
Abstract
The liver displays an outstanding wound healing and regenerative capacity unmatched by any other organ. This reparative response is governed by a complex network of inflammatory mediators, growth factors and metabolites that are set in motion in response to hepatocellular injury. However, when liver injury is chronic, these regenerative mechanisms become dysregulated, facilitating the accumulation of genetic alterations leading to unrestrained cell proliferation and the development of hepatocellular carcinoma (HCC). The epidermal growth factor receptor (EGFR or ErbB1) signaling system has been identified as a key player in all stages of the liver response to injury, from early inflammation and hepatocellular proliferation to fibrogenesis and neoplastic transformation. The EGFR system engages in extensive crosstalk with other signaling pathways, acting as a true signaling hub for other growth factors, cytokines and inflammatory mediators. Here, we briefly review essential aspects of the biology of the EGFR, the other ErbB receptors, and their ligands in liver injury, regeneration and HCC development. Some aspects of the preclinical and clinical experience with EGFR therapeutic targeting in HCC are also discussed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction: overview of the EGFR signaling system
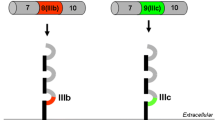
The epidermal growth factor receptor (EGFR), also known as ErbB1 or HER-1, is a transmembrane protein receptor endowed with tyrosine kinase activity [1]. While in lower organisms such as C. elegans and D. melanogaster only a single receptor type equivalent to EGFR has been described, in mammals there are three other related peptides called ErbB2/HER-2, ErbB3/HER-3 and ErbB4/HER-4 that jointly with ErbB1 constitute the known EGFR family [1–3]. This diversity parallels the complexity of systemic regulation in vertebrate organisms, and together with the broad variety of EGFR family of ligands discussed below; it is an indicator of the important role played by this signaling system in cell control. In general, all these EGFR related proteins share common structural features, which include an extracellular ligand-binding domain, a single-pass transmembrane domain and an intracellular domain that harbors the tyrosine kinase activity (Fig. 1) [4]. Following the tyrosine kinase domain there is a carboxy-terminal tail where tyrosine autophosphorylation sites are found [2, 4]. The intracellular domain of these receptors is highly conserved with the exception of ErbB3, in which amino acids important for the tyrosine kinase activity have been substituted [5]. This feature renders ErbB3 a tyrosine kinase-devoid receptor, although more recent studies could identify residual autophosphorylating capacity in ErbB3, which may be important for signaling [6]. Regarding the extracellular ligand-binding domain, two common cysteine-rich regions have been described, together with the presence of N-linked oligosaccharides in at least nine positions, which are essential for receptor function [7, 8]. Overall, the extracellular domain is less well conserved among the ErbB proteins, consistent with their differential specificity for ligand binding. ErbB2 has no known ligand; however, the rest of the ErbB receptors can be bound and activated by a family of growth factors encompassing 11 members [2, 5, 9]. Structurally these ligands are characterized by the presence of an EGF-like domain, which determines receptor-binding specificity, and additional motifs such as immunoglobulin domain, glycosylation sites and heparin-binding sites [5]. According to their receptor binding specificities, ErbB ligands can be classified in three different groups. EGF, transforming growth factor-α (TGFα), amphiregulin (AR) and epigen (EPG) bind specifically ErbB1, while betacellulin (BTC), heparin-binding EGF-like growth factor (HB-EGF) and epiregulin (EREG) bind ErbB1 and ErbB4. Neuregulins (NRG), which can be found in different alternatively spliced isoforms, bind ErbB3 and ErbB4 (Fig. 1) [3]. The complexity of this signaling system is further increased by the fact that after ligand binding ErbB receptors can homodimerize or heterodimerize, resulting in cross-phosphorylation reactions among these proteins. These interactions involve also the ErbB2 and ErbB3 receptors, even though they lack a known ligand and are almost devoid of kinase activity, respectively. In spite of these limitations, perhaps the most potent heterodimer in terms of triggering survival and proliferative signals is the ErbB2/ErbB3 complex [2, 5]. Upon ligand binding, ErbB proteins are auto-phosphorylated and cross-phosphorylated in specific tyrosine residues (Fig. 2), thus generating docking sites for a variety of signaling proteins like Shc, Grb2, Grb7, Crk, phospholipase Cγ, the kinases Src and phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3 K), the protein phosphatases SHP1 and SHP2, as well as the Cbl E3 ubiquitin ligase [2, 4, 5]. There are other proteins that do not directly bind the ErbB receptors but also participate in signaling, such as phospholipase D and the signal-transducer and activator of transcription (STAT) 1, 3 and 5 proteins [4, 10]. Downstream of activated ErbB receptors, we find several major intracellular signaling cascades such as the ras/raf/MEK/MAPK pathway (comprising the activation of extracellular signal-regulated kinase (ERK) and JUN N-terminal kinase), p38-MAPK, phospholipase C/protein kinase C pathway, the PI3 K/Akt–mTOR pathway (being PI3 K/Akt linked to NF-κB activation) and the STAT pathway [2, 4, 5, 9–11]. There is a considerable degree of crosstalk among these pathways, and they can lead to the activation of important transcriptional programs in the cell nucleus, governing the expression of genes involved in cell proliferation, survival, differentiation, adhesion, migration and metabolism [12]. Given the key effects of EGFR signaling on cellular homeostasis, important control mechanisms of the signal output are concomitantly activated upon receptor activation. These mechanisms include the immediate activation of parallel signals triggered also by EGFR involving methyl-transfer reactions impacting on the ras/raf/MEK/MAPK pathway [13], the de novo expression of EGFR inhibitory proteins, and the fine-tuning of EGFR endocytic degradation, among others [14–16].

General structure of the EGFR family of receptors and their cognate ligands, indicating their binding specificities. The constitutively active EGFRvIII mutant is also shown. Extracellular domains I and III participate in ligand binding, while domains II and IV are involved in homodimerization and heterodimerization upon ligand binding. TK tyrosine kinase domain, EGF epidermal growth factor, TGFα transforming growth factor α, AR amphiregulin, EPG epigen, BTC betacellulin, HB-EGF heparin-binding EGF, EREG epiregulin, NRGs neuregulins

Principal intracellular pathways triggered by ErbB receptor activation. Ligand binding induces a conformational change that allows receptor homodimerization or heterodimerization and functional activation. The intracellular tyrosine kinase (TK) domain and phosphorylation sites mediating interaction with adaptor proteins are schematically shown. Downstream signaling cascades and representative transcription factors engaged upon ErbB activation are indicated
The ErbB ligands are synthesized as membrane-anchored precursors. They are type I transmembrane proteins with an N-terminal extension, the EGF module and a short juxtamembrane stalk followed by a hydrophobic transmembrane domain and a carboxy-terminal or cytoplasmic tail [3]. These ligands display a low overall protein sequence identity (about 25 %), differing also in their glycosylation patterns and in the presence or absence of a heparin-binding domain [3, 17]. Release of the active factors from the cell surface is mediated by transmembrane proteases of the “a disintegrin and metalloproteinases” (ADAM) type, and ADAM17 also known as TNFα converting enzyme or TACE, together with ADAM10, are known to have an important effect on the shedding of ErbB ligands [18, 19]. Nevertheless, other types of enzymes may be involved in the shedding of ErbB ligands, as illustrated by the recent identification of rhomboid RHBDL2 intramembrane protease as a functional EGF sheddase in mammalian cells [20]. The soluble growth factors may bind their cognate receptors in an autocrine or paracrine manner, although membrane anchored ErbB ligands can also signal to adjacent cells in a juxtacrine fashion [3]. Ectodomain shedding also implies the generation of a carboxy-terminal cell-associated remnant of these growth factors, for which different regulatory functions are increasingly being recognized [3, 21, 22].
Importantly, the proteolytic activity of ADAMs is subject to regulation by multiple signals with consequences for downstream EGFR signaling. In particular, several G protein-coupled receptors (GPCRs) have been shown to activate EGFR/ErbB signal transduction even though GPCRs ligands do not directly interact with the EGFR [23–25]. This process, known as EGFR transactivation, places the EGFR signaling system at the center of converging signals with relevant biological consequences, including proliferative, survival, migratory and inflammatory cues [26]. This intricate, and not completely known network of direct and indirect EGFR activators, make this receptor family a remarkable signaling hub, with special significance in tumor development and cancer cell biology, and adds a significant degree of complexity to this signaling system [26–28]. It is important to highlight that the expression, maturation, trafficking to the cell surface, catalytic activity and substrate targeting of ADAMs are also subject to regulation, contributing additional regulatory points to the whole system [29, 30]. Moreover, in addition to EGFR transactivation by ADAM-mediated ligand release, this tyrosine kinase receptor can be engaged in other signaling pathways at different levels [31]. For instance, ErbB1 can heterodimerize with other tyrosine kinase receptors such as the platelet derived growth factor receptor (PDGFR), insulin like growth factor 1 receptor (IGF1-R) and hepatocyte growth factor (HGF) receptor c-MET, which play major roles in liver responses to injury and carcinogenesis [31]. More recently, ErbB1 has been shown to contribute to toll like receptor 3 (TLR3) signaling through its ability to phosphorylate, in cooperation with Src, specific tyrosine residues in TLR3 needed for the binding of TRIF adaptor protein [32], further illustrating the multifarious nature of EGFR family interactions in cellular signaling.
A key aspect not completely resolved is how signaling specificity is gained in this apparently redundant EGFR signaling system. One obvious level of specificity may be defined by the tissue and time-specific expression of the different ErbB receptors and ligands. There appears to be also different downstream inputs depending on the heterodimerization partners assembled. It is known that the recruitment of ErbB3 to heterodimers involves potent PI3 K activation, while no involvement of phospholipase C or the Ras-specific GTPase-activating protein (GAP) is observed, being these signal transducers actively implicated in ErbB1 signaling [2]. Another feature that may impart specificity within the system is the biological activity of the carboxy-terminal tails of the individual EGFR ligands, which are generated after ADAM-mediated shedding of growth factor ectodomains, an important aspect that certainly merits further studies [21, 22]. The mode of interaction of different ligands with the ErbB receptors, including ligand affinity, potency and intrinsic activity has been studied, and the observed differences may explain some of the diverse biological outcomes [33]. Also, it is clear that different ErbB ligands induce distinct phosphorylation patterns of the tyrosine residues within the receptors, leading to the activation of different intracellular signaling pathways [34, 35]. The kinetics of receptor phosphorylation, trafficking and degradation, or the differential temporal activation of downstream ERK phosphorylation elicited by different ErbB ligands has been also invoked as mechanisms able to impart specificity [36, 37]. The relevance of ErbB ligand-specific cellular effects, and, thus, the importance of further investigation on this topic, is evident in the field of cancer cell biology. For instance, it has been shown that in non-small-cell lung carcinoma (NSCLC) patients TGFα and AR serum concentrations positively correlate with tumor aggressiveness, while, conversely, EGF levels were higher in healthy individuals [38]. Similar observations have been made in breast cancer tissues, in which high EGF levels are associated with more favorable prognosis, while TGFα expression is linked to more aggressive tumors [39]. Conversely, and as will be discussed later, high circulating EGF levels are associated with increased risk of hepatocellular carcinoma (HCC) development in patients with liver cirrhosis [40, 41]. Although we have drawn here a far from comprehensive picture of the EGFR signaling system, this may suffice to appreciate the complexity and biological relevance of this family of growth factors and receptors and its potential impact in disease development.
The EGFR system in liver injury and repair
The liver possesses an extraordinary regenerative capacity upon parenchymal resection or necrotic tissue loss, and this response is tightly coordinated by an increasing list of cytokines, growth factors and metabolites [42–46]. Important evidence has accumulated on the central role of the EGFR signaling system in this reparative and regenerative response of the liver upon injury and inflammation, including the inhibition of intrahepatic lipid accumulation [47]. Hepatocytes display very high levels of ErbB1 [48], and the expression of most ErbB ligands is increased during liver tissue injury and regeneration, as observed for HB-EGF, TGFα, EREG and AR in rodent models of partial hepatectomy and experimental liver injury [49–53]. Interestingly, the expression of ADAM17 is also up-regulated in the liver after partial hepatectomy [54]. In vitro studies have demonstrated that ErbB ligands can convey strong proliferative and survival signals to hepatocytes, and these effects have been also confirmed in vivo upon intraperitoneal injection of these growth factors or through their transgenic overexpression [55–58]. Genetically modified mice lacking different members of the EGFR family of receptors and ligands have been generated over the past two decades. Different phenotypes were observed, mice lacking ErbB2, ErbB3 and ErbB4 were embryonic lethal, while ErbB1 deficient mice died during fetal development or survived a few days after birth depending on their genetic background [12]. Indeed these lethal phenotypes attested to the importance of this signaling system for embryonic development, but precluded further analysis of the implication of these receptors in hepatic responses. Insights into the involvement of ErbB1 in hepatocellular proliferation were gained by targeted deletion of this gene in the adult mouse liver. Hepatocyte-specific ErbB1 null mice showed delayed liver regeneration with a defective G1–S phase entry, liver injury and increased mortality after partial hepatectomy [59]. These observations were subsequently validated by ErbB1 knockdown using RNA interference in rats that were subjected to partial hepatectomy [60]. Furthermore, the profound alterations in liver regeneration found in growth hormone receptor deficient mice have been attributed to their lack of hepatocellular EGFR expression [61], and reduced EGFR levels are also found in hepatocyte-specific beta-catenin knockout mice, which also display suboptimal liver regeneration [62]. In the context of acute liver injury, a recent study has clearly established that ADAM17 and ErbB1 expression individually provide protection against hepatocellular apoptosis induced by Fas ligand. The defensive role of ADAM17 was attributed in part to its involvement in the generation of the soluble ErbB ligands AR, TGFα and HB-EGF [63].
Liver regeneration studies have been also performed in mice with genetic invalidation of different ErbB ligands. Before describing the phenotypes of these mice, it is worth mentioning that, in addition to their intra-hepatic autocrine or paracrine mode of action, early studies identified an important endocrine role for EGF in this process. Indeed, elimination of salivary glands, a major site for EGF production, markedly impaired liver regeneration [58], which attests to the importance of systemic mechanisms in this complex response [64]. Regarding specific ErbB ligands knockout mice, absence of TGFα or EREG did not significantly affect the course of liver regeneration after partial hepatectomy [52, 65], while lack of HB-EGF or AR resulted in a delayed hepatocyte progression through the cell cycle [53, 66]. AR seems to play a complex and important role in liver regeneration, in addition to serving as a mitogenic factor for hepatocytes it also seems important in the modulation of the liver acute phase response, a prerequisite for the normal progression through the cell cycle in hepatocytes [67, 68]. Importantly, a recent study demonstrated that AR neutralizing antibodies markedly inhibited regeneration in a clinically relevant model of small-for-size liver transplantation, while AR administration rescued liver grafts of 30 % the original size, highlighting both the biological relevance of this ErbB1 ligand and its potential therapeutic use [69]. In view of the different phenotypes of the respective knockout mice, it appears that not all ErbB ligands are functionally redundant during liver regeneration, and; therefore, the subtle differences in their signaling mechanisms mentioned before can be of biological relevance. In addition, differential expression and interaction of these growth factors in other liver cells types, including macrophages and stellate cells could also be important [26, 70].
The EGFR system in hepatocarcinogenesis
Persistent liver injury due to chronic alcohol consumption, bad dietary habits leading to intrahepatic fat accumulation as well as hepatotropic viral infections resulting in chronic liver injury and cirrhosis, are strongly linked to the development of liver tumors [71–73]. The intrahepatic microenvironment rich in inflammatory mediators and growth factors is part of the natural regenerative and reparative response of the liver [56]. However, when this response becomes chronic and injury and inflammation persist, important alterations occur. These include changes in the architecture of the organ due to excessive deposition of extracellular matrix (ECM) or fibrosis, uncontrolled cell proliferation allowing the fixation of genetic mutations, and the development of preneoplastic nodules, events commonly accompanied by loss of hepatic function [74–79]. These alterations pave the way for the development of HCC, being liver fibrosis a strong determinant for the appearance of liver tumors [80]. The EGFR system has been implicated in hepatocarcinogenesis from the early stages of the disease. Various ErbB ligands are overexpressed in chronic liver injury, as demonstrated both in experimental models and in liver tissue samples from cirrhotic patients, and their expression can be induced also by the predominant inflammatory signals found in acute and chronic liver injury [26, 53, 81, 82]. EGFR ligands are known to contribute to the phenotypic activation of ECM-producing cells, including stellate cells and liver myofibroblasts which express significant amounts of ErbB1 [26], eliciting migratory and proliferative responses in these cells [83–86]. Persistent activation of the EGFR signaling system is known to participate in the pathogenesis of fibrosis in other organs such as the kidney and lung [87–91]. However, further to the demonstration of the expression of EGFR ligands during chronic liver injury and the above-mentioned observations in cultured fibrogenic cells, the involvement of the EGFR system in the development of liver fibrosis is not completely known. One study has demonstrated that AR deficient mice develop less fibrosis in an experimental model of chronic CCl4 administration [86]. These mice showed less accumulation of collagen than wild type mice, and reduced expression of profibrogenic mediators such as transforming growth factor-β (TGFβ) and connective tissue growth factor (CTGF). Interestingly, AR behaved as a mitogen and pro-survival factor for cultured ECM-producing cells, where it also up-regulated CTGF expression [86]. As previously discussed, AR plays an important role in liver regeneration and is a potent hepatoprotectant; therefore, its involvement in liver fibrogenesis could be interpreted as a wound healing activity gone awry in chronic liver injury. The pro-fibrogenic role of AR has been also cogently demonstrated in a recent study on pulmonary fibrogenesis [87], suggesting that this ErbB1 ligand could be a potential therapeutic target in solid organ fibrosis. At variance with AR, a recent report demonstrated increased hepatic fibrosis in liver-specific HB-EGF deficient mice upon bile duct ligation, and showed that HB-EGF was able to antagonize the profibrogenic activity of TGFβ on ECM-producing cells [92]. These mice displayed increased liver injury, which may be an important determinant in their enhanced fibrogenic response. Moreover, this study also emphasized the remarkable hepatoprotective actions of HB-EGF [50, 93], as well as the diversity of actions of the different EGFR ligands.
Various studies evaluated ErbB1 status in HCC and found this receptor frequently overexpressed in tumoral tissues [94, 95]. ErbB1 expression correlated with high proliferating activity, intrahepatic metastasis, poor carcinoma differentiation and bad prognosis [94, 96, 97]. Overexpression of ErbB1 has been recently detected in a molecular subclass of HCC with chromosome 7 polysomy which is more frequent in male individuals [98]. In view of the above, this genetic alteration may contribute to explain the higher prevalence of HCC in the male population.
EGFRvIII is a naturally occurring EGFR mutant lacking 801 base pairs (exons 2–7) that exhibits ligand-independent constitutive activity (Fig. 1). This variant is not present in normal tissues, and pending further confirmation it has been found in up to 60 % of HCC tissues examined according to one study [99]. EGFRvIII is also detected in serum from patients with HCC, and its expression in HCC cells lines confers them enhanced growth and drug resistance properties [100, 101]. Regarding other members of the EGFR family, ErbB2 expression is not commonly found, and it is not related to any clinicopathological features, and similar observations have been made for ErbB4 [94]. The situation is different for ERbB3, which is frequently overexpressed and activated in cancerous lesions and is significantly related to important markers of disease progression [94, 102]. Interestingly, secreted isoforms of ErbB3 can be found in serum from HCC patients at early stages of the disease, correlating with portal vein invasion and metastasis [103].
Increased expression of ErbB ligands has also been observed in human HCC. In agreement with the role of the EGFR signaling system in tissue repair, the expression of most ligands is elevated in the chronically injured and cirrhotic liver, along with that of ADAM17 [31, 104–107]. Importantly, the mRNA levels EGF and TGFα in HCC tissues have been related to the prognosis of patients with liver cancer [108]. Most interestingly, increased EGF gene expression in cirrhotic non-tumoral liver tissues has been linked to reduced survival time after surgery [109]. Numerous experimental studies have demonstrated the functional significance of EGFR ligand expression in liver cancer cells. For instance, AR overexpression and ErbB1 define an autocrine loop capable of promoting cell proliferation, survival and resistance to cytotoxic drugs and TGFβ mediated apoptosis in human HCC cells [81, 106, 110–112]. Moreover, AR/ErbB1 signaling can in turn activate the expression of other growth factors such as CTGF, which has also been involved in hepatocarcinogenesis and has profound effects on the biology of HCC cells, including their proliferation and resistance to TNF-related apoptosis inducing ligand (TRAIL)-mediated apoptosis [113, 114]. Additionally, the AR/ErbB1 system may be also positioned downstream of other growth factors signaling systems, and serve to mediate and amplify their effects in HCC cells as described recently for fibroblast growth factor 19 (FGF19) [115]. Another recent report has also identified the overexpression of the ErbB3 ligands NRG in HCC tissues, and has demonstrated their ability to increase HCC cells' migratory and invasive properties [102].
Two recent reports have identified additional mechanisms for the dysregulation of EGFR activity in HCC. These involve the impaired expression in tumor cells of genes that behave as negative regulators of EGFR signaling. Mitogen-inducible gene-6 (MIG-6) is an adaptor protein and a regulator of receptor tyrosine kinase signaling. MIG-6 expression is up-regulated during liver regeneration, and hepatocytes lacking MIG-6 display enhanced cell proliferation after partial liver resection, coinciding with increased PI3K/Akt signaling [116]. Importantly, MIG-6 gene expression was found markedly reduced in a majority of tumors (64 %) inversely correlating with EGFR expression [116]. Another negative regulator of EGFR signaling lost in HCC is mucin-15 (MUC15). MUC15 is a transmembrane protein highly glycosylated that is normally expressed in epithelial cells. It was observed that MUC15 expression was aberrantly decreased in HCC tissues, correlating with a poor prognosis. MUC15 was found to interact physically with EGFR, enhancing EGF-induced endocytosis and degradation of the EGFR, and attenuating downstream PI3K–Akt signaling [117]. This work identified a tumor suppressor role for MUC15 in HCC through its ability to regulate EGFR signaling.
Finally, as further evidence of the implication of the EGFR system in the pathogenesis of HCC, we can mention two recent and very interesting studies identifying the association of a functional polymorphism in the 5′ untranslated region of the EGF gene with increased risk for development of HCC in liver cirrhotic patients [40, 41]. This polymorphism conferred an extended half-life to EGF mRNA transcripts and increased EGF expression, which correlated with increased phosphorylation of ErbB1 in cirrhotic liver tissue samples [40]. These findings, together with other observations summarized elsewhere [28, 110], have contributed to identify the EGFR signaling system as an ideal candidate for the targeted therapy of HCC.
Targeting the EGFR system in HCC: experimental success and clinical failure
Two types of molecules with different mechanisms of action have been so far used to target and inhibit the EGFR in HCC. These include small molecules able to compete with ATP and inhibit the tyrosine kinase activity of this receptor [small tyrosine kinase inhibitors (TKIs): gefitinib, erlotinib and lapatinib], and EGFR-targeted antibodies that prevent ligand binding, receptor activation and dimerization, ultimately inducing receptor downregulation (cetuximab or erbitux and panitumumab) [118–120]. Over the past decade, several studies performed both in cultured HCC cell lines and in vivo experimental models demonstrated a potent antitumoral activity for both types of molecules. Cell cycle arrest, apoptosis, enhanced sensitivity towards cytostatics and reduced in vivo HCC cell growth and metastasis formation were reported in response to these agents, either alone or in combination [121–127]. A noteworthy study demonstrated that the administration of the small molecule EGFR inhibitor gefitinib to rats undergoing chemical carcinogenesis had a clear inhibitory effect on the development of HCC nodules. Importantly, this investigation was performed in rats that developed HCC on a cirrhotic background, which is relevant to the clinical situation, and the antitumoral effects occurred in the absence of liver toxicity [128]. These encouraging studies, combined with the biological observations summarized in the previous section, paved the way for the development of several clinical trials. Unfortunately, no responses were found to cetuximab in two phase II studies in patients with advanced HCC (reviewed in [129]), while some activity was observed in a preliminary study combining cetuximab with gemcitabine plus oxaliplatin in poor prognosis patients [130]. Similar trials have tested the efficacy of small TKIs. Gefitinib showed no activity in a single-arm phase II study [129], while lapatinib, a dual inhibitor of ErbB1 and ErbB2, also demonstrated poor activity [131]. Erlotinib is the only agent that so far has shown a modest activity as a single agent in advanced HCC providing disease stabilization [132, 133]. Erlotinib has been also tested in combination with bevacizumab, an antibody that binds and neutralizes the pro-angiogenic factor vascular endothelial growth factor (VEGF). While two studies reported modest but clinically meaningful antitumor activity [134, 135], another indicated a minimal activity based on objective response and progression-free survival [136]. Similarly, a recent study concluded that the combination of erlotinib with sorafenib, a multikinase inhibitor and the only small molecule that has shown clinical benefit in advanced HCC so far [119], did not improve overall survival in comparison to sorafenib alone in patients with advanced HCC (reviewed in [137], [138]). These discouraging observations attest to the molecular complexity of HCC in humans, and the ability of liver cancer cells to develop mechanisms of resistance to these and other targeted therapies. Identification of these resistance mechanisms, and the development of molecular tools to counteract them, could open new avenues to improve the efficacy of EGFR inhibitors for HCC treatment.
Mechanisms of resistance to EGFR targeted therapies in HCC: identification of strategies to increase the efficacy of EGFR inhibitors
Targeted drug resistance has emerged as a major issue in HCC treatment. Even considering the efficacy of sorafenib, the overall clinical benefit of these new drugs is still far from satisfactory. This limited therapeutic success calls for a better understanding of the underlying mechanisms of resistance, and also for the identification of biomarkers that can select potentially responsive patients and monitor the response to treatment. The resistance mechanisms to EGFR-targeted drugs may operate at various cellular and molecular levels, and many of them have been already characterized in other types of solid tumors [139–142] (Fig. 3). For instance, it is becoming clear that the concomitant activation of other signaling systems also governed by growth factors may cause resistance to EGFR-targeted drugs. This has been demonstrated for the insulin-like growth factor 2 (IGF2) and IGF1-R pathway, which inhibition potentiated the antitumoral effect of gefitinib in HCC through the attenuation of IGF-1R-stimulated AR release [143]. Interestingly, this IGF-1R-mediated mechanism of resistance to EGFR targeted therapies also operates in a reciprocal fashion. It was subsequently shown that HCC cells overcame IGF-1R inhibition through ErbB3 activation in an EGFR-dependent manner, and that ErbB3 represented a critical mediator in acquired resistance to anti-IGF-1R therapy [144]. The HGF/c-MET pathway has emerged as another driver system in hepatocarcinogenesis [145]. Functional crosstalks between the EGFR and HGF/c-MET systems have been established for different cancers, providing a rationale for their combinatorial blockade [146]. As previously mentioned, physical interaction between EGFR and c-MET has been observed in tumoral cells, including HCC cells, leading to the activation of c-MET signaling [147]. In addition, a recent interesting study in which HCC cells were also included demonstrated that HGF induced profound changes in EGFR tyrosine kinase activity and EGFR protein interactions in the cell membrane [148]. HGF treatment induced EGFR tyrosine kinase inhibition, and forced this receptor to interact with and stabilize several cancer-related proteins, including the receptor tyrosine kinases EphA2 and Axl, which endow the cells with increased survival capacity even in the presence of anti-EGFR drugs. Importantly, gefitinib sensitivity was restored upon c-MET inhibition [148].

Overview of mechanisms implicated in the resistance to EGFR-targeted therapies. 1 Involvement of the c-MET/HGF system: c-MET amplification and/or c-MET/EGFR interaction, 2 IGF1-R activation and EGFR transactivation, 3 ADAM17 activation and EGFR ligand release (for instance Pro-AR: membrane anchored amphiregulin precursor), 4 increased expression of ErbB ligands, 5 crosstalk with other ErbB receptors, 6 expression of the EGFRvIII mutant, 7 overexpression of ILK and interaction with laminin 5 in the extracellular matrix (ECM), and induction of epithelial mesenchymal transition (EMT), 8 EGFR association with EphrinB1, 9 activation of Ras pathway, 10 overexpression of the phosphatase inhibitor CIP2A, 11 overexpression of the major vault protein (MVP), 12 nuclear activity of EGFR
Another recent study has identified the interaction of the EGFR with the complex system defined by the ephrin ligands and the Eph tyrosine kinase receptors [149]. This work was performed in head and neck squamous cell carcinoma (HNSCC) cells, and demonstrated that in the absence of the cellular protein tyrosine phosphatase PTPN13, EphrinB1, a target of PTPN13, can associate with EGFR promoting ERK1/2 signaling. Most importantly this association was favored in the presence of cetuximab [150]. Although this new EGFR/EphrinB1 crosstalk has not been evaluated in HCC cells, PTPN13 down-regulation and EphrinB1 overexpression are frequently found in HCC tissues [151, 152], suggesting the existence of this pro-tumorigenic and drug resistance mechanism in liver cancer, which may be worth exploring.
One increasingly recognized mechanism of drug resistance, including EGFR-targeted drug resistance, operating in a variety of cancers is the nuclear localization of the EGFR. In the nucleus this receptor has been reported to function not only as tyrosine kinase but also as a transcription factor, and to display pro-tumorigenic effects even in a kinase-independent fashion [153, 154]. Nuclear translocation of the EGFR has been observed in various carcinomas in association with the up-regulation of ErbB ligands and resistance to small TKIs and therapeutic antibodies, which can also promote EGFR nuclear traffic [154]. Interestingly, one of the earliest evidence of the nuclear localization of the EGFR was obtained in the regenerating liver [155, 156], and the nuclear presence of EGFR was associated with DNA synthesis [157]. In view of this, it would be interesting to examine thoroughly the subcellular distribution of EGFR in HCC cells and tissues, particularly in biopsies from patients that were treated with EGFR-inhibitory agents. New strategies are being developed to restore the therapeutic response of cancer cells to EGFR-directed drugs by targeting the nuclear receptor, and HCC treatment could also benefit from these approaches [153, 154].
Another phenomenon associated with resistance to EGFR-targeted agents is epithelial–mesenchymal transition (EMT). It was found that, depending on their EMT status, HCC cells were more or less susceptible to small TKIs and cetuximab, being cells classified as “epithelial” and more responsive to these agents [126]. The enhanced resistance of the “mesenchymal” cell lines was attributed to their ability to activate Akt signaling through alternative mechanisms, independent from EGFR and EGFR/Erbb3 heterodimers. One of the identified mechanisms was governed by the serine/threonine protein kinase integrin-linked kinase (ILK), and consistent inhibition of ILK reduced Akt activation and increased sensitivity to EGFR-targeted drugs [126]. Interestingly, and as pointed out by the authors, ECM components that may drive ILK activation such as laminin-5, have been reported to increase resistance to gefitinib in HCC cells [158]. These studies are illustrative of the role of intracellular signaling pathways activated in HCC, such as ILK [159], and ECM components present in the tumor microenvironment, like laminin-5 [160], in generating resistance towards EGFR-directed agents. Another recent study further confirmed the relevance of AKT activation in gefitinib resistance in HCC cells. In this case the authors identified the major vault protein (MVP), a protein overexpressed in a significant number of HCCs and previously involved in the regulation of intracellular signaling, as a mediator of AKT activation through mechanisms that may require further elucidation [161]. Also within AKT signaling, overexpression of CIP2A (cancerous inhibitor of protein phosphatase 2A) was found to up-regulate phospho-AKT levels and, thus, mediate erlotinib resistance in HCC cells [162]. The Ras and Jak/STAT are other important intracellular pathways downstream of the EGFR that are ubiquitously activated in HCC, providing growth and survival cues to the cancer cells. The persistent activation of these pathways can be mediated by the tumoral microenvironment rich in cytokines and growth factors, but has also been attributed to the epigenetic down-regulation of genes involved in their negative regulation [163]. This later mechanism of Ras and Jak/STAT activation can contribute to resistance towards EGFR inhibitory drugs.
Although it has not been tested in HCC, preclinical and clinical experience in other solid tumors has shown the enhanced antitumoral activity of combining two anti-EGFR agents (reviewed in [120]). Combination of a small EGFR TKI with an EGFR-targeted antibody results in a better inhibition of kinase activity, and a more efficient and broader blockade of downstream signaling pathways. The different mechanisms of action of these two types of agents contribute to explain this enhanced efficacy. This strategy also reduces the crosstalk with other ErbB family members, an important mechanism of resistance not only to ErbB-directed agents [164] but also to other targeted drugs like sorafenib [165].
As previously mentioned, EGFRvIII is a constitutively active EGFR mutant that has been detected in different solid tumors including HCC. Expression of EGFRvIII has been shown to confer resistance to chemotherapeutic agents and also to EGFR-targeted drugs in HNSCC cells [166]. Recent preclinical observations indicate that the EGFRvIII mutant could be a useful target in HCC. CH12, a monoclonal antibody directed to EGFRvIII elicited strong antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity in EGFRvIII expressing HCC cells and reduced the phosphorylation of EGFRvIII and downstream intracellular signaling, leading to cell growth inhibition and apoptosis [167]. Interestingly, EGFRvIII targeting with CH12 also potentiated the antitumoral effects of conventional cytotoxic agents as well as those of sorafenib, suggesting that combination therapies including this antibody could be of clinical value for HCCs that express this EGFR mutant, once this subgroup of patients is clearly identified [168, 169].
Complementary to the multiple targeting of the EGFR signaling system for cancer therapy would be the neutralization of ErbB ligands. Abundant experimental evidence indicates that interference with the shedding of these growth factors using ADAM inhibitors, knockdown of their gene expression or the use of neutralizing antibodies have potent antitumoral effects. This has been clearly demonstrated for ligands such as AR and HB-EGF in different types of tumors [170, 171], and particularly for AR in HCC [110]. Interestingly, interference with AR gene expression overcomes resistance to gefitinib in NSCLC cells [172]. The potential relevance of neutralizing AR in conjunction with EGFR may exceed the mere blockade of a protumorigenic autocrine loop. A recent report identified AR produced by mast cells as a critical factor for the optimal functioning of regulatory T (Treg) cells in vivo [173]. Both mast cells and Treg cells are known to accumulate in the peritumoral region during cancer development, and contribute to the immune-suppressive environment that fosters tumor growth. The finding that mast cells derived AR sustains the functioning of EGFR expressing Treg cells opens the possibility of targeting the AR/EGFR axis to enhance antitumor T cell responses, which could be interesting for tertiary prevention and for the potentiation of HCC immune-therapy [174, 175].
In spite of all these advancements, a key but as yet unresolved issue in the targeted treatment of HCC is the definition of useful biomarkers. These would allow the identification of patient subpopulations that could benefit from treatment, the prioritization of targeting strategies, and the monitoring of treatment responses [176]. Further work is definitively needed in this direction, particularly in a molecularly heterogeneous type of tumor such as HCC.
Abbreviations
- ADAM:
-
A disintegrin and metalloproteinases
- AR:
-
Amphiregulin
- BTC:
-
Betacellulin
- CIP2A:
-
Cancerous inhibitor of protein phosphatase 2A
- CTGF:
-
Connective tissue growth factor
- ECM:
-
Extracellular matrix
- EGFR:
-
Epidermal growth factor receptor
- EPG:
-
Epigen
- EREG:
-
Epiregulin
- ERK:
-
Extracellular signal-regulated kinase
- FGF19:
-
Fibroblast growth factor 19
- GAP:
-
GTPase-activating protein
- GPCR:
-
G protein-coupled receptors
- HB-EGF:
-
Heparin binding epidermal growth factor like
- HCC:
-
Hepatocellular carcinoma
- HGF:
-
Hepatocyte growth factor
- HNSCC:
-
Head and neck squamous cell carcinoma
- IGF1-R:
-
Insulin like growth factor 1 receptor
- IGF2:
-
Insulin like growth factor 2
- ILK:
-
Integrin linked kinase
- MIG-6:
-
Mitogen-inducible gene-6
- MUC15:
-
Mucin 15
- MVP:
-
Major vault protein
- NRG:
-
Neuregulin
- NSCLC:
-
Non-small-cell lung carcinoma
- PDGFR:
-
Platelet derived growth factor receptor
- PI3 K:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- PTPN13:
-
Protein tyrosine phosphatase non-receptor 13
- STAT:
-
Signal-transducer and activator of transcription
- TACE:
-
TNF-alpha converting enzyme
- TGFα:
-
Transforming growth factor-α
- TGFβ:
-
Transforming growth factor-β
- TKI:
-
Tyrosine kinase inhibitor
- TLR3:
-
Toll like receptor 3
- TRAIL:
-
TNF-related apoptosis inducing ligand
References
Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;20(110):669–72.
Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–16.
Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J Cell Physiol. 2009;218:460–6.
Jorissen RN, Walker F, Pouliot N, Garrett TPJ, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res. 2003;10(284):31–53.
Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;17(366):2–16.
Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci USA. 2010;27(107):7692–7.
Soderquist AM, Todderud G, Carpenter G. The role of carbohydrate as a post-translational modification of the receptor for epidermal growth factor. Adv Exp Med Biol. 1988;231:569–82.
Bishayee S. Role of conformational alteration in the epidermal growth factor receptor (EGFR) function. Biochem Pharmacol. 2000;15(60):1217–23.
Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol. 2004;44:195–217.
Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer. 2001;8:11–31.
Cavin LG, Wang F, Factor VM, Kaur S, Venkatraman M, Thorgeirsson SS, et al. Transforming growth factor-alpha inhibits the intrinsic pathway of c-Myc-induced apoptosis through activation of nuclear factor-kappaB in murine hepatocellular carcinomas. Mol Cancer Res. 2005;3:403–12.
Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation. 2007;75:770–87.
Andreu-Pérez P, Esteve-Puig R, de Torre-Minguela C, López-Fauqued M, Bech-Serra JJ, Tenbaum S, et al. Protein arginine methyltransferase 5 regulates ERK1/2 signal transduction amplitude and cell fate through CRAF. Sci Signal. 2011;4:ra58.
Segatto O, Anastasi S, Alemà S. Regulation of epidermal growth factor receptor signalling by inducible feedback inhibitors. J Cell Sci. 2011;1(124):1785–93.
Zhang C, Li A, Zhang X, Xiao H. A novel TIP30 protein complex regulates EGF receptor signaling and endocytic degradation. J Biol Chem. 2011;18(286):9373–81.
Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12:104–17.
Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res. 2003;10(284):2–13.
Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32–43.
Blobel CP, Carpenter G, Freeman M. The role of protease activity in ErbB biology. Exp Cell Res. 2009;315:671–82.
Adrain C, Strisovsky K, Zettl M, Hu L, Lemberg MK, Freeman M. Mammalian EGF receptor activation by the rhomboid protease RHBDL2. EMBO Rep. 2011;12:421–7.
Nishi E, Klagsbrun M. Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is a mediator of multiple physiological and pathological pathways. Growth Factors. 2004;22:253–60.
Stoll SW, Johnson JL, Li Y, Rittié L, Elder JT. Amphiregulin carboxy-terminal domain is required for autocrine keratinocyte growth. J Investig Dermatol. 2010;130:2031–40.
Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol. 2006;291:C1–10.
Almendro V, García-Recio S, Gascón P. Tyrosine kinase receptor transactivation associated to G protein-coupled receptors. Curr Drug Targets. 2010;11:1169–80.
Liebmann C. EGF receptor activation by GPCRs: an universal pathway reveals different versions. Mol Cell Endocrinol. 2011;331:222–31.
Berasain C, Perugorría MJ, Latasa MU, Castillo J, Goñi S, Santamaría M, et al. The epidermal growth factor receptor: a link between inflammation and liver cancer. Exp Biol Med (Maywood). 2009;234:713–25.
Rodland KD, Bollinger N, Ippolito D, Opresko LK, Coffey RJ, Zangar R, et al. Multiple mechanisms are responsible for transactivation of the epidermal growth factor receptor in mammary epithelial cells. J Biol Chem. 2008;14(283):31477–87.
Berasain C, Castillo J, Perugorría MJ, Latasa MU, Prieto J, Avila MA. Inflammation and liver cancer. Ann N Y Acad Sci. 2009;26(1155):206–21.
Murphy G. Regulation of the proteolytic disintegrin metalloproteinases, the ‘Sheddases’. Semin Cell Dev Biol. 2009;20:138–45.
Dang M, Armbruster N, Miller MA, Cermeno E, Hartmann M, Bell GW, et al. Regulated ADAM17-dependent EGF family ligand release by substrate-selecting signaling pathways. Proc Natl Acad Sci USA. 2013;11(110):9776–81.
Berasain C, Ujue Latasa M, Urtasun R, Goñi S, Elizalde M, Garcia-Irigoyen O, et al. Epidermal growth factor receptor (EGFR) crosstalks in liver cancer. Cancers. 2011;3:2444–61.
Yamashita M, Chattopadhyay S, Fensterl V, Saikia P, Wetzel JL, Sen GC. Epidermal growth factor receptor is essential for Toll-like receptor 3 signaling. Sci Signal. 2012;5:ra50.
Wilson KJ, Mill C, Lambert S, Buchman J, Wilson TR, Hernandez-Gordillo V, et al. EGFR ligands exhibit functional differences in models of paracrine and autocrine signaling. Growth Factors. 2012;30:107–16.
Saito T, Okada S, Ohshima K, Yamada E, Sato M, Uehara Y, et al. Differential activation of epidermal growth factor (EGF) receptor downstream signaling pathways by betacellulin and EGF. Endocrinology. 2004;145:4232–43.
Gilmore JL, Scott JA, Bouizar Z, Robling A, Pitfield SE, Riese DJ, et al. Amphiregulin-EGFR signaling regulates PTHrP gene expression in breast cancer cells. Breast Cancer Res Treat. 2008;110:493–505.
Stern KA, Place TL, Lill NL. EGF and amphiregulin differentially regulate Cbl recruitment to endosomes and EGF receptor fate. Biochem J. 2008;15(410):585–94.
Nakakuki T, Birtwistle MR, Saeki Y, Yumoto N, Ide K, Nagashima T, et al. Ligand-specific c-Fos expression emerges from the spatiotemporal control of ErbB network dynamics. Cell. 2010;28(141):884–96.
Lemos-González Y, Rodríguez-Berrocal FJ, Cordero OJ, Gómez C, Páez de la Cadena M. Alteration of the serum levels of the epidermal growth factor receptor and its ligands in patients with non-small cell lung cancer and head and neck carcinoma. Br J Cancer. 2007;21(96):1569–78.
Révillion F, Lhotellier V, Hornez L, Bonneterre J, Peyrat J-P. ErbB/HER ligands in human breast cancer, and relationships with their receptors, the bio-pathological features and prognosis. Ann Oncol. 2008;19:73–80.
Tanabe KK, Lemoine A, Finkelstein DM, Kawasaki H, Fujii T, Chung RT, et al. Epidermal growth factor gene functional polymorphism and the risk of hepatocellular carcinoma in patients with cirrhosis. JAMA. 2008;2(299):53–60.
Abu Dayyeh BK, Yang M, Fuchs BC, Karl DL, Yamada S, Sninsky JJ, et al. A functional polymorphism in the epidermal growth factor gene is associated with risk for hepatocellular carcinoma. Gastroenterology. 2011;141:141–9.
Michalopoulos GK. Liver regeneration after partial hepatectomy: critical analysis of mechanistic dilemmas. Am J Pathol. 2010;176:2–13.
Böhm F, Köhler UA, Speicher T, Werner S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol Med. 2010;22(2):294–305.
Gilgenkrantz H, de l’Hortet AC. New insights into liver regeneration. Clin Res Hepatol Gastroenterol. 2011;35:623–9.
Fausto N, Campbell JS, Riehle KJ. Liver regeneration. J Hepatol. 2012;57:692–4.
Uriarte I, Fernández-Barrena MG, Monte MJ, Latasa MU, Chang HCY, Carotti S, et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut. 2013;62:899–910.
Wang T, Takikawa Y, Tabuchi T, Satoh T, Kosaka K, Suzuki K. Carnosic acid (CA) prevents lipid accumulation in hepatocytes through the EGFR/MAPK pathway. J Gastroenterol. 2012;47:805–13.
Carver RS, Stevenson MC, Scheving LA, Russell WE. Diverse expression of ErbB receptor proteins during rat liver development and regeneration. Gastroenterology. 2002;123:2017–27.
Webber EM, FitzGerald MJ, Brown PI, Bartlett MH, Fausto N. Transforming growth factor-alpha expression during liver regeneration after partial hepatectomy and toxic injury, and potential interactions between transforming growth factor-alpha and hepatocyte growth factor. Hepatology. 1993;18:1422–31.
Kiso S, Kawata S, Tamura S, Higashiyama S, Ito N, Tsushima H, et al. Role of heparin-binding epidermal growth factor-like growth factor as a hepatotrophic factor in rat liver regeneration after partial hepatectomy. Hepatology. 1995;22:1584–90.
Stolz DB, Mars WM, Petersen BE, Kim TH, Michalopoulos GK. Growth factor signal transduction immediately after two-thirds partial hepatectomy in the rat. Cancer Res. 1999;15(59):3954–60.
Lee D, Pearsall RS, Das S, Dey SK, Godfrey VL, Threadgill DW. Epiregulin is not essential for development of intestinal tumors but is required for protection from intestinal damage. Mol Cell Biol. 2004;24:8907–16.
Berasain C, García-Trevijano ER, Castillo J, Erroba E, Lee DC, Prieto J, et al. Amphiregulin: an early trigger of liver regeneration in mice. Gastroenterology. 2005;128:424–32.
Lin X-M, Liu Y-B, Zhou F, Wu Y-L, Chen L, Fang H-Q. Expression of tumor necrosis factor-alpha converting enzyme in liver regeneration after partial hepatectomy. World J Gastroenterol. 2008;7(14):1353–7.
Berasain C, García-Trevijano ER, Castillo J, Erroba E, Santamaría M, Lee DC, et al. Novel role for amphiregulin in protection from liver injury. J Biol Chem. 2005;13(280):19012–20.
Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286–300.
Berasain C, Castillo J, Prieto J, Avila MA. New molecular targets for hepatocellular carcinoma: the ErbB1 signaling system. Liver Int. 2007;27:174–85.
Collin de L’hortet A, Gilgenkrantz H, Guidotti J-E. EGFR: a master piece in G1/S phase transition of liver regeneration. Int J Hepatol. 2012;2012:476910.
Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci USA. 2007;23(104):17081–6.
Paranjpe S, Bowen WC, Tseng GC, Luo J-H, Orr A, Michalopoulos GK. RNA interference against hepatic epidermal growth factor receptor has suppressive effects on liver regeneration in rats. Am J Pathol. 2010;25(176):2669–81.
Zerrad-Saadi A, Lambert-Blot M, Mitchell C, Bretes H, Collin de L’hortet A, Baud V, et al. GH receptor plays a major role in liver regeneration through the control of EGFR and ERK1/2 activation. Endocrinology. 2011;152:2731–41.
Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SPS. Conditional deletion of beta-catenin reveals its role in liver growth and regeneration. Gastroenterology. 2006;131:1561–72.
Murthy A, Defamie V, Smookler DS, Di Grappa MA, Horiuchi K, Federici M, et al. Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J Clin Investig. 2010;120:2731–44.
Avila MA. Long distance calling for liver regeneration: identification of neuroendocrine signalling pathways activated after partial hepatectomy. J Hepatol. 2011;54:403–5.
Russell WE, Kaufmann WK, Sitaric S, Luetteke NC, Lee DC. Liver regeneration and hepatocarcinogenesis in transforming growth factor-alpha-targeted mice. Mol Carcinog. 1996;15:183–9.
Mitchell C, Nivison M, Jackson LF, Fox R, Lee DC, Campbell JS, et al. Heparin-binding epidermal growth factor-like growth factor links hepatocyte priming with cell cycle progression during liver regeneration. J Biol Chem. 2005;28(280):2562–8.
Pardo-Saganta A, Pardo-Saganta A, Latasa MU, Latasa MU, Castillo J, Castillo J, et al. The epidermal growth factor receptor ligand amphiregulin is a negative regulator of hepatic acute-phase gene expression. J Hepatol. 2009;51:1010–20.
Santamaría M, Pardo-Saganta A, Alvarez-Asiain L, Di Scala M, Qian C, Prieto J, et al. Nuclear α1-antichymotrypsin promotes chromatin condensation and inhibits proliferation of human hepatocellular carcinoma cells. Gastroenterology. 2013;144:818–28.
Liu Q, Rehman H, Krishnasamy Y, Haque K, Schnellmann RG, Lemasters JJ, et al. Amphiregulin stimulates liver regeneration after small-for-size mouse liver transplantation. Am J Transplant. 2012;12(8):2052–61.
Sakuda S, Tamura S, Yamada A, Miyagawa J-I, Yamamoto K, Kiso S, et al. NF-kappaB activation in non-parenchymal liver cells after partial hepatectomy in rats: possible involvement in expression of heparin-binding epidermal growth factor-like growth factor. J Hepatol. 2002;36:527–33.
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;22(365):1118–27.
White DL, Kanwal F, El-Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol. 2012;10:1342–59.
Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47(Suppl):S2–6.
Avila MA, Berasain C, Torres L, Martin-Duce A, Corrales FJ, Yang H, et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol. 2000;33:907–14.
Berasain C, Herrero J-I, García-Trevijano ER, Avila MA, Esteban JI, Mato JM, et al. Expression of Wilms’ tumor suppressor in the liver with cirrhosis: relation to hepatocyte nuclear factor 4 and hepatocellular function. Hepatology. 2003;38:148–57.
Aravalli RN, Cressman ENK, Steer CJ. Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Arch Toxicol. 2013;87:227–47.
Gea VH, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144(3):512–27.
Nikolaou K, Sarris M, Talianidis I. Molecular pathways: the complex roles of inflammation pathways in the development and treatment of liver cancer. Clin Cancer Res. 2013;1(19):2810–6.
Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int. 2013;2013:187204.
Zhang DY, Friedman SL. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology. 2012;56:769–75.
Castillo J, Goñi S, Latasa MU, Perugorría MJ, Calvo A, Muntané J, et al. Amphiregulin induces the alternative splicing of p73 into its oncogenic isoform DeltaEx2p73 in human hepatocellular tumors. Gastroenterology. 2009;137:1805–15.
Fujii T, Fuchs BC, Yamada S, Lauwers GY, Kulu Y, Goodwin JM, et al. Mouse model of carbon tetrachloride induced liver fibrosis: histopathological changes and expression of CD133 and epidermal growth factor. BMC Gastroenterol. 2010;10:79.
Pinzani M, Gesualdo L, Sabbah GM, Abboud HE. Effects of platelet-derived growth factor and other polypeptide mitogens on DNA synthesis and growth of cultured rat liver fat-storing cells. J Clin Investig. 1989;84:1786–93.
Gressner AM. Cytokines and cellular crosstalk involved in the activation of fat-storing cells. J Hepatol. 1995;22:28–36.
Yang C, Zeisberg M, Mosterman B, Sudhakar A, Yerramalla U, Holthaus K, et al. Liver fibrosis: insights into migration of hepatic stellate cells in response to extracellular matrix and growth factors. Gastroenterology. 2003;124:147–59.
Perugorría MJ, Perugorría MJ, Latasa MU, Latasa MU, Nicou A, Nicou A, et al. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology. 2008;48:1251–61.
Zhou Y, Lee JY, Lee CM, Cho WK, Kang MJ, Koff JL, et al. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-β-induced pulmonary fibrosis. J Biol Chem. 2012;7(287):41991–2000.
Liu N, Guo J-K, Pang M, Tolbert E, Ponnusamy M, Gong R, et al. Genetic or pharmacologic blockade of EGFR inhibits renal fibrosis. J Am Soc Nephrol. 2012;23:854–67.
Samarakoon R, Dobberfuhl AD, Cooley C, Overstreet JM, Patel S, Goldschmeding R, et al. Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal. 2013;25:2198–209.
Tang J, Liu N, Tolbert E, Ponnusamy M, Ma L, Gong R, et al. Sustained activation of EGFR triggers renal fibrogenesis after acute kidney injury. Am J Pathol. 2013;183:160–72.
Tzouvelekis A, Ntolios P, Karameris A, Vilaras G, Boglou P, Koulelidis A, et al. Increased expression of epidermal growth factor receptor (EGF-R) in patients with different forms of lung fibrosis. Biomed Res Int. 2013;2013:654354.
Takemura T, Yoshida Y, Kiso S, Kizu T, Furuta K, Ezaki H, et al. Conditional loss of heparin-binding EGF-like growth factor results in enhanced liver fibrosis after bile duct ligation in mice. Biochem Biophys Res Commun. 2013;26(437):185–91.
Takemura T, Yoshida Y, Kiso S, Saji Y, Ezaki H, Hamano M, et al. Conditional knockout of heparin-binding epidermal growth factor-like growth factor in the liver accelerates carbon tetrachloride-induced liver injury in mice. Hepatol Res. 2013;43:384–93.
Ito Y, Takeda T, Sakon M, Tsujimoto M, Higashiyama S, Noda K, et al. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br J Cancer. 2001;18(84):1377–83.
Buckley AF, Burgart LJ, Sahai V, Kakar S. Epidermal growth factor receptor expression and gene copy number in conventional hepatocellular carcinoma. Am J Clin Pathol. 2008;129:245–51.
Kira S, Nakanishi T, Suemori S, Kitamoto M, Watanabe Y, Kajiyama G. Expression of transforming growth factor alpha and epidermal growth factor receptor in human hepatocellular carcinoma. Liver. 1997;17:177–82.
Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;26(25):3787–800.
Keng VW, Sia D, Sarver AL, Tschida BR, Fan D, Alsinet C, et al. Sex bias occurrence of hepatocellular carcinoma in Poly7 molecular subclass is associated with EGFR. Hepatology. 2013;57:120–30.
Ou C, Wu F-X, Luo Y, Cao J, Zhao Y-N, Yuan W-P, et al. Expression and significance of epidermal growth factor receptor variant type III in hepatocellular carcinoma. Ai Zheng. 2005;24:166–9.
Wang H, Jiang H, Zhou M, Xu Z, Liu S, Shi B, et al. Epidermal growth factor receptor vIII enhances tumorigenicity and resistance to 5-fluorouracil in human hepatocellular carcinoma. Cancer Lett. 2009;28(279):30–8.
Zhou M, Gong B, Gu J, Li Z. EGFRvIII mRNA detection in the serum of patients with hepatocellular carcinoma. Liver Int. 2010;30:925–7.
Hsieh S-Y, He J-R, Hsu C-Y, Chen W-J, Bera R, Lin K-Y, et al. Neuregulin/erythroblastic leukemia viral oncogene homolog 3 autocrine loop contributes to invasion and early recurrence of human hepatoma. Hepatology. 2011;53:504–16.
Hsieh S-Y, He J-R, Yu M-C, Lee W-C, Chen T-C, Lo S-J, et al. Secreted ERBB3 isoforms are serum markers for early hepatoma in patients with chronic hepatitis and cirrhosis. J Proteome Res. 2011;7(10):4715–24.
Harada K, Shiota G, Kawasaki H. Transforming growth factor-alpha and epidermal growth factor receptor in chronic liver disease and hepatocellular carcinoma. Liver. 1999;19:318–25.
Kömüves LG, Feren A, Jones AL, Fodor E. Expression of epidermal growth factor and its receptor in cirrhotic liver disease. J Histochem Cytochem. 2000;48:821–30.
Castillo J, Erroba E, Perugorría MJ, Santamaría M, Lee DC, Prieto J, et al. Amphiregulin contributes to the transformed phenotype of human hepatocellular carcinoma cells. Cancer Res. 2006;15(66):6129–38.
Moon WS, Park HS, Yu KH, Park MY, Kim KR, Jang KY, et al. Expression of betacellulin and epidermal growth factor receptor in hepatocellular carcinoma: implications for angiogenesis. Hum Pathol. 2006;37:1324–32.
Daveau M, Scotte M, François A, Coulouarn C, Ros G, Tallet Y, et al. Hepatocyte growth factor, transforming growth factor alpha, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol Carcinog. 2003;36:130–41.
Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;6(359):1995–2004.
Berasain C, Castillo J, Perugorría MJ, Prieto J, Avila MA. Amphiregulin: a new growth factor in hepatocarcinogenesis. Cancer Lett. 2007;254:30–41.
Ortiz C, Caja L, Sancho P, Bertran E, Fabregat I. Inhibition of the EGF receptor blocks autocrine growth and increases the cytotoxic effects of doxorubicin in rat hepatoma cells: role of reactive oxygen species production and glutathione depletion. Biochem Pharmacol. 2008;15(75):1935–45.
Sancho P, Bertran E, Caja L, Carmona-Cuenca I, Murillo MM, Fabregat I. The inhibition of the epidermal growth factor (EGF) pathway enhances TGF-beta-induced apoptosis in rat hepatoma cells through inducing oxidative stress coincident with a change in the expression pattern of the NADPH oxidases (NOX) isoforms. Biochim Biophys Acta. 2009;1793:253–63.
Mazzocca A, Fransvea E, Dituri F, Lupo L, Antonaci S, Giannelli G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology. 2010;51:523–34.
Urtasun R, Urtasun R, Latasa MU, Latasa MU, Demartis MI, Demartis MI, et al. Connective tissue growth factor autocriny in human hepatocellular carcinoma: oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology. 2011;54:2149–58.
Latasa MU, Salis F, Urtasun R, Garcia-Irigoyen O, Elizalde M, Uriarte I, et al. Regulation of amphiregulin gene expression by β-catenin signaling in human hepatocellular carcinoma cells: a novel crosstalk between FGF19 and the EGFR system. PLoS ONE. 2012;7:e52711.
Reschke M, Ferby I, Stepniak E, Seitzer N, Horst D, Wagner EF, et al. Mitogen-inducible gene-6 is a negative regulator of epidermal growth factor receptor signaling in hepatocytes and human hepatocellular carcinoma. Hepatology. 2010;51:1383–90.
Wang R-Y, Chen L, Chen H-Y, Hu L, Li L, Sun H-Y, et al. MUC15 inhibits dimerization of EGFR and PI3K–AKT signaling and is associated with aggressive hepatocellular carcinomas in patients. Gastroenterology. 2013.
Avila MA, Berasain C, Sangro B, Prieto J. New therapies for hepatocellular carcinoma. Oncogene. 2006;26(25):3866–84.
Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140:1410–26.
Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer. 2013;13:663–73.
Matsuo M, Sakurai H, Saiki I. ZD1839, a selective epidermal growth factor receptor tyrosine kinase inhibitor, shows antimetastatic activity using a hepatocellular carcinoma model. Mol Cancer Ther. 2003;2:557–61.
Huether A, Höpfner M, Baradari V, Schuppan D, Scherübl H. EGFR blockade by cetuximab alone or as combination therapy for growth control of hepatocellular cancer. Biochem Pharmacol. 2005;25(70):1568–78.
Huether A, Höpfner M, Sutter AP, Schuppan D, Scherübl H. Erlotinib induces cell cycle arrest and apoptosis in hepatocellular cancer cells and enhances chemosensitivity towards cytostatics. J Hepatol. 2005;43:661–9.
Liu Y, Poon RTP, Shao W, Sun X, Chen H, Kok TW, et al. Blockage of epidermal growth factor receptor by quinazoline tyrosine kinase inhibitors suppresses growth of human hepatocellular carcinoma. Cancer Lett. 2007;8(248):32–40.
Giannelli G, Sgarra C, Porcelli L, Azzariti A, Antonaci S, Paradiso A. EGFR and VEGFR as potential target for biological therapies in HCC cells. Cancer Lett. 2008;18(262):257–64.
Fuchs BC, Fujii T, Dorfman JD, Goodwin JM, Zhu AX, Lanuti M, et al. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008;1(68):2391–9.
Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972–83.
Schiffer E, Housset C, Cacheux W, Wendum D, Desbois-Mouthon C, Rey C, et al. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology. 2005;41:307–14.
Zhu AX. New agents on the horizon in hepatocellular carcinoma. Ther Adv Med Oncol. 2013;5:41–50.
Asnacios A, Fartoux L, Romano O, Tesmoingt C, Louafi SS, Mansoubakht T, et al. Gemcitabine plus oxaliplatin (GEMOX) combined with cetuximab in patients with progressive advanced stage hepatocellular carcinoma: results of a multicenter phase 2 study. Cancer. 2008;15(112):2733–9.
Ramanathan RK, Belani CP, Singh DA, Tanaka M, Lenz H-J, Yen Y, et al. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer Chemother Pharmacol. 2009;64:777–83.
Philip PA, Mahoney MR, Allmer C, Thomas J, Pitot HC, Kim G, et al. Phase II study of Erlotinib (OSI-774) in patients with advanced hepatocellular cancer. J Clin Oncol. 2005;20(23):6657–63.
Thomas MB, Chadha R, Glover K, Wang X, Morris J, Brown T, et al. Phase 2 study of erlotinib in patients with unresectable hepatocellular carcinoma. Cancer. 2007;1(110):1059–67.
Thomas MB, Morris JS, Chadha R, Iwasaki M, Kaur H, Lin E, et al. Phase II trial of the combination of bevacizumab and erlotinib in patients who have advanced hepatocellular carcinoma. J Clin Oncol. 2009;20(27):843–50.
Hsu C-H, Kang YK, Yang T-S, Shun C-T, Shao Y–Y, Su W-C, et al. Bevacizumab with erlotinib as first-line therapy in Asian patients with advanced hepatocellular carcinoma: a multicenter phase II study. Oncology. 2013;85:44–52.
Philip PA, Mahoney MR, Holen KD, Northfelt DW, Pitot HC, Picus J, et al. Phase 2 study of bevacizumab plus erlotinib in patients with advanced hepatocellular cancer. Cancer. 2012;1(118):2424–30.
Dekervel J, van Pelt J, Verslype C. Advanced unresectable hepatocellular carcinoma: new biologics as fresh ammunition or clues to disease understanding? Curr Opin Oncol. 2013;25:409–16.
Finn RS. Emerging targeted strategies in advanced hepatocellular carcinoma. Semin Liver Dis. 2013;33(Suppl 1):S11–9.
Ratushny V, Astsaturov I, Burtness BA, Golemis EA, Silverman JS. Targeting EGFR resistance networks in head and neck cancer. Cell Signal. 2009;21:1255–68.
Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol. 2010;7:493–507.
Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther. 2011;1(11):777–92.
Yamaguchi H, Chang S–S, Hsu JL, Hung M-C. Signaling cross-talk in the resistance to HER family receptor targeted therapy. Oncogene. 2013.
Desbois-Mouthon C, Cacheux W, Blivet-Van Eggelpoël M-J, Barbu V, Fartoux L, Poupon R, et al. Impact of IGF-1R/EGFR cross-talks on hepatoma cell sensitivity to gefitinib. Int J Cancer. 2006;1(119):2557–66.
Desbois-Mouthon C, Baron A, Blivet-Van Eggelpoël M-J, Fartoux L, Venot C, Bladt F, et al. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin Cancer Res. 2009;1(15):5445–56.
Giordano S, Columbano A. Met as a therapeutic target in HCC: facts and hopes. J Hepatol. 2013.
Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG. Targeting MET as a strategy to overcome crosstalk-related resistance to EGFR inhibitors. Lancet Oncol. 2009;10:709–17.
Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem. 2000;24(275):8806–11.
Gusenbauer S, Vlaicu P, Ullrich A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene. 2013;15(32):3846–56.
Lisabeth EM, Falivelli G, Pasquale EB. Eph receptor signaling and ephrins. Cold Spring Harbor Perspect Biol. 2013;5(9):1–20.
Vermeer PD, Colbert PL, Wieking BG, Vermeer DW, Lee JH. Targeting ERBB receptors shifts their partners and triggers persistent ERK signaling through a novel ERBB/EFNB1 complex. Cancer Res. 2013;15(73):5787–97.
Yeh S-H, Wu D-C, Tsai C-Y, Kuo T-J, Yu W-C, Chang Y-S, et al. Genetic characterization of fas-associated phosphatase-1 as a putative tumor suppressor gene on chromosome 4q21.3 in hepatocellular carcinoma. Clin Cancer Res. 2006;15(12):1097–108.
Sawai Y, Tamura S, Fukui K, Ito N, Imanaka K, Saeki A, et al. Expression of ephrin-B1 in hepatocellular carcinoma: possible involvement in neovascularization. J Hepatol. 2003;39:991–6.
Han W, Lo H-W. Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett. 2012;28(318):124–34.
Brand TM, Iida M, Luthar N, Starr MM, Huppert EJ, Wheeler DL. Nuclear EGFR as a molecular target in cancer. Radiother Oncol. 2013.
Raper SE, Burwen SJ, Barker ME, Jones AL. Translocation of epidermal growth factor to the hepatocyte nucleus during rat liver regeneration. Gastroenterology. 1987;92:1243–50.
Marti U, Burwen SJ, Wells A, Barker ME, Huling S, Feren AM, et al. Localization of epidermal growth factor receptor in hepatocyte nuclei. Hepatology. 1991;13:15–20.
Schausberger E, Eferl R, Parzefall W, Chabicovsky M, Breit P, Wagner EF, et al. Induction of DNA synthesis in primary mouse hepatocytes is associated with nuclear pro-transforming growth factor alpha and erbb-1 and is independent of c-jun. Carcinogenesis. 2003;24:835–41.
Giannelli G, Azzariti A, Fransvea E, Porcelli L, Antonaci S, Paradiso A. Laminin-5 offsets the efficacy of gefitinib (“Iressa”) in hepatocellular carcinoma cells. Br J Cancer. 2004;29(91):1964–9.
Chan J, Ko FCF, Yeung Y-S, Ng IO-L, Yam JWP. Integrin-linked kinase overexpression and its oncogenic role in promoting tumorigenicity of hepatocellular carcinoma. PLoS ONE. 2011;6:e16984.
Bergamini C, Sgarra C, Trerotoli P, Lupo L, Azzariti A, Antonaci S, et al. Laminin-5 stimulates hepatocellular carcinoma growth through a different function of alpha6beta4 and alpha3beta1 integrins. Hepatology. 2007;46:1801–9.
Losert A, Lötsch D, Lackner A, Koppensteiner H, Peter-Vörösmarty B, Steiner E, et al. The major vault protein mediates resistance to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Lett. 2012;28(319):164–72.
Yu H-C, Chen H-J, Chang Y-L, Liu C-Y, Shiau C-W, Cheng A-L, et al. Inhibition of CIP2A determines erlotinib-induced apoptosis in hepatocellular carcinoma. Biochem Pharmacol. 2013;1(85):356–66.
Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee J-S, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130:1117–28.
Huang S, Li C, Armstrong EA, Peet CR, Saker J, Amler LC, et al. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2013;15(73):824–33.
Blivet-Van Eggelpoël MJ, Chettouh H, Fartoux L, Aoudjehane L, Barbu V, Rey C, et al. Epidermal growth factor receptor and HER-3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J Hepatol. 2012;57(1):108–15.
Sok JC, Coppelli FM, Thomas SM, Lango MN, Xi S, Hunt JL, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res. 2006;1(12):5064–73.
Jiang H, Wang H, Tan Z, Hu S, Wang H, Shi B, et al. Growth suppression of human hepatocellular carcinoma xenografts by a monoclonal antibody CH12 directed to epidermal growth factor receptor variant III. J Biol Chem. 2011;18(286):5913–20.
Yang Y, Jiang H, Gao H, Kong J, Zhang P, Hu S, et al. The monoclonal antibody CH12 enhances the sorafenib-mediated growth inhibition of hepatocellular carcinoma xenografts expressing epidermal growth factor receptor variant III. Neoplasia. 2012;14:509–18.
Jiang H, Dong Q, Luo X, Shi B, Wang H, Gao H, et al. The monoclonal antibody CH12 augments 5-fluorouracil-induced growth suppression of hepatocellular carcinoma xenografts expressing epidermal growth factor receptor variant III. Cancer Lett. 2013.
Miyamoto S, Fukami T, Yagi H, Kuroki M, Yotsumoto F. Potential for molecularly targeted therapy against epidermal growth factor receptor ligands. Anticancer Res. 2009;29:823–30.
Busser B, Sancey L, Brambilla E, Coll J-L, Hurbin A. The multiple roles of amphiregulin in human cancer. Biochim Biophys Acta. 2011;1816:119–31.
Busser B, Sancey L, Josserand V, Niang C, Favrot MC, Coll J-L, et al. Amphiregulin promotes BAX inhibition and resistance to gefitinib in non-small-cell lung cancers. Mol Ther. 2010;18:528–35.
Zaiss DMW, van Loosdregt J, Gorlani A, Bekker CPJ, Gröne A, Sibilia M, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity. 2013;38:275–84.
Hoshida Y, Fuchs BC, Tanabe KK. Prevention of hepatocellular carcinoma: potential targets, experimental models, and clinical challenges. Curr Cancer Drug Targets. 2012;1(12):1129–59.
Pardee AD, Butterfield LH. Immunotherapy of hepatocellular carcinoma: unique challenges and clinical opportunities. Oncoimmunology. 2012;1(1):48–55.
Villanueva A. Rethinking future development of molecular therapies in hepatocellular carcinoma: a bottom-up approach. J Hepatol. 2013;59:392–5.
Acknowledgments
Work in the authors’ laboratory is supported by the agreement between FIMA and the “UTE project CIMA”; RTICC-RD06 00200061; CIBEREhd; and FIS Grants PI10/02642 and PI10/00038 from Instituto de Salud Carlos III, Spain.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Berasain, C., Avila, M.A. The EGFR signalling system in the liver: from hepatoprotection to hepatocarcinogenesis. J Gastroenterol 49, 9–23 (2014). https://doi.org/10.1007/s00535-013-0907-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-013-0907-x