
Abstract
Interleukin-33 (IL-33), IL-36, and IL-38 are members of the IL-1 cytokine family. The expression of each cytokine has been reported to be increased in the inflamed mucosa of patients with inflammatory bowel disease (IBD). IL-33 and IL-36 have been studied for pro- and anti-inflammatory functions, and IL-38 has been characterized as an anti-inflammatory cytokine by antagonizing the IL-36 receptor (IL-36R). IL-33 is a nuclear cytokine constitutively expressed by certain cell types such as epithelial, endothelial, and fibroblast-like cells and released on necrotic cell death. IL-33 mainly induces type 2 immune response through its receptor suppression tumorigenicity 2 (ST2) from Th2 cells and type 2 innate lymphoid cells (ILC2s), but also by stimulating Th1 cells, regulatory T cells, and CD8+ T cells. IL-36 cytokines consist of three agonists: IL-36α, IL-36β, and IL-36γ, and two receptor antagonists: IL-36R antagonist (IL-36Ra) and IL-38. All IL-36 cytokines bind to the IL-36R complex and exert various functions through NF-κB and mitogen-activated protein kinase (MAPK) pathways in inflammatory settings. IL-33 and IL-36 also play a crucial role in intestinal fibrosis characteristic manifestation of CD. In this review, we focused on the current understanding of the pro- and anti-inflammatory roles of IL-33, IL-36, and IL38 in experimental colitis and IBD patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inflammatory bowel diseases (IBDs), including ulcerative colitis (UC) and Crohn’s disease (CD), are chronic inflammatory disorders characterized by uncontrolled innate and adaptive immune responses against dietary antigens and gut microbiota leading to sustained mucosal inflammation [1]. Cytokines mediate the crosstalk between innate and adaptive immune responses, and disruption of this interaction leads to the initiation and perpetuation of mucosal inflammation.
The interleukin (IL)-1 family cytokines consist of seven agonists (IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, and IL-36γ), three receptor antagonists (IL-1Ra, IL-36Ra, and IL-38), and IL-37 as an anti-inflammatory molecule [2]. There are four receptor complexes for these molecules: the IL-1 receptor (R) [IL-1R1 and IL-1 receptor accessory protein (IL-1RAcP)], the IL-33 receptor [suppression of tumorigenicity 2 (ST2) and IL-1RAcP], the IL-18 receptor (IL-18Ra and IL-18Rb), and the IL-36 receptor (IL-36R and IL-1RAcP) [2]. These cytokines mediate inflammation and tissue repair through activation of innate immunity at the forefront of local defense and are deeply involved in the pathogenesis of IBD [3]. They play situation-associated functions in intestinal homeostasis and inflammation [3]. In this review, we focus on the latest information on IL-33, IL-36, and IL-38, and provide an overview of their complex roles in IBD.
Interleukin-33
IL-33 and ST2 signaling
IL-33 (IL-1F11) is a ligand for the IL-1R family member ST2 [4]. IL-33 is a 30 kDa protein, composed of an N-terminal nuclear domain containing a chromatin-binding motif and an 18 kDa C-terminal IL-1-like cytokine domain rich in β sheets, separated by a divergent central part [5] (Fig. 1). The IL-33 injection to mice induces multiple proinflammatory effects, such as eosinophilia, splenomegaly, goblet cell hyperplasia, and mucous production, and serum IL-5 and IgE elevation [4]. IL-33 also stimulates type 2 innate lymphoid cells (ILC2s) and induces extremely high amounts of the Th2 cytokines IL-5 and IL-13 [6, 7]. Based on these findings, IL-33 has been regarded as an inducer of type 2 immune responses. However, it became clear that IL-33 plays a crucial role in both innate and adaptive immunity, since various immune cells such as ILC2s, mast cells, regulatory T cells (Tregs), Th2 cells, Th1 cells, and CD8+ T cells express IL-33 receptor ST2 [5, 8, 9]. IL-33 is a key immune modulator with pleiotropic activities in type 2, type 1, and regulatory immune responses and involved in the pathophysiology of allergic, fibrotic, and infectious diseases [5, 10].

Release and proteolytic activation of IL-33. IL-33 is constitutively expressed in endothelial cells, epithelial cells, and fibroblasts, and is accumulated in the nucleus. Full-length IL-33 is a 30 kDa protein, composed of an N-terminal nuclear domain containing a chromatin-binding motif and an 18 kDa the C-terminal IL-1-like cytokine domain rich in b sheets, separated by a divergent central part [5]. Full-length IL-33 is released as a bioactive molecule mainly upon necrotic cell death. Unlike IL-1β and IL-18, IL-33 exerts biological activity as a full-length molecule. Inflammatory proteases such as calpain, the neutrophil cathepsin G and elastase, and the mast cell chymase and tryptase cleave IL-33 within its central domain and generate more potent mature forms (18–21 kDa) [5]. In contrast, IL-33 is inactivated by caspase-mediated cleavage at the IL-1-like domain during apoptosis [5]. IL-33 stimulates ST2-expressing cells such as ILC2s, mast cells, Tregs, Th2 cells, Th1 cells, and CD8+ T cells. IL-33 binds to ST2, and this complex activates IL-1RAcP, a key molecule of IL-33 signaling shared with other IL-1 family members (IL-1α, IL-1β, IL-36) [12]. The complex of IL-33/ST2/IL1RAcP stimulates the signal through MyD88 adaptor, IRAK1 and IRAK4 kinases, and TRAF6, leading to the activation of MAP kinases and NF-κB [4, 9]
ST2 is a full-length, membrane-spanning molecule and a soluble ST2 decoy variant antagonizes the function of IL-33 [11]. Many (but not all) mast cells, ILC2s, and Tregs express high levels of ST2 [5]. IL-33 binds to ST2, and this complex activates IL-1RAcP, a key molecule of IL-33 signaling shared with other IL-1 family members (IL-1α, IL-1β, IL-36) [12]. The complex of IL-33/ST2/IL1RAcP stimulates the signal through MyD88 adaptor, IRAK1 and IRAK4 kinases, and TRAF6, leading to the activation of MAP kinases and NF-κB [4, 9]. The IL-33-induced intracellular signal pathways resembles to those activated by IL-1 and IL-18. We have previously demonstrated that IL-4 and IFN-g are strong inducers of ST2 in human myofibroblasts [13], and pretreatment with these factors induced a marked enhancement of IL-33-asscoiated responses [13].
IL-33 expression and intracellular localization
IL-33 has been demonstrated to have a dual character, an extracellular ligand, and an intracellular signaling molecule [14, 15]. IL-33 is constitutively and/or inducively expressed in certain cell types such as endothelial, epithelial, and fibroblast-like cells. Newly synthesized full-length IL-33 translocates into the nucleus using their nuclear localization sequence and bind to heterochromatin [16, 17] and is accumulated in the nucleus [8, 9]. IL-33 is released mainly upon necrotic cell death as a full-length bioactive form (Fig. 1). Unlike IL-1β and IL-18, and similar to IL-1α, IL-33 has biological activity as a full-length molecule [5]. Shorter mature forms encompassing the IL-1-like cytokine domain are produced during inflammation. Inflammatory proteases such as calpain, the neutrophil cathepsin G and elastase, and the mast cell chymase and tryptase cleave a central part of IL-33 and generate more potent mature forms (18–21 kDa) [2, 9, 18, 19] (Fig. 1). In contrast, caspases (caspase 3 and 7) cleave and inactivate IL-33 during apoptosis [5]. This process is important to prevent the excessive immune response after physiological programmed cell death (apoptosis), as opposed to pathological cell death (necrosis) [20].
Although nucleus IL-33 has been considered to behave as a transcriptional repressor [21], Bessa et al. found a significant role of nuclear localization of IL-33 in maintaining systemic immune homeostasis. They generated mice harboring the mutation of the N-terminus of IL-33 (IL33tm1/+ mice) [22]. These mice lack nuclear localization of IL-33 and consequent chromatin association, and IL-33 is released from producing cells and influences systemic organs. IL33tm1/+ mice exhibited lethal inflammation characterized by eosinophil-dominated immune cell infiltration of multiple organs. The profound inflammatory responses were absent in ST2-deficient mice. These indicated that nuclear localization of IL-33 is an important cellular mechanism for preventing systemic inflammations induced by IL-33 release. In contrast, it is likely that disruption of proper localization of IL-33 in the nucleus may lead to chronic, non-resolving inflammation like IBD [22].
IL-33 does not possess a signal sequence which mediates cytokine secretion through the ER–Golgi secretory pathway [5]. However, several studies have demonstrated that IL-33 is released from cells after exposure to extracellular stimuli such as ATP, uric acid or intracellular calcium inducer [23,24,25]. We observed that IL-1β and tumor necrosis factor (TNF)-α induced a time- and dose-dependent release and intracellular accumulation of IL-33 in human colonic myofibroblasts without an increase of cell death [26]. In this study, IL-1β- and TNF-α-induced IL-33 release was completely eliminated by treatment with caspase-1 siRNA. These observations suggest that excessively produced IL-33 may be intracellularly processed by caspases and processed (inactive) IL-33 may be released from the cells. This may be one of the mechanisms preventing systemic and excessive immune responses.
IL-33 and IBD
The immunological backgrounds of ulcerative colitis (UC) and Crohn’s disease (CD) have been reported to differ somewhat [27], although some overlaps exist in immunological responses in these diseases. CD is characterized by cytokines indicative of Th1 and Th17 polarization, whereas UC has a cytokine profile more characteristic for Th2 polarization [27, 28]. Regarding IL-33, there are several reports demonstrating that expression of IL-33 protein and transcripts is upregulated preferentially in the inflamed mucosa of UC patients [26, 29,30,31,32], suggesting an active involvement of IL-33 in a prominent Th2 response of UC patients. On the other hand, mucosal IL-33 expression of CD patients has been reported to be comparable to that observed in controls [26, 29, 31]. Thus, IL-33 may more significantly contribute to UC than CD, but this should be further investigated.
Although expression of IL-33 is increased in the active mucosa of UC patients, it remains unclear whether IL-33 plays a causative role or not. Clinical observations suggest that IL-33 plays a primarily role in the mucosal inflammation of IBD as an alarmin which evokes a proinflammatory response. In this regard, previous studies of experimental colitis have shown pathogenic and protective roles of IL-33.
Sadhom et al. demonstrated that inhibition of IL-33 signaling by anti-ST2 antibodies ameliorated dextran sodium sulfate (DSS)-induced colitis, and IL-33 injection led to increase in epithelial permeability [33]. In other studies, experimental colitis was exacerbated by IL-33 signaling and subsequent IL-33-induced Th2 responses [34,35,36]. IL-33 injection exacerbated DSS-colitis via Th2 immune responses and inhibition of Th1 polarization [34]. The IL-33-induced aggravation of DSS-colitis was not observed in IL-4-deficient mice, indicating an important role of Th2 immune response in proinflammatory effect of IL-33 [34]. IL-33 is a promoter of the differentiation of Th9 cells, and IL-9 derived from Th9 cells may drive the pathogenic Th2 immune responses associated with UC [37].
On the other hand, several studies have demonstrated a protective role of IL-33 in experimental colitis. Duan et al. demonstrated that IL-33 injection substantially ameliorated trinitrobenzene sulfonic acid (TNBS)-induced colitis. In this study, the protective effect of IL-33 was partly associated with prominent upregulation of Tregs, and depletion of Tregs significantly abrogated the impact of the protective effects [38], indicating that the protective role of IL-33 is mediated by Treg induction. In other studies, IL-33 was protective in colitis by inducing M2 macrophages which generate high amounts of anti-inflammatory cytokines IL-10 and TGF-β [39, 40]. In these mice, goblet cell differentiation was also induced and this may be one of factors associating with amelioration of colitis [40]. IL-33 stimulates ILC2s and induces production of amphiregulin, a growth factor of the EGF family, which intensifies epithelial barrier and promotes tissue repair [41]. Thus, these observations suggest that IL-33 may play a protective role in experimental colitis via activation of type 2 immune responses and may lead to resolution of mucosal inflammation.
Collectively, experimental studies in IBD models have demonstrated both pathogenic and protective roles of IL-33. Furthermore, it is unclear whether abnormal IL-33 signaling primarily drives IBD pathophysiology or is a consequent phenomenon to the mucosal inflammation. As such, several matters concerning the basic and clinical roles of IL-33 in the pathophysiology of IBD are yet to be clarified.
IL-33 and intestinal fibrosis
Intestinal fibrosis is a long-term complication of IBD, particularly of CD, frequently leading to narrowing of the lumen that needs endoscopic dilatation or surgery [42]. IL-33 activates key players in tissue fibrosis, such as Th2 cells and ILC2 cells [9]. Type 2 cytokines derived from Th2 cells, such as IL-4, IL-5, and IL-13, induce various pathological conditions of eosinophil infiltration, elevation of mucous secretion, and tissue fibrosis [43]. ILC2 cells also produce type 2 cytokines in an antigen independent manner and participate in tissue fibrosis [6, 7]. IL-33-induced IL-13 promotes collagen accumulation in IBD via inhibition of matrix metalloproteinase synthesis in myofibroblasts [44]. In addition, we have previously observed that IL-33 directly stimulates proliferation of human myofibroblasts [13]. Consequently, IL-33 is involved in intestinal fibrosis through induction of type 2 cytokines and direct stimulation of myofibroblast growth.
Interleukin-36
IL-36 cytokine subfamily
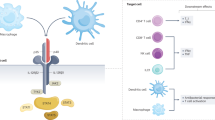
IL-36 was initially discovered 20 years ago as a member of the IL-1 cytokine superfamily [45,46,47]. It was identified through the use of DNA database searches for homologs to IL-1 [48, 49]. The IL-36 subfamily consists of four members: IL-36α (IL-1F6), IL-36β (IL-1F8), IL-36γ (IL-1F9), and IL-36R antagonist (IL-36Ra, IL-1F5) [45,46,47]. IL-36α, IL-36β, and IL-36γ are agonistic cytokines and IL-36Ra is a specific receptor antagonist [50]. Similar to other IL-1 family members such as IL-1b and IL-33, IL-36 cytokines (IL-36α, IL-36β, and IL-36γ) are secreted as a biologically inactive pro-IL-36 that changes to an active form by proteolytic processing by neutrophil-derived proteases and exerts its proinflammatory activity [50] (Fig. 2). IL-36α is activated by elastase and cathepsin G, whereas IL-36β is preferentially activated by cathepsin G and IL-36γ by either elastase or proteinase-3 [51]. Cleavage of their N-termini by neutrophil-derived proteases results in a greater than 1000-fold increase in their activity [52, 53]. IL-36Ra is also processed by elastase and facilitates its anti-inflammatory activities [54].

Activation and IL-36R signaling. The IL-36 family comprises three agonistic ligands (IL-36α, IL-36β, and IL-36γ) and one specific receptor antagonist (IL-36Ra). IL-36 cytokines (IL-36α, IL-36β, and IL-36γ) are secreted as a biologically inactive pro-IL-36 that is activated by proteolytic processing by neutrophil-derived proteases. IL-36α is activated by elastase and cathepsin G, whereas IL-36β is preferentially activated by cathepsin G and IL-36 γ by either elastase or proteinase-3 [51]. Cleavage of their N-termini results in a greater than 1000-fold increase in their activity. IL-36Ra is also processed by elastase and facilitates its anti-inflammatory activities. The complex of IL-36/IL-36R/IL1RAcP induces the activation MyD88 adaptor, leading to the activation of MAP kinases and NF-κB [4, 9]. IL-38 shares 43% homology with IL-36Ra, and also exerts anti-inflammatory effects by antagonizing IL-36R
However, like other IL-1 cytokine families, IL-36 lacks a signal sequence that can direct the newly synthesized cytokines to the endoplasmic reticulum [55], and the molecular mechanism underlying IL-36 secretion has not fully been identified. Recent studies have reported that IL-36 cytokines use a non-conventional secretion pathway involving P2X7 receptor (R) and Gasdermin D pores [55, 56]. Prolonged activation of P2X7R by ATP opens a non-selective pore in the plasma membrane, allowing the passage of cytokines [56].
Each member of the IL-36 family binds to the IL-36R. This engages IL-1RAcP as a co-receptor, and the IL-36R/IL-1RAcP complex facilitates downstream signaling via NF-κB and mitogen-activated protein kinase (MAPK) pathways [45, 48, 50, 57] (Fig. 2). Inhibitory molecules of IL-36 signaling such as IL-36Ra and IL-38 also binds to IL-36R and block IL-1RAcP recruitment, thereby inhibiting activation of IL-36-mediated signaling cascades [49, 58].
Biology of IL-36 cytokines
Under homeostasis, IL-36 cytokines are constitutively expressed at a low level in various organs, including the skin, intestine, lungs, and brain [50, 59, 60]. Under inflammatory condition, IL-36 cytokines are mainly secreted by a variety of cells including keratinocytes, T and B cells, monocytes/macrophages, epithelial and fibroblastic cells [50, 61,62,63,64]. We have previously reported that human colonic myofibroblasts are cellular source of IL-36γ in the intestine [64]. In these cells, IL-36γ was strongly induced by the combination of IL-1β and TNF-α via activation of MAPKs and transcription factors, NF-κB and AP-1 [64]. In macrophages/monocytes, IL-36 cytokines were induced in response to toll-like receptor ligands, including lipopolysaccharide (LPS), flagellin, CpG and poly I:C or pro-inflammatory cytokines including IL-1α, IL-1β, interferon (IFN)-γ or IL-18 [50, 59, 65, 66]. Furthermore, epidermal growth factors (EGFs) and fibroblast growth factors (FGFs) have been shown as inducers of IL-36 cytokines from epithelial and T cells [59, 67].
IL-36 cytokines have been reported to be involved in both innate and adaptive immune responses. Previous studies have shown that IL-36 cytokines stimulate dendritic cells (DCs), macrophages, T cells, keratinocytes, epithelial cells, and myofibroblasts [50, 57, 68, 69]. Murine and human DCs express IL-36R and respond to IL-36 cytokines (IL-36α, IL-36β, and IL-36γ) and produce a wide range of proinflammatory cytokines including IL-2, IL-23, IL-6, IL-12, granulocyte/macrophage-colony stimulating factor (GM-CSF) and TNF-α [70]. IL-36 cytokines also enhance the expression of CD80, CD86, and MHC class II of DCs [71]. Furthermore, IL-36 cytokines stimulate CD4+ T cells and induce the production of IFN-γ, IL-4, and IL-17 [70]. IL-36α and IL-36γ have been shown to promote the differentiation of naïve CD4+ T cells into Th1, Th17, and Th9 cells while simultaneously suppressing the development of Tregs. [69, 72, 73].
In colonic epithelial cells, IL-36α and IL-36γ, but not IL-36β, induced expression of CXC chemokines (CXCL1, CXCL2, CXCL3, CXCL6, and CXCL8 etc.), other inflammatory cytokines and mediators (IL-1β, IL-32, and complement C3) [74]. These responses were mediated by MyD88 adaptor proteins (MyD88, TRAF6, IRAK1, and TAK1), transcription factors (NF-κB and AP-1), and MAPKs [74]. In human colonic myofibroblasts, IL-36α and IL-36γ stimulated the secretion of IL-6 and CXC chemokines (CXCL1, CXCL2, and CXCL8) [75]. In addition, the combination of IL-36 cytokines plus IL-17A or of IL-36 cytokines plus TNF-α showed a synergistic effect on the induction of IL-6 and CXC chemokines [75].
IL-36 and IBD
In the last 10 years, there has been an increasing number of reports demonstrating the crucial role of IL-36 cytokines in the context of IBD. These studies suggest that IL-36 cytokines are involved in the pathophysiology of IBD as not only a pathogenic factor but also a protective factor according to the phase of disease.
We observed upregulation of IL-36α and IL-36γ, but not IL-36β, in biopsy samples from the inflamed colonic mucosa of patients with IBD, in particular UC [74]. Immunohistochemical analysis showed that T cells, monocytes, and plasma cells were the cellular source of IL-36α and IL-36γ in colonic mucosa [74]. Similar observations have been made by other groups [72, 76], while another group demonstrated an elevation of all IL-36 cytokines in the inflamed colonic mucosa of UC patients [77]. Scheibe et al. reported that IL-36α was mainly produced by macrophages in active IBD mucosa, while the intestinal epithelial cells expressed predominantly IL-36γ [78]. In the samples obtained from fibrotic tissue of IBD patients, a strong elevation of IL-36α expression was observed in correlation with the degree of inflammation [79]. Taken together, these results indicated that expression of IL-36, in particular IL36α and IL-36γ, is increased in the inflamed mucosa of patients with IBD.
Multiple studies of experimental colitis models have provided evidence of a crucial role of IL-36 in IBD. DSS-colitis was weakened in both IL-36R−/− mice and anti-IL-36R neutralizing antibody-treated mice and led to an increased survival rate, indicating a proinflammatory role of IL-36R signaling [78]. Russell et al. reported that DSS-colitis was improved in IL-36R−/− mice and this was accompanied by reduced Th1 response and enhanced Th17 response [72]. Similarly, a recent study reported that intraperitoneal injection of IL-36β exacerbated DSS-colitis via inhibition of Treg induction and increase of Th2 response [80].
In contrast, recovery of DSS-colitis was defective in IL-36R−/− mice with a marked reduction of IL-22 expression [81]. Defective recovery in this model was overcome by IL-22 restoration with an aryl hydrocarbon receptor agonist [81]. Since IL-22 mediates epithelial proliferation, restitution, and mucosal protection by mucin secretion [82], IL-36R signaling may be critical in the resolution of mucosal damage via induction of IL-22 response. Consistently, IL-36R- and IL-36γ-deficient mice exhibited dramatically reduced IL-23, IL-22, and antimicrobial peptide (AMP) levels, and consequently failed to recover from acute intestinal damage [83]. Impaired recovery of these mice could be overcome by treatment with exogenous IL-23 with a restoration of IL-22 and AMP expression in the colon [83], indicating a protective cytokine network involving IL-36γ, IL-23, and IL-22 in the gut. Thus, IL-36 signaling has provided conflicting results with protective as well as proinflammatory effects in acute DSS-colitis models.
In the chronic colitis models induced by DSS and 2,4,6-trinitrobenzene sulfonic acid (TNBS), mucosal inflammation was improved in IL-36R−/− mice or by neutralizing anti-IL-36R antibodies [79]. Oxazolone-induced colitis, which is a Th2 colitis model mediated mainly by Th9 response, was improved in IL-36R−/− and/or IL-36γ−/− mice with reduction of mucosal IL-9-producing cells and with an increased number of Tregs [73]. The observation of chronic colitis models suggests a proinflammatory role of IL-36 cytokines.
IL-36 and intestinal fibrosis
Recent studies have suggested that IL-36 cytokines actively participate in intestinal fibrosis by regulating immune cell recruitment and activation [50, 57, 79]. Scheibe et al. have extensively investigated the association of IL-36 signaling with intestinal fibrosis [79], and have shown an increased expression of type I and VI collagens in the inflamed mucosa of IBD patients [79]. Fibrotic changes were associated with higher expression of IL-36α and a greater number of myofibroblasts. RNA sequencing revealed the ability of IL-36 signaling to induce expression of genes involved in the regulation of fibrosis and tissue remodeling [79]. They also demonstrated that fibrotic changes as well as colitis activity was less severe in IL-36R−/− mice or in mice treated with anti-IL-36R antibodies [79]. These findings collectively indicate a close relationship between IL-36R signaling and chronic inflammation and fibrosis in the gut.
The cellular source of IL-36 has been reported to be intestinal fibroblasts, myofibroblasts, epithelial cells, and macrophages [74, 78, 79]. Among them, macrophages are considered to be its major source [79]. However, we have previously shown that human colonic myofibroblasts produce high amounts of IL-36γ in response to the stimuli of IL-1β or IL-1β plus TNF-α [64], suggesting an active role of intestinal myofibroblasts in fibrosis via IL-36 secretion.
Recent studies have suggested that IL-36R signaling attenuates acute inflammation but may drive fibrosis in chronic inflammation. Inflammation in chronic colitis may be carried by activated fibroblasts exhibiting the myofibroblast phenotype, which produce profibrotic/inflammatory cytokines, chemokines, and large amounts of extracellular matrix [84].
Therapeutic approaches targeting IL-36 signaling
The success of anti-TNF-α antibodies in the treatment of IBD triggered a subsequent search and development of new biologics targeting cytokines or cytokine signaling. However, some clinical trials of biologics targeting a single cytokine, such as anti-IFN-γ, anti-IL-17A, and anti-IL-13 antibodies, have unfortunately failed in IBD [85,86,87]. This suggests that inhibition of a single cytokine may be insufficient to induce resolution of all cases of IBD, whereas strategies targeting multiple inflammatory mediators may be more effective and clinically beneficial to IBD.
In this regard, IL-36 may be an attractive candidate as a therapeutic target of IBD. As previously mentioned, IL-36 actively participate in the pathogenesis of IBD, with increased expression in the inflamed mucosa of both CD and UC patient. A blockade of IL-36R signaling effectively suppressed severity of inflammation and fibrosis in experimental colitis in mice. Furthermore, favorable therapeutic effects of spesolimab (BI655130), a monoclonal antibody against the IL-36R, have been demonstrated in patients with generalized pustular psoriasis [88]. Based on these backgrounds, several clinical studies of spesolimab in IBD are ongoing. In the first study, the efficacy of spesolimab on anal fistulas was investigated in CD patients (NCT03752970) [89]. The efficacy of spesolimab is being evaluated in UC patients who experienced primarily or secondary failure to TNF antagonists or vedolizumab (NCT03482635) [90]. In addition, the efficacy of a combination of spesolimab and TNFα antagonists on achievement of mucosal healing is under investigation in UC patients (NCT03123120) 91. Furthermore, an open-labeled phase 2 trial evaluating long-term safety and efficacy of spesolimab is ongoing in UC patients (NCT03648541) 92.
Simultaneous blocking of multiple IL-1 family cytokines may be more appropriate for preventing compensatory responses induced by single cytokine blocking. Recently, it has been reported that a monoclonal antibody against human IL-1RAcP (IL-1R3) can block the signaling of six IL-1 family members including IL-1β and IL-36 [93]. Blocking IL-1R3 inhibited IL-36-driven Th1 responses and revealed a wide-range anti-inflammatory effect. Since several IL-1 family members are involved in the pathogenesis of IBD, IL-1R3 blockade may be a potential option for therapeutic intervention [93].
Broad-range blocking of IL-36R signaling may possess pros and cons. Inhibition of their proinflammatory activity is beneficial, but there are potentially detrimental effects, such as disruption of mucosal barrier, impairment of tissue repair and host protection. Therefore, the biologics that targeting a single IL-36 ligand may be ideal, since the roles of IL-36 cytokines are different in early and late stages of IBD.
Interleukin-38
IL-38 (IL-1F10) shares 37% of DNA sequence with IL-1R antagonist (IL-1Ra) and 43% with IL-36R antagonist (IL-36Ra) [94, 95]. Anti-inflammatory effects of IL-38 are associated with its antagonizing activity to IL-36R and IL-1R [96] (Fig. 2). IL-38 has been reported to be involved in the pathophysiology of various inflammatory disorders [97,98,99,100,101,102,103]. IL-38 precursor is a molecular weight of 16.9 kD [96], and its biological activity is dependent on N-terminal processing [104]. IL-36-induced CXCL8 secretion is inhibited by IL-38 in mononuclear cells and keratinocytes [98, 105].
We have recently demonstrated that the IL-38 expression is increased in the active mucosa of UC patients [106] and a similar observation has been made by Xie et al. [77]. The major cellular source of IL-38 was B cells in the active mucosa of UC patients [77, 106] (Fig. 3). IL-38 suppressed the IL-36γ-induced mRNA expression of chemokines (CXCL1, CXCL2, and CXCL8) in colonic epithelial cell lines with blockade of activation of NF-κB and MAPKs [106]. Furthermore, DSS-colitis was deteriorated in IL-38−/− mice [106, 107], and this was supported by Xie et al. where IL-38 injection leads to a suppression of DSS-colitis with reduced mucosal expression of IL-1β and TNF-α [77]. IL-38 is an anti-inflammatory cytokine and may be a protective factor in the pathogenesis of IBD.

Immunofluorescence pictures for IL-38 protein expression in the active mucosa of UC patients. IL-38 was green and CD19 (B cells) was red. Double positive cells were detected as yellow in the merged panel. Original magnification is × 400. This picture is a modified version of our previous report [106] and used under the permission by the society for free radical research Japan
Conclusion
Recent progress in biomedical research has introduced several biological tools targeting cytokines and cytokine signaling in the actual clinical setting of IBD. However, cytokine networks involved in the pathogenesis of IBD may be extremely complicated and even new biologics exert favorable clinical effects only in certain groups of IBD patients. The IL-1 family cytokines described in this review are involved in the pathogenesis of IBD as potent modulators of mucosal immunity. These cytokines are characterized by the dichotomous roles as exacerbators and inhibitors of inflammation, and may be factors contributing to a complexity of the cytokine networks in IBD. For the realization of novel biologics targeting these cytokines, further studies are required to define the molecular mechanisms determining their behavior in mucosal inflammation across different stages of disease.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. 2015;12:720–7.
Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–18.
Williams MA, O’Callaghan A, Corr SC. IL-33 and IL-18 in inflammatory bowel disease etiology and microbial interactions. Front Immunol. 2019;10:1091.
Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90.
Cayrol C, Girard JP. Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev. 2018;281:154–68.
Eberl G, Colonna M, Di Santo JP, Mckenzie AN. Innate lymphoid cells Innate lymphoid cells: a new paradigm in immunology. Science. 2015;348:aaa6566.
Nussbaum JC, Van Dyken SJ, von Moltke J, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. 2013;502:245–8.
Hodzic Z, Schill EM, Bolock AM, Good M. IL-33 and the intestine: the good, the bad, and the inflammatory. Cytokine. 2017;100:1–10.
Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16:676–89.
Luthi AU, Cullen SP, McNeela EA, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98.
Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1-and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. 2008;20:1019–30.
Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev. 2008;223:20–38.
Nishida A, Andoh A, Imaeda H, Inatomi O, Shiomi H, Fujiyama Y. Expression of interleukin 1-like cytokine interleukin 33 and its receptor complex (ST2L and IL1RAcP) in human pancreatic myofibroblasts. Gut. 2010;59:531–41.
Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nat Immunol. 2016;17:122–31.
Martin MU. Special aspects of interleukin-33 and the IL-33 receptor complex. Semin Immunol. 2013;25:449–57.
Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS ONE. 2008;3: e3331.
Sundlisaeter E, Edelmann RJ, Hol J, et al. The alarmin IL-33 is a notch target in quiescent endothelial cells. Am J Pathol. 2012;181:1099–111.
Lefrancais E, Roga S, Gautier V, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA. 2012;109:1673–8.
Lefrancais E, Duval A, Mirey E, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci USA. 2014;111:15502–7.
Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA. 2009;106:9021–6.
Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. 2014;31:31–7.
Bessa J, Meyer CA, de Vera Mudry MC, et al. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun. 2014;55:33–41.
Hudson CA, Christophi GP, Gruber RC, Wilmore JR, Lawrence DA, Massa PT. Induction of IL-33 expression and activity in central nervous system glia. J Leukoc Biol. 2008;84:631–43.
Hara K, Iijima K, Elias MK, et al. Airway uric acid is a sensor of inhaled protease allergens and initiates type 2 immune responses in respiratory mucosa. J Immunol. 2014;192:4032–42.
Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011;186:4375–87.
Kobori A, Yagi Y, Imaeda H, et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol. 2010;45:999–1007.
Muzes G, Molnar B, Tulassay Z, Sipos F. Changes of the cytokine profile in inflammatory bowel diseases. World J Gastroenterol. 2012;18:5848–61.
Nemeth ZH, Bogdanovski DA, Barratt-Stopper P, Paglinco SR, Antonioli L, Rolandelli RH. Crohn’s disease and ulcerative colitis show unique cytokine profiles. Cureus. 2017;9: e1177.
Beltran CJ, Nunez LE, Diaz-Jimenez D, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1097–107.
Gundersen MD, Goll R, Hol J, et al. Loss of interleukin 33 expression in colonic crypts—a potential marker for disease remission in ulcerative colitis. Sci Rep. 2016;6:35403.
Pastorelli L, Garg RR, Hoang SB, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci USA. 2010;107:8017–22.
Sponheim J, Pollheimer J, Olsen T, et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am J Pathol. 2010;177:2804–15.
Sedhom MA, Pichery M, Murdoch JR, et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut. 2013;62:1714–23.
Pushparaj PN, Li D, Komai-Koma M, et al. Interleukin-33 exacerbates acute colitis via interleukin-4 in mice. Immunology. 2013;140:70–7.
Seidelin JB, Coskun M, Kvist PH, Holm TL, Holgersen K, Nielsen OH. IL-33 promotes GATA-3 polarization of gut-derived T cells in experimental and ulcerative colitis. J Gastroenterol. 2015;50:180–90.
Imaeda H, Andoh A, Aomatsu T, et al. Interleukin-33 suppresses notch ligand expression and prevents goblet cell depletion in dextran sulfate sodium-induced colitis. Int J Mol Med. 2011;28:573–8.
Hufford MM, Kaplan MH. A gut reaction to IL-9. Nat Immunol. 2014;15:599–600.
Duan L, Chen J, Zhang H, et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3 (+) regulatory T-cell responses in mice. Mol Med. 2012;18:753–61.
Tu L, Chen J, Xu D, et al. IL-33-induced alternatively activated macrophage attenuates the development of TNBS-induced colitis. Oncotarget. 2017;8:27704–14.
Seo DH, Che X, Kwak MS, et al. Interleukin-33 regulates intestinal inflammation by modulating macrophages in inflammatory bowel disease. Sci Rep. 2017;7:851.
Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci USA. 2015;112:10762–7.
Hirai F, Andoh A, Ueno F, et al. Efficacy of endoscopic balloon dilation for small bowel strictures in patients With Crohn’s disease: a nationwide, multi-centre, open-label prospective cohort study. J Crohns Colitis. 2018;12:394–401.
Nakayama T, Hirahara K, Onodera A, et al. Th2 cells in health and disease. Annu Rev Immunol. 2017;35:53–84.
Bailey JR, Bland PW, Tarlton JF, et al. IL-13 promotes collagen accumulation in Crohn’s disease fibrosis by down-regulation of fibroblast MMP synthesis: a role for innate lymphoid cells? PLoS ONE. 2012;7: e52332.
Dinarello C, Arend W, Sims J, et al. IL-1 family nomenclature. Nat Immunol. 2010;11:973.
Tripodi D, Conti F, Rosati M, et al. IL-36 a new member of the IL-1 family cytokines. J Biol Regul Homeost Agents. 2012;26:7–14.
Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10:89–102.
Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem. 2004;279:13677–88.
Sims JE, Nicklin MJ, Bazan JF, et al. A new nomenclature for IL-1-family genes. Trends Immunol. 2001;22:536–7.
Elias M, Zhao S, Le HT, et al. IL-36 in chronic inflammation and fibrosis—bridging the gap? J Clin Invest. 2021. https://doi.org/10.1172/JCI144336.
Clancy DM, Sullivan GP, Moran HBT, et al. Extracellular neutrophil proteases are efficient regulators of IL-1, IL-33, and IL-36 cytokine activity but poor effectors of microbial killing. Cell Rep. 2018;22:2937–50.
Towne JE, Renshaw BR, Douangpanya J, et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J Biol Chem. 2011;286:42594–602.
Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, Martin SJ. Neutrophil-derived proteases escalate inflammation through activation of IL-36 family cytokines. Cell Rep. 2016;14:708–22.
Macleod T, Doble R, McGonagle D, et al. Neutrophil Elastase-mediated proteolysis activates the anti-inflammatory cytokine IL-36 receptor antagonist. Sci Rep. 2016;6:24880.
Martin U, Scholler J, Gurgel J, Renshaw B, Sims JE, Gabel CA. Externalization of the leaderless cytokine IL-1F6 occurs in response to lipopolysaccharide/ATP activation of transduced bone marrow macrophages. J Immunol. 2009;183:4021–30.
Dos Santos JPS, Ribeiro RCB, Faria JV, et al. Synthesis, biological evaluation and molecular modeling studies of novel triazole-linked menadione-furan derivatives as inhibitors. J Bioenerg Biomembr. 2022. https://doi.org/10.1007/s10863-022-09947-2.
Ngo VL, Kuczma M, Maxim E, Denning TL. IL-36 cytokines and gut immunity. Immunology. 2021;163:145–54.
Mulero JJ, Pace AM, Nelken ST, et al. IL1HY1: a novel interleukin-1 receptor antagonist gene. Biochem Biophys Res Commun. 1999;263:702–6.
Bassoy EY, Towne JE, Gabay C. Regulation and function of interleukin-36 cytokines. Immunol Rev. 2018;281:169–78.
Yuan ZC, Xu WD, Liu XY, Liu XY, Huang AF, Su LC. Biology of IL-36 signaling and its role in systemic inflammatory diseases. Front Immunol. 2019;10:2532.
Smith DE, Renshaw BR, Ketchem RR, Kubin M, Garka KE, Sims JE. Four new members expand the interleukin-1 superfamily. J Biol Chem. 2000;275:1169–75.
Frey S, Derer A, Messbacher ME, et al. The novel cytokine interleukin-36alpha is expressed in psoriatic and rheumatoid arthritis synovium. Ann Rheum Dis. 2013;72:1569–74.
Blumberg H, Dinh H, Trueblood ES, et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med. 2007;204:2603–14.
Takahashi K, Nishida A, Shioya M, et al. Interleukin (IL)-1beta Is a strong inducer of IL-36gamma expression in human colonic myofibroblasts. PLoS ONE. 2015;10: e0138423.
Bachmann M, Scheiermann P, Hardle L, Pfeilschifter J, Muhl H. IL-36gamma/IL-1F9, an innate T-bet target in myeloid cells. J Biol Chem. 2012;287:41684–96.
Lian LH, Milora KA, Manupipatpong KK, Jensen LE. The double-stranded RNA analogue polyinosinic-polycytidylic acid induces keratinocyte pyroptosis and release of IL-36gamma. J Invest Dermatol. 2012;132:1346–53.
Gabay C, Towne JE. Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J Leukoc Biol. 2015;97:645–52.
Leon G, Hussey S, Walsh PT. The diverse roles of the IL-36 family in gastrointestinal inflammation and resolution. Inflamm Bowel Dis. 2021;27:440–50.
Leon G, Hernandez Santana YE, Irwin N, et al. IL-36 cytokines imprint a colitogenic phenotype on CD4(+) T helper cells. Mucosal Immunol. 2022;15:491–503.
Vigne S, Palmer G, Lamacchia C, et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood. 2011;118:5813–23.
Foster AM, Baliwag J, Chen CS, et al. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J Immunol. 2014;192:6053–61.
Russell SE, Horan RM, Stefanska AM, et al. IL-36alpha expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol. 2016;9:1193–204.
Harusato A, Abo H, Ngo VL, et al. IL-36gamma signaling controls the induced regulatory T cell-Th9 cell balance via NFkappaB activation and STAT transcription factors. Mucosal Immunol. 2017;10:1455–67.
Nishida A, Hidaka K, Kanda T, et al. Increased expression of interleukin-36, a member of the interleukin-1 cytokine family, in inflammatory bowel disease. Inflamm Bowel Dis. 2016;22:303–14.
Kanda T, Nishida A, Takahashi K, et al. Interleukin (IL)-36alpha and IL-36gamma induce proinflammatory mediators from human colonic subepithelial myofibroblasts. Front Med (Lausanne). 2015;2:69.
Boutet MA, Bart G, Penhoat M, et al. Distinct expression of interleukin (IL)-36alpha, beta and gamma, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin Exp Immunol. 2016;184:159–73.
Xie C, Yan W, Quan R, et al. Interleukin-38 is elevated in inflammatory bowel diseases and suppresses intestinal inflammation. Cytokine. 2020;127: 154963.
Scheibe K, Backert I, Wirtz S, et al. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut. 2017;66:823–38.
Scheibe K, Kersten C, Schmied A, et al. Inhibiting interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation. Gastroenterology. 2019;156(1082–97): e11.
Zhu J, Xu Y, Li Z, Liu S, Fu W, Wei Y. Interleukin-36beta exacerbates DSS-induce acute colitis via inhibiting Foxp3(+) regulatory T cell response and increasing Th2 cell response. Int Immunopharmacol. 2022;108: 108762.
Medina-Contreras O, Harusato A, Nishio H, et al. Cutting edge: IL-36 receptor promotes resolution of intestinal damage. J Immunol. 2016;196:34–8.
Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J Gastroenterol. 2018;53:465–74.
Ngo VL, Abo H, Maxim E, et al. A cytokine network involving IL-36gamma, IL-23, and IL-22 promotes antimicrobial defense and recovery from intestinal barrier damage. Proc Natl Acad Sci USA. 2018;115:E5076–85.
Mao R, Rieder F. Cooling down the hot potato: anti-interleukin 36 therapy prevents and treats experimental intestinal fibrosis. Gastroenterology. 2019;156:871–3.
Reinisch W, Panes J, Khurana S, et al. Anrukinzumab, an anti-interleukin 13 monoclonal antibody, in active UC: efficacy and safety from a phase IIa randomised multicentre study. Gut. 2015;64:894–900.
Reinisch W, de Villiers W, Bene L, et al. Fontolizumab in moderate to severe Crohn’s disease: a phase 2, randomized, double-blind, placebo-controlled, multiple-dose study. Inflamm Bowel Dis. 2010;16:233–42.
Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–700.
Bachelez H, Choon SE, Marrakchi S, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. New Engl J Med. 2019;380:981–3.
Boehringer Ingelheim. NCT03752970: A study testing how BI655130 works in patients with fistulizing Crohn's disease. https://clinicaltrials.gov/ct2/show/NCT03752970.
Boehringer Ingelheim. NCT03482635: BI655130 (SPESOLIMAB) induction treatment in patients with moderate-to-severe ulcerative colitis. https://clinicaltrials.gov/ct2/show/NCT03482635.
Boehringer Ingelheim. NCT03123120: A Study in patients with mild or moderate ulcerative colitis who take a TNF inhibitor. The study investigates whether bowel inflammation improves when patients take BI655130 in addition to their current therapy. https://www.clinicaltrials.gov/ct2/show/NCT03123120.
Boehringer Ingelheim. NCT03648541: BI655130 long-term treatment in patients with moderate-to severe ulcerative colitis. https://clinicaltrials.gov/ct2/show/NCT03648541.
Hojen JF, Kristensen MLV, McKee AS, et al. IL-1R3 blockade broadly attenuates the functions of six members of the IL-1 family, revealing their contribution to models of disease. Nat Immunol. 2019;20:1138–49.
Bensen JT, Dawson PA, Mychaleckyj JC, Bowden DW. Identification of a novel human cytokine gene in the interleukin gene cluster on chromosome 2q12-14. J Interferon Cytokine Res. 2001;21:899–904.
de Graaf DM, Teufel LU, Joosten LAB, Dinarello CA. Interleukin-38 in health and disease. Cytokine. 2022;152: 155824.
Xu WD, Huang AF. Role of interleukin-38 in chronic inflammatory diseases: a comprehensive review. Front Immunol. 2018;9:1462.
Han Y, Mora J, Huard A, et al. IL-38 ameliorates skin inflammation and limits IL-17 production from gammadelta T Cells. Cell Rep. 2019;27(835–46): e5.
Mercurio L, Morelli M, Scarponi C, et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis. 2018;9:1104.
Xu WD, Su LC, He CS, Huang AF. Plasma interleukin-38 in patients with rheumatoid arthritis. Int Immunopharmacol. 2018;65:1–7.
Takenaka SI, Kaieda S, Kawayama T, et al. IL-38: a new factor in rheumatoid arthritis. Biochem Biophys Rep. 2015;4:386–91.
Matsuoka M, Kawayama T, Tominaga M, et al. Attenuated airway eosinophilic inflammations in IL-38 knockout mouse model. Kurume Med J. 2019;65:37–46.
Rudloff I, Godsell J, Nold-Petry CA, et al. Brief report: interleukin-38 exerts antiinflammatory functions and is associated with disease activity in systemic lupus erythematosus. Arthritis Rheumatol. 2015;67:3219–25.
Ciccia F, Accardo-Palumbo A, Alessandro R, et al. Interleukin-36alpha axis is modulated in patients with primary Sjogren’s syndrome. Clin Exp Immunol. 2015;181:230–8.
Mora J, Schlemmer A, Wittig I, et al. Interleukin-38 is released from apoptotic cells to limit inflammatory macrophage responses. J Mol Cell Biol. 2016;8:426–38.
van de Veerdonk FL, Stoeckman AK, Wu G, et al. IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc Natl Acad Sci USA. 2012;109:3001–5.
Ohno M, Imai T, Chatani M, et al. The anti-inflammatory and protective role of interleukin-38 in inflammatory bowel disease. J Clin Biochem Nutr. 2022;70:64–71.
de Graaf DM, Wang RX, Amo-Aparicio J, et al. IL-38 gene deletion worsens murine colitis. Front Immunol. 2022;13: 840719.
Acknowledgements
This work was supported by the Japan Agency for Medical Research and Development (AMED) under Grant number JP20gm1010008h9904 (AA), and in part by a Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan under Grant number 22K07664(AA), and in part by a Health and Labour Sciences Research Grants for Research on Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan under Grant number 20FC1037 (AA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Andoh, A., Nishida, A. Pro- and anti-inflammatory roles of interleukin (IL)-33, IL-36, and IL-38 in inflammatory bowel disease. J Gastroenterol 58, 69–78 (2023). https://doi.org/10.1007/s00535-022-01936-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-022-01936-x