-
Gastroesophageal reflux disease
- nCRT:
-
Neoadjuvant chemoradiotherapy
- nCT:
-
Neoadjuvant chemotherapy
- OAd:
-
Oncolytic adenovirus
References
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
National Cancer Institute. Surveillance, Epidemiology, and End Results Program. SEER*Explorer: an interactive website for SEER cancer statistics. https://seer.cancer.gov. Accessed April 22, 2022.
Peng D, Zaika A, Que J, El, et al. The antioxidant response in Barrett’s tumorigenesis: a double-edged sword. Redox Biol. 2021;41: 101894.
Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33.
Joseph A, Raja S, Kamath S, et al. Esophageal adenocarcinoma: a dire need for early detection and treatment. Cleve Clin J Med. 2022;89:269–79.
Donlon NE, Moran B, Kamilli A, et al. CROSS versus FLOT regimens in esophageal and esophagogastric junction adenocarcinoma: a propensity-matched comparison. Ann Surg. 2022;276:792–8.
Eyck BM, Van Lanschot JJ, Hulshof MCCM, CROSS Study Group, et al. Ten-year outcome of neoadjuvant chemoradiotherapy plus surgery for esophageal cancer: the randomized controlled CROSS trial. J Clin Oncol. 2021;39:1995–2004.
Elliott JA, Klevebro F, Mantziari S, ENSURE Study Group, et al. Neoadjuvant chemoradiotherapy versus chemotherapy for the treatment of locally advanced esophageal adenocarcinoma in the european multicenter ENSURE study. Ann Surg. 2023;278:692–700.
Howells A, Marelli G, Lemoine NR, et al. Oncolytic viruses-interaction of virus and tumor cells in the battle to eliminate cancer. Front Oncol. 2017;7:195.
Yamamoto M, Alemany R, Adachi Y, et al. Characterization of the cyclooxygenase-2 promoter in an adenoviral vector and its application for the mitigation of toxicity in suicide gene therapy of gastrointestinal cancers. Mol Ther. 2001;3:385–94.
Yamamoto M, Davydova J, Wang M, Siegal GP, Krasnykh V, Vickers SM, Curiel DT. Infectivity enhanced, cyclooxygenase-2 promoter-based conditionally replicative adenovirus for pancreatic cancer. Gastroenterology. 2003;125:1203–18.
Davydova J, Le LP, Gavrikova T, et al. Infectivity-enhanced cyclooxygenase-2-based conditionally replicative adenoviruses for esophageal adenocarcinoma treatment. Cancer Res. 2004;64:4319–27.
Sato-Dahlman M, Miura Y, Huang JL, et al. CD133-targeted oncolytic adenovirus demonstrates anti-tumor effect in colorectal cancer. Oncotarget. 2017;8:76044–56.
Li S, Hoefnagel SJM, Krishnadath KK. Molecular biology and clinical management of esophageal adenocarcinoma. Cancers (Basel). 2023;15:5410.
Silberg DG, Swain GP, Suh ER, et al. Cdx1 and cdx2 expression during intestinal development. Gastroenterology. 2000;119:961–71.
Eda A, Osawa H, Satoh K, et al. Aberrant expression of CDX2 in Barrett’s epithelium and inflammatory esophageal mucosa. J Gastroenterol. 2003;38:14–22.
Lord RV, Brabender J, Wickramasinghe K, et al. Increased CDX2 and decreased PITX1 homeobox gene expression in Barrett’s esophagus and Barrett’s-associated adenocarcinoma. Surgery. 2005;138:924–31.
Hong J, Behar J, Wands J, et al. Bile acid reflux contributes to development of esophageal adenocarcinoma via activation of phosphatidylinositol-specific phospholipase Cgamma2 and NADPH oxidase NOX5-S. Cancer Res. 2010;70:1247–55.
Xia HH-X, Zhang ST, Lam SK, et al. Expression of macrophage migration inhibitory factor in esophageal squamous cell carcinoma and effects of bile acids and NSAIDs. Carcinogenesis. 2005;26:11–5.
Kazumori H, Ishihara S, Rumi MAK, et al. Bile acids directly augment caudal related homeobox gene Cdx2 expression in oesophageal keratinocytes in Barrett’s epithelium. Gut. 2006;55:16–25.
Roman S, Petre A, Thepot A, et al. Downregulation of p63 upon exposure to bile salts and acid in normal and cancer esophageal cells in culture. Am J Physiol. 2007;293:G45-53.
Morrow DJ, Avissar NE, Toia L, et al. Pathogenesis of Barrett’s esophagus: bile acids inhibit the Notch signaling pathway with induction of CDX2 gene expression in human esophageal cells. Surgery. 2009;146:714–21.
Huang J, Liu H, Sun T, et al. Omeprazole prevents CDX2 and SOX9 expression by inhibiting hedgehog signaling in Barrett’s esophagus cells. Clin Sci (Lond). 2019;133:483–95.
Huo X, Zhang HY, Zhang XI, et al. Acid and bile salt-induced CDX2 expression differs in esophageal squamous cells from patients with and without Barrett’s esophagus. Gastroenterology. 2010;139:194–203.
Yoshida H, Sato-Dahlman M, Hajeri P, et al. Mutant myogenin promoter-controlled oncolytic adenovirus selectively kills PAX3-FOXO1-positive rhabdomyosarcoma cells. Transl Oncol. 2021;14: 100997.
Coleman HG, Xie SH, Lagergren J. The epidemiology of esophageal adenocarcinoma. Gastroenterology. 2018;154:390–405.
Sjoquist KM, Burmeister BH, Smithers BM, Australasian Gastro-Intestinal Trials Group, et al. Survival after neoadjuvant chemotherapy or chemoradiotherapy for resectable oesophageal carcinoma: an updated meta-analysis. Lancet Oncol. 2011;12:681–92.
Sihag S, Nobel T, Hsu M, et al. Survival after trimodality therapy in patients with locally advanced esophagogastric adenocarcinoma: does only a complete pathologic response matter? Ann Surg. 2022;276:1017–22.
LaRocca CJ, Salzwedel AO, Sato-Dahlman M, et al. Interferon alpha-expressing oncolytic adenovirus for treatment of esophageal adenocarcinoma. Ann Surg Oncol. 2021;28:8556–64.
Villanacci V, Rossi E, Zambelli C, et al. COX-2, CDX2, and CDC2 immunohistochemical assessment for dysplasia-carcinoma progression in Barrett’s esophagus. Dig Liver Dis. 2007;39:305–11.
Brabender J, Marjoram P, Lord RV, et al. The molecular signature of normal squamous esophageal epithelium identifies the presence of a field effect and can discriminate between patients with Barrett’s esophagus and patients with Barrett’s-associated adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2005;14:2113–7.
Silberg DG, Furth EE, Taylor JK, et al. CDX1 protein expression in normal, metaplastic, and neoplastic human alimentary tract epithelium. Gastroenterology. 1997;113:478–86.
Moons LM, Bax DA, Kuipers EJ, Siersema PD, Kusters JG, et al. The homeodomain protein CDX2 is an early marker of Barrett’s oesophagus. J Clin Pathol. 2004;57:1063–8.
Groisman GM, Amar M, Meir A. Expression of the intestinal marker Cdx2 in the columnar-lined esophagus with and without intestinal (Barrett’s) metaplasia. Mod Pathol. 2004;17:1282–8.
Yu JH, Zheng JB, Qi J, et al. Bile acids promote gastric intestinal metaplasia by upregulating CDX2 and MUC2 expression via the FXR/NF-κB signalling pathway. Int J Oncol. 2019;54:879–92.
Cobler L, Pera M, Garrido M, et al. CDX2 can be regulated through the signalling pathways activated by IL-6 in gastric cells. Biochim Biophys Acta. 2014;1839:785–92.
Kauer WK, Peters JH, DeMeester TR, et al. Composition and concentration of bile acid reflux into the esophagus of patients with gastroesophageal reflux disease. Surgery. 1997;122:874–81.
Gotley DC, Morgan AP, Cooper MJ. Bile acid concentrations in the refluxate of patients with reflux oesophagitis. Br J Surg. 1988;75:587–90.
Iftikhar SY, Ledingham S, Steele RJ, et al. Bile reflux in columnar-lined Barrett’s oesophagus. Ann R Coll Surg Engl. 1993;75:411–6.
Al-Batran SE, Homann N, Pauligk C, FLOT4-AIO Investigators, et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): a randomised, phase 2/3 trial. Lancet. 2019;393:1948–57.
Leitner S, Sweeney K, Oberg D, et al. Oncolytic adenoviral mutants with E1B19K gene deletions enhance gemcitabine-induced apoptosis in pancreatic carcinoma cells and anti-tumor efficacy in vivo. Clin Cancer Res. 2009;15:1730–40.
Bhattacharyya M, Francis J, Eddouadi A, et al. An oncolytic adenovirus defective in pRb-binding (dl922-947) can efficiently eliminate pancreatic cancer cells and tumors in vivo in combination with 5-FU or gemcitabine. Cancer Gene Ther. 2011;18:734–43.
Boyer J, Rohleder K, Ketner G. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology. 1999;263:307–12.
Hayes S, Ahmed S, Clark P. Immunohistochemical assessment for Cdx2 expression in the Barrett metaplasia-dysplasia-adenocarcinoma sequence. J Clin Pathol. 2011;64(2):110–3.
Acknowledgements
The study was supported by NIH/NCI R01CA276480 and the Department of Surgery Research Support funding to M.Y.
Author information
Authors and Affiliations
Contributions
M.Y. and N.N. contributed to the conceptualization of the study. S.S., M.D., B.R., and K.J. were involved in the construction of the adenovirus. N.N. carried out the investigation. M.Y. provided supervision. N.N wrote the original draft, and M.Y. contributed to the review and editing process. All listed authors discussed the results and reviewed the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nakamura, N., Shinoda, S., Sato-Dahlman, M. et al. Development of a novel oncolytic adenovirus controlled by CDX2 promoter for esophageal adenocarcinoma therapy. J Gastroenterol (2024). https://doi.org/10.1007/s00535-024-02147-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00535-024-02147-2