
Abstract
Background
We investigated the effects of dental infection with Porphyromonas gingivalis (P.g.), an important periodontal pathogen, on NASH progression, by feeding mice a high fat diet (HFD)and examining P.g. infection in the liver of NASH patients.
Methods
C57BL/6J mice were fed either chow-diet (CD) or HFD for 12 weeks, and then half of the mice in each group were infected with P.g. from the pulp chamber (HFD-P.g.(−), HFD-P.g.(+), CD-P.g.(−) and CD-P.g.(+)). Histological and immunohistochemical examinations, measurement of serum lipopolysaccharide (LPS) levels and ELISA for cytokines in the liver were performed. We then studied the effects of LPS from P.g. (P.g.-LPS) on palmitate-induced steatotic hepatocytes in vitro, and performed immunohistochemical detection of P.g. in liver biopsy specimens of NASH patients.
Results
Serum levels of LPS are upregulated in P.g.(+) groups. Steatosis of the liver developed in HFD groups, and foci of Mac2-positive macrophages were prominent in HFD-P.g.(+). P.g. was detected in Kupffer cells and hepatocytes. Interestingly, areas of fibrosis with proliferation of hepatic stellate cells and collagen formation were only observed in HFD-P.g.(+). In steatotic hepatocytes, expression of TLR2, one of the P.g.-LPS receptors, was upregulated. P.g.-LPS further increased mRNA levels of palmitate-induced inflammasome and proinflammatory cytokines in steatotic hepatocytes. We demonstrated for the first time that P.g. existed in the liver of NASH patients with advanced fibrosis.
Conclusions
Dental infection of P.g. may play an important role in NASH progression through upregulation of the P.g.-LPS-TLR2 pathway and activation of inflammasomes. Therefore, preventing and/or eliminating P.g. infection by dental therapy may have a beneficial impact on management of NASH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is one of the major chronic liver diseases in adults and children, affecting over 30 % of the population in Western countries [1–3]. NAFLD is strongly associated with obesity, metabolic syndrome, and diabetes mellitus type 2 [1, 2, 4]. NAFLD shows a wide histopathological spectrum of liver injury including steatosis and steatohepatitis with or without fibrosis (non-alcoholic steatohepatitis; NASH) [5]. In 1998, the two-hit hypothesis of NASH pathogenesis was proposed [6]. The first hit involves fat accumulation in the liver as a result of excessive delivery of free fatty acids (FFA) and imbalance of lipid synthesis and export in hepatocytes. The second hit involves oxidative stress caused by factors that enhance the production of reactive oxygen species [6, 7]. Growing evidence indicates that lipopolysaccharides (LPS) originating from the gastrointestinal tract can act as a second hit [8–12].
Porphyromonas gingivalis (P.g.), one of the most important periodontal pathogens, is detected not only in biofilm in periodontal pockets but also as a major bacteria in infected pulp chambers with periapical periodontal diseases [13, 14]. P.g. is known to enter the blood circulation and is disseminated throughout the body. It is considered to be a confounding risk factor for systemic diseases such as cardiovascular disease, diabetes mellitus, preterm birth and rheumatoid arthritis [15–18]. However there is no report showing a relationship between P.g. and liver diseases such as NAFLD/NASH.
Here we postulate that dental infection of P.g. is a critical risk factor for NASH progression, which acts as a “second hit” to induce inflammation and fibrogenic responses in steatosis. In this study, we demonstrated that P.g. exacerbated diet-induced steatohepatitis via the induction of inflammasomes and inflammatory cytokines in the liver. Furthermore, the infection of P.g. was demonstrated for the first time in the liver of NASH patients. These results indicate that dental infection of P.g. promotes the progression of NASH. Accordingly, preventing and/or treating dental infection and decolonization of P.g. in the liver could have a beneficial impact on NASH.
Methods
Animal studies
The experimental protocol described below was approved by the animal care committee of Hiroshima University.
A total of 24, 5-week-old male C57BL/6J mice (Charles River Japan, Inc., http://www.crj.co.jp/) were randomly divided into two groups. One was fed a high fat diet (HFD-60; Oriental Yeast Co., Ltd., http://www.oyc-bio.jp/) (HFD group), the other was fed a chow-diet (CD group). After development of fatty liver for 12 weeks of HFD feeding, the mice were divided into two subgroups (N = 6), with and without dental infection of P.g., named HFD-P.g.(+) and HFD-P.g.(−), respectively. CD-P.g.(+) and CD-P.g.(−) were also prepared to serve as controls. For dental infection of P.g., the roof of the pulp chamber of the right and left first molars of the upper jaw was removed with a #1/2 round bar. After removing the coronal pulp, a small cotton swab including 1 μl of phosphate buffered saline (PBS) containing 107 cells of P.g. W83 strain was inserted into the pulp chamber and sealed with Caviton (GC Co., http://www.gcdental.co.jp/index.html). After 6 weeks, tissue samples were taken from molar regions and the liver for histological evaluation. Blood serum and liver tissues from the animals were collected and stored at −80 °C prior to examining LPS levels and cytokine expression, respectively.
Immunohistochemistry
Immunolocalization of P.g. in the periodontal tissue and liver was examined using two antisera; rat antiserum against LPS obtained from P.g. (P.g.-LPS) (1:1,000 dilution) and rabbit antiserum against whole P.g. (1:1,000 dilution). The rabbit antisera against whole P. gingivalis were kindly provided by Professor Kazuyuki Ishihara (Tokyo Dental College). Monoclonal antibodies; Mac2 (1:500 dilution; BioLegend, http://www.biolegend.com/), MCA771GA (1/5,000 dilution; serotec, http://www.abdserotec.com/) and α-smooth muscle actin (α-SMA) (1:500 dilution; DAKO Japan, http://www.dako.jp/) were used for immunohistochemical detection of macrophages, neutrophils and hepatic myofibroblasts, respectively. For detection of TLR2 in mouse liver tissue, sc-16237 (1/100 dilution; SANTA CRUZ BIOTECHNOLOGY, INC. http://www.scbt.jp/) was used.
Specificity was ascertained by substituting PBS and normal serum for each antibody.
Histomorphometric analysis of liver tissues
For grading inflammatory reaction in the liver, five different fields were randomly selected under 100× magnification, and the number of foci with more than 10 Mac2-positive macrophages was counted. Stages of fibrosis were evaluated with Azan-Mallory staining sections as no fibrosis (stage 0), only zone 3 perisinusoidal fibrosis (stage 1), as above with portal fibrosis (stage 2); as above with bridging fibrosis (stage 3), and cirrhosis (stage 4) according to generally accepted definitions [19].
Identification of living P.g. in periodontal tissue after dental infection of P.g.
After P.g. infection, extracted teeth with periodontal tissue were washed in sterilized PBS. The washing solution was plated on anaerobic blood agar and cultured in a 5 % CO2 atmosphere using the Anaeropack system (Mitsubishi Gas Chemical Co., http://www.mgc.co.jp) at 37 °C. After a 4-day-cultivation, colonies were collected. DNA was extracted from colonies using the DNeasy Tissue Kit (QIAGEN Science, http://www.qiagen.com/). To identify the P.g. W83 strain, mgl gene that encodes l-methionine-α-deamino-γ-mercaptomethane-lyase, which is specifically expressed in the P.g. [20], was amplified by PCR analysis. PCR primer pairs for mgl are listed in Table 1. DNA from the P.g. W83 strain was used as a positive control and those from Escherichia coli (E.c.) and Aggregatibacter actinomycetemcomitans (A.a.) were used as negative controls.
Concentration of LPS and titer of antibody against P.g. in serum
Serum concentration of LPS was measured using Endospecy ES-50M Set (SEIKAGAKU BIOBUSINESS Co., http://www.seikagakubb.co.jp/). The colorimetric analysis was performed as described by the manufacturer’s protocol. The IgG antibodies that reacted to the bacterial antigens were measured by the ELISA method published in a previous study [21].
Cytokine levels in liver tissues
Fifty μg of protein in diluent (Bio-Plex Diluent kit # 171-305-008, Bio-Rad, http://www.bio-rad.com/) from each liver sample was adjusted to examine cytokine levels of MCP-1, IL-1β, IL-6, IL-8, IL-17, IFN-γ, and TNF-α using a multiplex bead-based immunoassay kit (# 171-K11070, Bio-Rad).
Cell culture and palmitate treatment in vitro
Immortalized human fetal hepatocytes (Hc3716-hTERT), kindly provided by Prof. Hidetoshi Tahara (Hiroshima University), were maintained in hepatocyte basal medium (HBM; Takara Bio Co. http://www.takara-bio.co.jp/) as described previously [22]. The 5 × 105 of Hc3716-hTERT were seeded in a 35 mm collagen coated dish and cultured for 1 day. 10 mM palmitate (SIGMA-ALDRICH, http://www.sigmaaldrich.com/)/1 % Bovine Serum Albumin (BSA) stock solution was prepared according to the method of Wobser [23]. The Hc3716-hTERT cells were cultured in HBM containing 0.4 mM palmitate for 18 h to induce accumulation of lipids. FFA-free-BSA-treated cells were used as a control. The palmitate-treated cells and control cells were incubated in fresh medium with or without P.g.-LPS (1 μg/ml). The culture cells were harvested 6 h after stimulation.
RNA extraction and RT-PCR analysis
Total RNA was extracted from the mouse liver tissue and the harvested cells using TRIzol® reagent (Invitrogen Co., http://www.invitrogen.jp). One μg of total RNA was used for cDNA synthesis using the ReverTra Dash kit (TOYOBO Co., Ltd., http://www.toyobo.co.jp/). Aliquots of total cDNA were amplified using KOD-Plus-DNA Polymerase (TOYOBO Co.). Amplification for Toll-like receptor (TLR) 2, TLR4, inflammatory cytokines (IL-1β, IL-6, IL-8 and TNF-α) and inflammasomes (Nod-like receptor 3, NLRP3 and caspase-1, Casp-1) was performed using a MyCycler™ thermal cycler (Bio-Rad). PCR primer pairs are listed in Table 1. PCR products were electrophoresed on 1.5 % agarose gels at 100 mV and visualized by ethidium bromide.
Quantitative real time PT-PCR was performed in the Applied Biosystem StepOnePlus™ (Applied Biosystems, http://www.appliedbiosystems.jp) using TaqMan® Fast Advanced Master Mix (Applied Biosystems) and specific primers and probe for TLR2 (Forward: ATGAGAACAATGATGCTGCCATT, Reverse; ACTCCAGGTAGGTCTTGGTGTTCA, Probe; AAAAAAGCCATTCCCCAGCGCTTCT). The reaction product was quantified with 18S (X03205.1 Eukaryotic 18S rRNA; Applied Biosystems) as the reference gene.
Human liver samples
Formalin-fixed, paraffin-embedded human liver biopsy samples from 40 patients with NASH were retrieved from the pathological file of Hiroshima University Hospital. This study meets the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Hiroshima University Committee for the Protection of Human Subjects in Research. The liver biopsy specimens of NASH patients (14 males and 26 females; age 53.7 ± 13.8 years) included various degrees of fibrosis stages. Histological features related to fibrosis such as zone 3 perisinusoidal (pericentral vein) fibrosis (score 0/1/2/3, n = 7/18/12/3), periportal fibrosis (score 0/1/2/3, 1/21/9/9/), bridging fibrosis (score 0/1/2/3, n = 7/7/24/2) and total fibrosis score (sum of each score, score 0/1/2/3/4/5/6/7/8/9, n = 0/0/8/3/14/4/4/3/4/0) were correlated with the presence of P.g. infection in the liver.
Statistical analysis
Results are reported as mean ± standard deviation. Differences between groups were compared using the Mann–Whitney U test. P < 0.05 was considered statistically significant.
Results
P.g. infected from pulp establishes periapical granuloma and can be detected in periapical granuloma after 6 weeks (Fig. 1)
In contrast to normal periodontal tissues (Fig. 1a), all the animals of CD-P.g.(+) and HFD-P.g.(+) showed total pulp necrosis and periapical granuloma with infiltration of neutrophils and macrophages (Fig. 1b). P.g. was immunodetected in the pulp chamber and in neutrophils and macrophages in the periapical granuloma (Fig. 1c).

P.g. infected from pulp establishes periapical granuloma and can be detected in periapical granuloma after 6 weeks. a, b Histological findings of periodontal tissue in a uninfected (P.g.(−)) and b infected (P.g.(+)) mice. Periapical granuloma with bone destruction was developed in the periapical periodontal area of P.g.(+) mice. H&E, Scale bars 100 μm. c Immunolocalization of P.g. (brown particles) was observed in neutrophils (also positively stained with mouse neutrophils antibody) and macrophages (also positive for Mac2) in periapical granuloma. Scale bars 100 μm. d Gene expression for the mgl gene, which encodes l-methionine-α-deamino-γ-mercaptomethane-lyase of P.g. was detected in colony obtained from periapical area. Escherichia coli (E.c.) and Aggregatibacter actinomycetemcomitans (A.a.) were used as negative controls. e Serum concentration of LPS was upregulated both in CD-P.g.(+) and HFD-P.g.(+). *P < 0.05. f Serum antibody titer of P.g. was upregulated in CD-P.g.(+) and HFD-P.g.(+). *P < 0.05, **P < 0.01. P.g. Porphyromonas gingivalis, CD cho diet, HFD high fat diet
Expression of the mgl gene was detected in DNA extracts of colonies grown from the extracted molars with P.g. infection. Expression of mgl was seen in DNA extracts of the P.g. W83 strain used in this study but not in those of E.c. and A.a., which were used as negative controls (Fig. 1d).
Serum LPS levels (Fig. 1e) and serum titer of P.g. antibody (Fig. 1f) in CD-P.g.(+) and HFD-P.g.(+) were significantly higher than those in CD-P.g.(−) and HFD-P.g.(−) (P < 0.05) There was no significant difference in LPS levels and P.g. antibody titers between the CD-P.g.(+) and the HFD-P.g.(+).
Dental infection of P.g. induces significant upregulation of cytokine levels in the liver of HFD-P.g.(+) (Fig. 2)
Production levels of MCP-1 (P < 0.01), TNF-α, IL-17 and IL-1β (P < 0.05) were significantly increased in the liver of HFD-P.g.(−) compared to CD-P.g.(−) at week 0. There was no difference in production levels between CD-P.g.(+) and CD-P.g.(−) at week 6. In both HFD groups at week 6, the cytokine production levels were significantly higher than those in the CD groups. Remarkably, significant increases of MCP-1, TNF-α and IL-17 production were observed in HFD-P.g.(+) compared with HFD-P.g.(−) (P < 0.05).

Dental infection of P.g. induces significant upregulation of cytokine levels in the liver of HFD-P.g.(+). Bio-Plex inflammatory cytokine assays were used to profile expression of inflammatory mediators, MCP-1 (a), TNF-α (b), IL-17 (c) and IL-1β (d). Mean ± SEM, *P < 0.05, **P < 0.01. P.g. Porphyromonas gingivalis, CD cho diet, HFD high fat diet
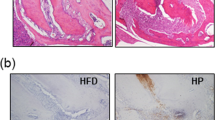
Dental infection of P.g. promotes pathological progression of HFD-induced NASH (Fig. 3)
No obvious pathological changes were observed in CD-P.g.(−) (Fig. 3a). In contrast, there were small foci of inflammatory cell infiltration in CD-P.g.(+) (Fig. 3b). In HFD-P.g.(−), marked steatosis developed, particularly in the central vein area, and fatty degeneration of hepatocytes was prominent (Fig. 3c). Small foci of Mac2-positive macrophages (Fig. 3c; arrows) and periportal inflammation were seen. In HFD-P.g.(+), the amount of fat deposited in hepatocytes shows a tendency to increase in comparison with that in HFD-P.g.(−) (Fig. 3d). Mallory bodies were seen in a ballooning hepatocyte. Infiltration of Mac2-positive macrophages were more prominent in HFD-P.g.(+). Interestingly, focal fibrosis of the liver was observed only in HFD-P.g.(+). Spindle cells in the fibrotic area were immunopositive for α-SMA, indicating their myofibroblastic nature (so-called hepatic stellate cells). Accumulation of collagen around the hepatic stellate cells was revealed by Azan-Mallory staining. Immunohistochemical staining revealed that P.g.-positive particles were detected in hepatocytes (Fig. 3e) and Kupffer cells.

Dental infection of P.g. promotes pathological progression of HFD-induced NASH. a No obvious histologic changes were seen in liver of CD-P.g.(−) (H&E, Scale bar 10 μm). b Some small foci of inflammation were observed in CD-P.g.(+) (H&E, Scale bar 10 μm). c Fatty degeneration of hepatocytes was prominent in HFD-P.g.(−) (H&E, Scale bar 100 μm). Arrow shows inflammatory cell infiltration. The foci of Mac2-positive macrophages (upper, middle H&E, right immunohistochemistry, Scale bar 10 μm) were detected. Portal inflammation was also observed (right lower, H&E, Scale bar 10 μm). d In HFD-P.g.(+), the amount of fat deposited in hepatocytes was increasing. Number of foci of Mac2-positive cells (arrows) also increased (H&E, Scale bar 100 μm). Ballooning hepatocytes with Mallory bodies were prominent, (right upper, H&E, Scale bar 10 μm). Mac2-positive macrophages increased, (right lower, Immunohistochemistry, Scale bar 10 μm). Lower line shows an area of fibrosis seen in HFD-P.g.(+). In this area, α-SMA-positive hepatic myofibroblasts associated with collagen-formation were proliferated (left; H&E, center; Immunohistochemistry of α-SMA, right; Azan Mallory staining, Scale bars 10 μm). e Immunolocalization of P.g. in hepatocyte of HFD-P.g.(+), Scale bar 10 μm. P.g. Porphyromonas gingivalis, CD cho diet, HFD high fat diet. f The number of foci of Mac2 positive cells *P < 0.05, **P < 0.01
The number of foci of Mac2-positive cells in HFD-P.g.(−) and HFD-P.g.(+) were significantly higher than those in CD groups (P < 0.01). The number of foci of Mac2-positive cells in the HFD-P.g.(+) was higher than that in HFD-P.g.(−) (P < 0.05) (Fig. 3f). Table 2 shows staging of fibrosis. Most cases in CD groups were in stage 0. In HFD-P.g.(−), there were two of stage 2 and four of stage 3. Moreover, HFD-P.g.(+) included four of stage 3 and two of stage 4, indicating greater progression of fibrosis in HFD-P.g.(+) than in the other experimental groups, including HFD-P.g.(−).
Palmitate promotes inflammasome and LPS-induced proinflammatory cytokine mRNA expression in human hepatocytes in vitro (Fig. 4)
Palmitate induced marked accumulation of cytoplasmic lipid droplets in human hepatocytes (Fig. 4a). In the present study, we used the palmitate-treated cells as an in vitro steatotic hepatocyte model. The human hepatocyte constitutively showed strong expression of TLR4. Interestingly, palmitate treatment induced upregulation of TLR2 expression (Fig. 4b). In the steatotic hepatocytes, mRNA expressions of IL-1β, IL-6 and TNF-α were slightly upregulated. Stimulation of P.g.-LPS dramatically upregulated mRNA expression of inflammasomes including NLRP3 and Casp-1, in addition to proinflammatory cytokines, including IL-1β, IL-6, IL-8 and TNF-α in steatotic hepatocytes (Fig. 4c).

Palmitate promotes inflammasome and LPS-induced proinflammatory cytokine mRNA expression in human hepatocytes in vitro. Human hepatocyte cell line (Hc3716-hTERT) was treated with 0.4 mM palmitate for 18 h. a Palmitate induced accumulation of lipid droplets in human hepatocytes (Oil Red O staining, Scale bars 10 μm). b mRNA expression of P.g.-LPS receptors. In steatotic hepatocytes TLR2-mRNA expression level was upregulated. c mRNA expression of inflammatory cytokines (IL-1β, IL-6, IL-8 and TNF-α) and inflammasome (NLRP3 and casp-1) in human hepatocytes with and without palmitate treatment at 6 h after P.g.-LPS stimulation. P.g.-LPS synergistically upregulated palmitate-induced cytokines and inflammasome expression
TLR2 expression in liver tissue is significantly upregulated in HFD-groups (Fig. 5)
During in vitro study, palmitate treatment induced upregulation of TLR2 expression in hepatocytes. Therefore TLR2 expression in liver tissue was analyzed in CD- and HFD-groups. In CD-P.g.(-), TLR2-positive hepatocytes are scattered in liver (Fig. 5a), whereas in HFD-P.g.(+), numerous hepatocytes having lipid droplets strongly positively expressed TLR2 (Fig. 5b). TLR2-mRNA expression levels in HFD groups are significantly higher than those of CD groups, 2.5-fold between HFD-P.g.(−) and CD-P.g.(−) and 3.3-fold between HFD-P.g.(+) and CD-P.g.(+), respectively (Fig. 5c). There is no difference in TLR2-mRNA expression level between CD-P.g.(−) and CD-P.g.(+) and between HFD-P.g.(−) and HFD-P.g.(+).

TLR2 expression in liver tissue is significantly upregulated in HFD-groups. a In CD-P.g.(−). TLR2-positive hepatocytes are scattered in liver (Scale bar 10 μm). b Numerous hepatocytes having lipid droplets positively expressed TLR2 in HFD-P.g.(+) liver strongly (Scale bar 10 μm). c TLR2-mRNA expression levels in HFD groups are significantly higher than those of CD groups, *P < 0.05
P.g. infection is detected in the liver biopsy specimens from patients with NASH (Fig. 6)
The immunolocalization of P.g. was detected in 21 of 40 biopsy specimens (52.5 %). P.g. was identified as single and aggregated brown particles in hepatocytes (Fig. 6a). Interestingly, the P.g.-positive cases showed significantly higher fibrosis scores including perisinusoidal fibrosis score (P = 0.018), periportal fibrosis score (P = 0.049) and total fibrosis score, (a sum of each score, P = 0.007) than the P.g.-negative cases (Fig. 6b).

P.g. infection is detected in the liver biopsy specimens from patients with NASH. a Liver tissues from NASH patient were stained with H&E, Azan-Mallory staining, and immunohistochemistry for P.g. P.g. was detected in hepatocytes associated with fibrosis (Scale bars 10 μm). b P.g. infection and fibrosis scores in NASH patient. The line in vertical dot plot indicates the median value. NASH patients with P.g.-infection had higher fibrosis scores
Discussion
The liver is constantly exposed to microbial products from the enteric microflora. Recently, accumulating data have demonstrated that gut-derived LPS can act as a second hit and results in progressive liver injury leading to NASH [24, 25]. It is also reported that steatosis sensitizes the liver for NASH [26, 27]. However, whether LPS derived from periodontal pathogens amplifies the inflammation and fibrosis in the liver has been unclear.
P.g., a gram-negative anaerobic bacterium, is considered to be a primary causative bacterium of chronic periodontitis [28]. P.g. is detected not only in biofilm in periodontal pockets but also as a major bacterium in infected pulp chambers with periapical periodontal diseases [13, 14]. Therefore, in the present study, to obtain anaerobic conditions suitable for P.g. growth we applied P.g. from pulp chamber. In this animal model, we confirmed that P.g. infected into the pulp chamber remained alive and grew continuously in the pulp chamber over a long period of time, resulting in development of periapical granuloma. These observations indicate that the periapical granuloma is a persistant and sustainable supply source of the P.g. itself and its products, and can lead to infection and responses in liver tissues due to their spread through the bloodstream. Therefore we considered that the animal model used in the present study closely reproduced dental infection of P.g. in NASH patients. In 2012, using an HFD-induced NASH model, Yoneda et al. [29] reported the effects of direct injection of P.g. into the jugular vein on liver tissue of HFD-induced steatosis mice model. It was reported that marked increase in body and liver weight, increase in accumulation of lipid in liver, and increase in ALT and TG levels were observed. In the present study, we could demonstrate additional new information including significant upregulation of LPS levels and titer levels of P.g. antibody in serum and cytokine and TLR2 expression levels in liver in HFD-P.g.(+). Moreover, in liver of HFD-P.g.(+) massive fibrotic areas were developed and P.g. was immunohistochemically detected in hepatocytes and Kupffer cells. The present results would provide speculation of the underlining mechanism.
Our new findings, which include detection of elevated levels of serum LPS and demonstration of P.g. infection in liver tissues in infected mice, are indicating that P.g. and its products may enter from the dental lesion into blood circulation and affect the liver condition. Gibson et al. [30] reported that DNA of P.g. that was orally challenged to mice was detected in blood by PCR. Boggess et al. [31] reported that P.g. DNA was detected in the maternal livers and placenta in rabbits with chronic maternal exposure using a subcutaneous steel chamber. Because of abundant sinusoidal blood flow, the liver is likely one of the organs where P.g. is easily able to reach and colonize.
It is well known that many of characteristic pathophysiological features of human NASH are induced by HFD feeding in rodents [10, 32]. Similarly, histopathological findings such as steatosis, lobular inflammation and scant perisinusoidal fibrosis indicating the early stage of NASH were observed in HFD-P.g.(−) mice in our study. Interestingly, fibrosis with proliferation of hepatic stellate cells was only observed in HFD-P.g.(+) mice. These findings suggest that dental infection of P.g. accelerates pathological progression of NASH. Accumulating evidence indicates the importance of TLR4 signaling in the pathogenesis of NASH. Hepatic lipid accumulation and mRNA levels of hepatic fibrogenic markers were reduced in methionine choline-deficient diet-induced steatohepatitis of TLR4-mutant mice [33]. Csak et al. [24] also reported that fibrosis markers such as α-SMA, procollagen-1 and TGF-β were attenuated in livers of NASH model in MD-2 and TLR4 KO mice. P.g.-LPS is an agonist both for TLR2 and TLR4 [28]. Both TLR2 and TLR4 stimulation induce proinflammatory cytokine production via the NF-κB signaling pathway [28, 34]. Therefore, we propose that P.g.-LPS plays a critical role in progression of fibrosis in steatotic liver through the LPS-TLR pathway.
FFAs appear to be the major mediator of excessive fat accumulation in the liver. In patients with NASH, serum FFA levels are commonly elevated, which is related to disease severity and considered a potential endogenous danger signal [35, 36]. The levels of serum FFAs in NASH models induced by HFD were elevated [37]. In the present study, treatment with palmitate induced fat accumulation, upregulated expression of TLR2, proinflammatory cytokines and NLRP3-inflammasome, and enhanced sensitivity to P.g.-LPS in hepatocytes. Actually TLR2 expression was upregulated in steatotic liber of HFD groups at protein and mRNA levels. Our results lead to two potential mechanisms in which P.g.-LPS dramatically upregulated expression of proinflammatory cytokines in steatotic hepatocytes. In control hepatocytes, P.g.-LPS slightly induces proinflammatory cytokines mainly via TLR4-pathway (Fig. 7a). In steatotic hepatocyte, the amplification through the TLR2-pathway is evident (Fig. 7b). It was reported that TLR4-signaling induces TLR2-expression [38]. Therefore FFA, a potential TLR4 agonist [39], may upregulate TLR2-expression in hepatocytes via TLR4-signaling, resulting in upregulation of cytokine expression caused by P.g.-LPS (an agonist for both TLR4 and TLR2). The second mechanism is activation of the IL-1β pathway through the NLRP3 inflammasome (Fig. 7c). Recently, it has been reported that the NLRP3 inflammasome senses obesity-associated danger signals like FFA and contributes to obesity-induced inflammation and insulin resistance [40, 41]. The NLRP3 inflammasome is a member of the Nod-like receptor family of innate immune cell sensors. It leads to Casp-1 cleavage and subsequent activation of IL-1β and IL-18 in adipose tissue [40] and hematopoietic cells [41]. Csak et al. [36] demonstrated that long-term HFD-feeding induced steatohepatitis associated with increased IL-1β production and NLRP3 inflammasome activation. As a result, upregulation of serum LPS and hepatocyte infection associated with dental infection of P.g. promotes production of proinflammatory cytokines and subsequently leads to pathological progression of NASH.
Finally we examined the presence of P.g. in the liver of NASH patients. Surprisingly, P.g. was detected in 52.5 % of the liver biopsy specimens, and NASH patients with P.g. infection showed higher fibrosis scores than the NASH patients without P.g. infection. In subsequent studies, we will examine a larger number of liver biopsy specimens from NASH patients to clarify the importance of P.g. infection in pathological progression of NASH. We are also going to examine periodontal conditions of NASH patients in correlation with the presence of P.g. infection in the liver.

The proposed mechanisms responsible for P.g.-LPS induced dramatic upregulation of proinflammatory cytokines expression in steatotic hepatocytes. a P.g.-LPS induces proinflammatory cytokine production mainly through the TLR4 pathway in control hepatocytes. b Amplification through TLR2-pathway. In steatotic hepatocytes, TLR2-expression is upregulated. As an antagonist of TLR2, P.g.-LPS understandably show effective upregulation of proinflammatory cytokine production via the TLR2-pathway. c Amplification through the IL-1β pathway activated by NLRP3 inflammasomes. In steatotic hepatocytes, the NLRP3 inflammasome is upregulated. This inflammasome converts inactive pro-IL-1β to active IL-1β. Then IL-1β induces additive proinflammatory cytokine production
Furthermore, chronic periodontitis, one type of dental infection, is among the most prevalent of microbial diseases in human and occurs worldwide with a prevalence rate of more than 70 % [42]. Recently, it has become well accepted that control of periodontitis can improve status of systemic diseases including type 2 diabetes and cardiovascular disease [15, 16]. Thus, further study is also needed on the efficiency of dental therapy and/or elimination of liver infection by antibiotics in liver function and condition.
In summary, we first demonstrated that the dental infection of P.g. exacerbated the pathological progression of NASH from simple steatohepatitis to steatohepatitis with fibrosis by way of a mechanism that involves synergistic interaction between FFA-induced NLRP3 inflammasome activation and the LPS-TLR pathway. Our findings suggest that preventing and/or eliminating P.g. infection by dental therapy may have a beneficial impact on NASH.
Abbreviations
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Non-alcoholic steatohepatitis
- FFA:
-
Free fatty acid
- LPS:
-
Lipopolysaccharides
- P.g. :
-
Porphyromonas gingivalis
- HFD:
-
High fat diet
- CD:
-
Chow diet
- H&E:
-
Hematoxylin and Eosin
- α-SMA:
-
α-smooth muscle actin
- TLR:
-
Toll-like receptor
- NLRP3:
-
Nod-like receptor 3
- Casp-1:
-
Caspase-1
References
Sanyal AJ. NASH: a global health problem. Hepatol Res. 2011;41:670–4.
Malaguarnera M, Rosa MD, Nicoletti F, Malaguarnera L. Molecular mechanisms involved in NAFLD progression. J Mol Med. 2009;87:679–95.
Kojima S, Watanabe N, Numata M, et al. Increase in the prevalence of fatty liver in Japan over the past 12 years: analysis of clinical background. J Gastroenterol. 2003;38:954–61.
Leite NC, Villela-Nogueira CA, Pannain VLN, Bottino AC, Resende GFM, Cardoso CRL, et al. Histopathological stages of nonalcoholic fatty liver disease: prevalences and correlated factors. Liver Int. 2011;31(5):700–6.
Tannapfel A, Denk H, Dines H-P, Langer C, Schirmacher P, Trauner M, et al. Histopathological diagnosis of nonalcoholic and alcoholic fatty liver disease. Virchows Arch. 2011;458:511–23.
Day CP, Jamcs O. Steatohepatitis: a talc of two “hits”? Gastroenterology. 1988;114:842–5.
Day CP. Pathogenesis of steatohepatitis. Best Pract Res Clin Gastroenterol. 2002;16(5):663–78.
Sakaguchi S, Takahashi S, Sasaki T, Kumagai T, Nagata K. Progression of alcoholic and non-alcoholic steatohepatitis: common metabolic aspects of innate immune system and oxidative stress. Drug Metab Pharmacokinet. 2011;26(1):30–46.
Guo J, Friedman SL. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair. 2010;3:21.
Gabele E, Dostert K, Patsenker E, Stickel F, Hellerbrand C. A new model of interactive effects of alcohol and high-fat diet on hepatic fibrosis. Alchohorism Clin Extern Res. 2011;35(7):1361–7.
Wigg AJ, Roberts-Thomson IC, Dymock PB, McCharthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxemia, and tumor necrosis factor alpha in the pathogenesis of nonalcoholic steatohepatitis. Gut. 2001;48:206–11.
Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, et al. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008;28(7):1026–33.
Saito D, Coutinbo LL, Saito CPB, Tsai SM, Hoflinf JF, Goncalves RB. Real-time polymerase chain reaction quantification of Porphyromonas gingivalis and Tannerella forsythia in primary endodontic infections. J Endod. 2009;35:1518–24.
Pereira CV, Stipp RN, Fonseca DC, Pereira LJ, Hofling JF. Detection and clonal analysis of anaerobic bacteria associated to endodontic-periodontal lesions. J Periodontol. 2011;82(12):1767–75.
Seymour GJ, Ford PJ, Cullinan MP, Leishman S, Yamazaki K. Relationship between periodontal infections and systemic disease. Clin Microbiol Infect. 2007;13(Suppl 4):3–10.
Pizzo G, Guiglia R, Russo LL, Campisi G. Dentistry and internal medicine: from the focal infection theory to the periodontal medicine concept. Europ J Intern Med. 2011;21:496–502.
Wada K, Kamisaki Y. Roles of oral bacteria in cardiovascular diseases—from molecular mechanisms to clinical cases: involvement of Porphyromonas gingivalis in the development of human aortic aneurysm. J Pharmacol Sci. 2010;113:115–9.
Figuero E, Sanchez-Beltran M, Cuesta-Frecheso S, Tejerina JM, del Castro JA, Gutierrez JM, et al. Detection of periodontal bacteria in atheromatous plaques by nested polymerase chain reaction. J Periodontol. 2011;82(10):1469–77.
Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94(9):2467–74.
Yoshimura M, Nakano Y, Yamashita Y, Oho T, Saito T, Koga T. Formation of methyl mercaptan from l-methionine by Porphyromonas gingivalis. Infect Immun. 2000;68(12):6912–6.
Kawai T, Paster BJ, Komatsuzawa H, Ernst CW, Goncalves RB, Sasaki H, et al. Cross-reactive adaptive immune response to oral commensal bacteria results in an induction of receptor activator of nuclear factor-kappaB ligand (RANKL)-dependent periodontal bone resorption in a mouse model. Oral Microbiol Immunol. 2007;22(3):208–15.
Waki K, Anno K, Ono T, Ide T, Chayama K, Tahara H. Establishment of functional telomerase immortalized human hepatocytes and a hepatic stellate cell line for telomere-targeting anticancer drug development. Cancer Sci. 2010;101:1678–85.
Wobser H, Dorn C, Weiss TS, Amann T, Bollheimer C, Büttner R, et al. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 2009;19:996–1005.
Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D, et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. 2011;300:G433–41.
Aoyama T, Paik Y-H, Seki E. Toll-like receptor signaling and liver fibrosis. Gastroenterology Res Pract 2010; pii:192543. Epub 2010 Jul 25.
Gentile CL, Pagliassotti MJ. The role of fatty acids in the development and progression of nonalcoholic fatty liver disease. J Nutr Biochem. 2008;19:567–76.
Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V, et al. The role of lipid droplets in metabolic disease in rodent and humans. J Clin Invest. 2011;121:2102–10.
Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev. 2010;8:481–90.
Yoneda M, Naka S, Nakano K, Wada K, Endo H, Mawatari H, et al. Involvement of a periodontal pathogen, Porphyromonas gingivalis on the pathogenesis of non-alcoholic fatty liver disease. BMC Gastroenterol 2012;12(1):16. [Epub ahead of print].
Gibson FC 3rd, Hong C, Chou HH, Yumoto H, Chen J, Lien E, et al. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109(22):2801–6.
Boggess KA, Madianos PN, Preisser JS, Moise KJ, Offenbacher S. Chronic maternal and fetal Porphyromonas gingivalis exposure during pregnancy in rabbits. Am J Obstet Gynecol. 2005;192:554–7.
Sundaresan S, Vijayagopal, Mills N, Prasad C. A mouse model for nonalcoholic steatohepatitis. J Nutrit Biochem. 2011;22:979–84.
Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M, et al. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47(4):571–9.
Kocgozlu L, Elkaim R, Tenenbaum H, Werner S. Variable cell responses to P. gingivalis lipopolysaccharide. J Dent Res. 2009;88(8):741–5.
Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology. 2009;50(6):1827–38.
Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54(1):133–44.
Xu ZJ, Fan JG, Ding XD, Qiao L, Wang GL. Characterization of high-fat, diet-induced, non-alcoholic steatohepatitis with fibrosis in rats. Dig Dis Sci. 2010;55(4):931–40.
Fan J, Frey RS, Malik AB. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J Clin Invest. 2003;112(8):1234–43.
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–25.
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–88.
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12(5):408–15.
Albandar JM. Epidemiology and risk factors of periodontal diseases. Dent Clin North Am. 2005;49(3):517–32.
Acknowledgments
The authors thank Ryo Matsuda, Mao Muroi and Shinnichi Sakamoto for their support of this project. We are also grateful to Professor Kazuyuki Ishihara (Tokyo Dental College) for providing P.g.-specific polyclonal antibodies and Prof. Hidetoshi Tahara (Hiroshima University) for providing Hc3716-hTERT cells.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Furusho, H., Miyauchi, M., Hyogo, H. et al. Dental infection of Porphyromonas gingivalis exacerbates high fat diet-induced steatohepatitis in mice. J Gastroenterol 48, 1259–1270 (2013). https://doi.org/10.1007/s00535-012-0738-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-012-0738-1