
Abstract
Background
MicroRNAs can promote or suppress the evolution of malignant behaviors by regulating multiple targets. We aimed to determine the expression of miR-301a recently screened in gastric cancer, to investigate the biological effects of miR-301a and to identify the specific miR-301a target gene.
Methods
Quantitative real-time RT-PCR was used to test miR-301a expression. Functional effects were explored by a water-soluble tetrazolium salt assay, a colony formation assay in soft agar, a migration assay, an invasion assay and cytometry used to determine apoptosis and cell cycle. Nude mice were inoculated subcutaneously by retrovirus-mediated stably expressed SGC-7901 cells. The target gene was determined by bioinformatic algorithms, dual luciferase reporter assay and Western blot.
Results
Firstly, we found that miR-301a was significantly upregulated both in cells and tissues of gastric cancer. The expression level of miR-301a was inversely correlated with tumor differentiation of gastric cancer tissues. Secondly, miR-301a promoted cell growth, soft agar clonogenicity, migration, invasion, and decreased cell apoptosis induced by cisplatin in vitro, while blockage of miR-301a reduced the percentage of G2/M phase cells via flow cytometry in gastric cancer cells. Ectopic expression of miR-301a enhanced the subcutaneous tumorigenesis in vivo. Finally, miR-301a directly downregulated RUNX3 expression post-transcriptionally in gastric cancer.
Conclusion
Our results demonstrate that miR-301a plays important roles in the development of gastric cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer is the fourth most common cancer with the second highest mortality rate worldwide [1]. It is generally accepted that the development of gastric cancer, like other cancers, involves multiple steps, including the accumulation of genetic and epigenetic changes [2, 3]. However, the precise mechanism underlying gastric carcinogenesis remains unclear. MicroRNAs (miRNAs) are small non-coding RNAs that act as post-transcriptional regulators by targeting mRNAs for translational repression or cleavage [4–6]. A great number of studies have demonstrated that various miRNAs function as potential oncogenes or tumor suppressor genes during the initiation and progression of cancer [7]. Previous studies have identified numerous cancer specific miRNAs in various cancer types, such as colorectal cancer [8, 9], B cell chronic lymphocytic leukemia [10], breast cancer [11], lung cancer [12], papillary thyroid cancer [13], and hepatocellular carcinoma [14].
By comparing the miRNA expression profile, we found that miR-301a was one of the most significantly upregulated in gastric cancer cells and tissues, compared with its non-tumor tissues. The miR-301a is processed from the first intron of SKA2 transcript located at 17q22–23 in the human genome. Overexpression of miR-301a has been previously demonstrated in hepatocellular carcinoma, pancreatic tumor tissue, and small cell lung cancer compared with adjacent benign tissues [15–17]. However, the biological processes and molecular mechanisms underlying miR-301a remain unclear in gastric cancer. In this study we aimed to investigate the expression, biological functions and mechanisms of miR-301a in gastric cancer.
Materials and methods
Cell lines
The gastric cancer cell lines SNU-1, SNU-16, and KATO III were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). The SGC-7901, NCI-N87, MKN-28, BGC-823, MKN-45 and AGS were purchased from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. The immortalized normal gastric mucosal epithelial cell line (GES-1) was a gift from Prof. Feng Bi (Sichuan University, PR China). The embryonic kidney cell line 293T was preserved in our institute. Cells were grown in RPMI-1640 or Dulbecco’s modified Eagle’s medium (DMEM; Sigma, St Louis, MO, USA) supplemented with 10 % fetal bovine serum at 37 °C in a humidified atmosphere with 5 % CO2.
Tissue samples
Primary tumor tissues and their adjacent non-tumor tissues were collected from patients with gastric cancer undergoing radical gastrectomy at the Department of Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. Tissue samples were immediately snap-frozen in liquid nitrogen and stored in a deep freezer at −80 °C. All samples were collected with patients’ informed consent and confirmed by the pathological examination.
RNA isolation and qRT-PCR
Total RNA was extracted from tissue samples or cell lines using Trizol Agent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The expression level of miRNAs were assessed by the TaqMan stem-loop RT-PCR method (Applied Biosystems, Foster city, CA). The U6 small nuclear RNA (RNU6B; Applied Biosystems) was used for normalization. The relative expression ratio of miR-301a in each paired tumor and non-tumor tissue was calculated by the 2−ΔΔCT method. The miR-301a expression level was defined as upregulated when the relative expression ratio was >1, and defined as down-regulated when the relative expression ratio was <1. The mRNA expression level of RUNX3, Bim, MMP9 and SKA2 were measured by qRT-PCR and normalized using the GAPDH mRNA level. The expression level of RUNX3 mRNA relative to GAPDH mRNA was calculated using the 2−ΔΔCT method. Primers for qRT-PCR were as follows: RUNX3 F 5′-AGCACCACAAGCCACTTCAG-3′, R 5′-GGGAAGGAGCGGTCAAACTG-3′, Bim F 5′-TAAGTTCTGAGTGTGACCGAGA-3′, R 5′-GCTCTGTCTGTAGGGAGGTAGG-3′, MMP9 F 5′-TGTACCGCTATGGTTACACTCG-3′, R 5′-GGCAGGGACAGTTGCTTCT-3′, SKA2 F 5′-GCCGCATTTGTGCTACTGTG-3′, R 5′-CTCTGCCGCAGTTTTCTCTT-3′.
Transient transfection
Oligonucleotide hsa-miR-301a mimics (miR-301a), negative control (miR-control), hsa-miR-301a inhibitors (anti-miR-301a), inhibitor negative control (anti-miR-control) and siRNA for RUNX3 were purchased from GenePharma (Shanghai, PR China). Transfection of cells with oligonucleotides was performed using Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA, USA) at a final concentration of 100 nM. Transfection efficiency was monitored by qRT-PCR.
Cell proliferation assay
Cell proliferation was measured by the colorimetric water-soluble tetrazolium salt (WST) assay with a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). The cells were seeded onto 96-well plates (2 × 103 cells/well) 24 h post-transfection, and cell proliferation was examined every 24 h for fourdays. The number of viable cells was determined by measurement of the optical density difference between 450 and 600 nm using a Safire2 microplate reader (TECAN, Switzerland).
Soft agar colony formation assay
Cells were resuspended with 0.3 % soft agar 24 h post-transfection with oligonucleotides (100 nM) and layered onto 0.6 % solidified agar in RPMI 1640 containing 10 % FBS in 6-well plates (2 × 103 cells/well). The plates were then incubated for two weeks. Colonies that contained a minimum of 50 cells were counted under a microscope.
Apoptosis, cell cycle analysis and cell synchronization
Cells were collected following the addition of trypsin and washed with PBS 48 h post-transfection with oligonucleotides. For apoptosis analysis, cells were resuspended in Binding Buffer (Annexin V-FITC Apoptosis Detection Kit I; BD Pharmingen, San Diego, USA) at a concentration of 1 × 106 cells/ml. Next, 5 µl of FITC AnnexinV and 5 µl Propidium Iodide (BD Pharmingen) were added to 100 µl of the cell suspension. After incubation for 15 min in the dark, 400 µl Binding Buffer was added. Apoptosis was analysed by flow cytometry (FACS Calibur, Becton–Dickinson, Franklin Lakes, NJ, USA) using Cell Quest software (Becton–Dickinson). Apoptotic cells with Annexin V-FITC-positive and PI-negative label were detected.
For cell cycle analysis, the cells were fixed with 70 % ethanol and stored at 4 °C overnight. The following day, the fixed cells were washed with PBS, treated with RNase A (50 µg/ml), and stained with PI (50 µg/ml) for 30 min in the dark. Then the stained cells were analyzed by flow cytometry (FACS Calibur, Becton–Dickinson). At least 10,000 cells in each sample were analyzed to obtain a measurable signal.
For synchronization experiments, SGC-7901 cells were synchronized in early S phase by a double thymidine block (2 mM) for 12 h and subsequently released for 10 h in fresh medium. The SGC-7901 cells were synchronized in M phase by colchicine (0.1 µl/ml) and released into G1 phase.
Migration and invasion assay
Migration of the cells was assessed using 8 µm, 24-well format transwell chambers (Corning, NY, USA) according to the manufacturer’s instructions. For the invasion assay, the insert membranes were coated with diluted Matrigel (BD Biosciences, San Jose, CA, USA). Cells (3 × 104) in 300 µl serum-free medium were added to the upper chamber and cultured for 72 h. Finally, the insert membranes were stained with 0.04 % crystal violet.
Construction of the reporter gene system containing RUNX3 3′UTR
The wild-type Runx3 3′-UTR containing the predicted miR-301a binding site was synthesized using the following sequences: 5′-ctagtATGGAGCTGGGTGGAAACTGCTTTGCACTATCGTTTGCTTGGTGTTTGTTTTa-3′ (forward) and 5′-cgcgtAAAACAAACACCAAGCAAACGATAGTGCAAAGCAGTTTCCACCCAGCTCCATa-3′ (reverse). The mutant Runx3 3′-UTR construct was designed to mutate three intermittent nucleotides complementary to the miR-301a seed-region. The mutant sequences were: 5′-ctagtATGGAGCTGGGTGGAAACTGCTTaGgAgTATCGTTTGCTTGGTGTTTGTTTTa-3′ (forward) and 5′-cgcgtAAAACAAACACCAAGCAAACGATAcTcCtAAGCAGTTTCCACCCAGCTCCATa-3′ (reverse). Both strands were annealed and cloned into the SpeI-MluI sites of the pMIR-REPORT miRNA expression reporter vector (Applied Biosystems).
Luciferase activity assay
The 293T cells were cultured in 24-well plates and transfected with 0.2 µg of either wild-type or mutant pMIR/RUNX3 plasmid containing firefly luciferase, together with 0.01 µg of the pRL-TK vector (Promega, Wisconsin, USA) containing renilla luciferase and 1 µg oligonucleotides. Transfection was performed using Lipofectamine 2000 reagent (Invitrogen). Relative luciferase activity was calculated 36 h post-transfection by the Dual Luciferase Reporter Assay (Promega).
Western blot analysis
The RUNX3 protein level were quantified by standard western blotting procedures, using rabbit anti-human Runx3 antibody (Bioworld, Atlanta, Georgia, USA). Protein level were normalized to total GAPDH, using a rabbit monoclonal anti-GAPDH antibody (Abcam, UK).
Retrovirus production
A ~400 bp genomic region that included the primary transcript of miR-301a was cloned into the EcoRI–XhoI site of the modified pMSCV-GW-RfA-PGK-EGFP retroviral vector. Negative control vectors had no insert. For each cultured 293T plate (10 cm), a plasmid mixture containing 10 µg of miR-301a retroviral vector, 10 µg of gag/pol vector and 10 µg of VSVG vector was co-transfected with 90 µl FuGENE6 transfection reagent (Roche, Basel, Switzerland) added directly to 0.6 ml of serum-free medium. The plasmid/medium/FuGENE6 mix was added drop-wise to the cultured 293T plate. After 12 h, a 15 ml viral collection medium was added to the transfected cells. We then harvested the virus twice a day for two days. Then SGC-7901 cells were transfected by the retrovirus.
Tumor xenograft model and tumorigenicity assay
SGC-7901 cells (1.5 × 105 cells/mouse) stably transfected with retrovirus-miR-301a (SGC-7901/retro-miR-301a) or retrovirus-control vector (SGC-7901/vector) were subcutaneously injected into 4-week-old male BALB/C-nu/nu nude mice (Shanghai Laboratory Animal Center of China). The mice were raised for 28 days following injection, and tumors were measured every four days.
Statistical analysis
The relationship between the miR-301a expression level and clinicopathologic parameters was analyzed using the Pearson Chi-square test. When comparisons were made between two different groups, statistical significance was determined using the Student’s t test. All statistical analyses were performed using SPSS15.0 software (SPSS Inc., USA). A two-tailed value of p < 0.05 was considered to indicate a statistically significant result.
Results
miR-301a is aberrantly overexpressed in gastric cancer and is inversely associated with tumor differentiation
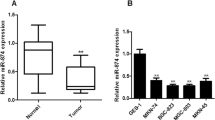
To explore miR-301a expression, quantitative real-time RT-PCR (qRT-PCR) analysis was performed. We found that miR-301a was greatly upregulated in nine gastric cancer cell lines compared with GES-1 (Fig. 1a). We also found an average of 2.29-fold overexpression of miR-301a in gastric cancer tissues compared with adjacent non-tumor tissues (p < 0.001, 45 of 51 patients; Fig. 1b and Online Resource S1).

miR-301a Expression in gastric cancer cell lines and tissues. a miR-301a expression in nine gastric cancer cell lines versus GES-1. b miR-301a expression in gastric cancer tissues versus adjacent non-tumor tissues. *p < 0.05, **p < 0.01, ***p < 0.001
We then investigated the relationship between an abnormal expression level of miR-301a and clinicopathologic factors in 51 patients. Based upon relative expression ratios of miR-301a/U6 > 3, the cases were divided into two groups: miR-301a high-expression group (n = 34) and miR-301a low-expression group (n = 17). The miR-301a high-expression group exhibited significantly lower tumor differentiation (p = 0.009). However, the miR-301a expression level did not show any relationship with age, gender, tumor site, local invasion, lymph node metastasis, and TNM stage (Table 1).
miR-301a promotes growth and clonogenicity of gastric cancer cells in vitro
Considering next overexpression and knockdown of miR-301a and a high oncogenicity in vivo, SGC-7901 cells were selected for further investigation. To investigate the biological function of miR-301a in the development and progression of gastric cancer, we transfected SGC-7901 cells with miR-301a mimic (SGC-7901/miR-301a) or anti-miR-301a (SGC-7901/anti-miR-301a) and confirmed that the expression of both was effectively altered (Online Resource S2 and S3). We next examined the effects of miR-301a overexpression or knockdown on gastric cancer cell proliferation. Using a WST assay, we observed that overexpression of miR-301a increased the growth rate of SGC-7901 cells compared to miR-control transfected cells at the third and fourth days following transfection (p = 0.033 and p = 0.002, respectively), whereas anti-miR-301a reduced cell growth activities at the same time points (p = 0.001 and p = 0.005, respectively; Fig. 2a).

Effects of miR-301a on growth activity of SGC-7901 cells. a Proliferation of SGC-7901 cells detected by WST assay. b, c Long-term growth activity of SGC-7901 cells detected by soft agar colony formation assay. The colonies were counted and identified. *, # p < 0.05, **, ## p < 0.01
To examine the effect of miR-301a on long-term and independent growth activity of SGC-7901 cells, we performed a colony forming assay in soft agar. The colony number of SGC-7901/miR-301a was higher compared with SGC-7901/miR-control (109 ± 11 vs. 154 ± 11, p = 0.002), whereas SGC-7901/anti-miR-301a showed lower colony-forming ability compared with SGC-7901/anti-miR-control (94 ± 8 vs. 50 ± 10, p = 0.003; Fig. 2b, c). These findings indicate that miR-301a promotes the colony-forming ability of gastric cancer cells.
miR-301a affected the apoptosis of gastric cancer cells treated with CDDP, while miR-301a inhibition induced cell cycle arrest in G2/M phase
Given cancer cell growth is affected by apoptosis or cell mitosis, we examined the effects of miR-301a on apoptosis or cell mitosis. Apoptosis of cancer cells can be induced by cisplatin (CDDP), a first-line chemical for gastric cancer treatment. Considering rare spontaneous apoptosis in SGC-7901 cells, SGC-7901 transfected with oligonucleotides was treated with 5 ng/ml CDDP. We found that the ectopic expression of miR-301a inhibited the apoptosis of SGC-7901 cells induced by CDDP, whereas knockdown of miR-301a augmented the apoptosis of SGC-7901 cells induced by CDDP (Fig. 3a, b).

miR-301a regulates apoptosis and cell cycle of SGC-7901. a, b FACS analysis of apoptosis by propidium iodide and annexin V-FITC staining of SGC-7901 cells treated by CDDP for 24 h. Reduced cell apoptosis was observed in miR-301a compared with control and vice versa. c, d Knockdown of miR-301a increased the percentage of G2/M phase SGC-7901 cells, but the G2/M phase did not change in the overexpressed miR-301a cells in FACS analysis. *p < 0.05, **p < 0.01, ***p < 0.001
The miR-301a is located at the first intron of SKA2 whose depletion affects cell cycle by metaphase-like delay [18]. Therefore, we hypothesized that the cell cycle might regulate miR-301a expression. To further elucidate the mechanism of miR-301a-mediated growth promotion in gastric cancer cells, cell cycle analysis was then performed (Fig. 3c, d). The results showed that, when miR-301a was knockdowned, the percentage of cells in G2/M phase was increased from 21.7 to 30.37 % (p = 0.013), whereas the percentage of cells in S phase and G0/G1 phase was decreased from 78.3 to 69.63 %. However, there was no significant difference in the percentage of cells in the G2/M phase when miR-301a is overexpressed in SGC-7901 cells. Furthermore, to examine whether mitosis affects miR-301a expression, miR-301a expression level was measured in diverse phases in the cell cycle of SGC-7901 cells (Online Resource S4). The results showed that miR-301a expression in S phase at 0 h, M phase and G1 phase was about two times higher than the lowest value in S phase at 8 h. It suggests that cell cycle affects miR-301a expression as well.
miR-301a promotes the migration and invasion of gastric cancer cells
To detect cell invasion abilities, cell migration and invasion assays were performed. Ectopic expression of miR-301a led to significantly enhanced cell migration (SGC-7901/miR-301a group vs. control group is 168 ± 8 vs. 85 ± 6 cells per field; p = 0.008) and invasion (SGC-7901/miR-301a group vs. control group is 132 ± 7 vs. 71 ± 6 cells per field; p = 0.031) (Fig. 4a–c), Whereas knockdown miR-301a significantly inhibited cell migration (SGC-7901/anti-miR-301a group vs. control group is 41 ± 4 vs. 80 ± 5 cells per field; p = 0.016) and invasion (SGC-7901/anti-miR-301a group vs. control group is 41 ± 5 vs. 70 ± 6 cells per field; p = 0.034).

miR-301a promotes migration and invasion of SGC-7901 cells. a Representative fields of SGC-7901 cells in the migration and invasion assays. b, c Average number of migration and invasion cells per field from three independent experiments. *p < 0.05, **p < 0.01
miR-301a promotes tumorigenicity in vivo
We further examined whether altered expression of miR-301a could affect tumor growth in vivo. Retrovirus-mediated SGC-7901/retro-miR-301a cells were verified to express miR-301a approximately 16-fold higher than SGC-7901/vector (p = 0.007; Online Resource S5). Then these two cell lines were injected subcutaneously into nude mice, and tumour formation was monitored every four days. Tumors grew faster in the SGC-7901/retro-miR-301a group than that in the group of the SGC-7901/vector. The average tumor volume in mice inoculated with SGC-7901/retro-miR-301a at day 28 was significantly larger than that in the mice inoculated with SGC-7901/vector (2.63 ± 0.98 vs. 1.45 ± 0.92 cm3; p = 0.029) (Fig. 5a, b).

miR-301a promotes tumorigenesis in vivo. a Tumorigenesis curves. Rapid tumor growth was observed in the SGC-7901/retro-miR-301a group. b Photographs of tumors derived from SGC-7901/retro-miR-301a or SGC-7901/vector cells in nude mice
RUNX3 is at least one of target genes of miR-301a that directly inhibits the expression of RUNX3 through its 3′UTR
To explore how miR-301a functions, we searched for its candidate target genes by bioinformatics analysis. Predicted miRNA targets were determined using the TargetScan, PicTar, and Miranda algorithms. We noticed that the tumor suppressor RUNX3 is a potential target gene of miR-301a in gastric cancer cells (Fig. 6a). To investigate whether the 3′UTR of RUNX3 mRNA is a functional target of miR-301a in gastric cancer cells, we first evaluated the reporter activity in 293T cells which co-transfected miR-301a (or miR-control), Luc-RUNX3 plasmid (or Luc-RUNX3-mut plasmid) and pRL-TK plasmid (containing the Renilla luciferase gene as an internal control). As shown in Fig. 6b, 293T cells co-transfected with miR-301a demonstrated a significant decrease of luciferase activity compared to the miR-control group. However, 293T cells co-transfected with miR-301a with Luc-RUNX3-mut plasmid and pRL-TK plasmid showed no significant difference in reporter activity as compared with cells co-transfected with miR-control group. Likewise, anti-miR-301a increased the luciferase activity of wild-type Luc-RUNX3, but had no effect on the mutant (Fig. 6c).

miR-301a binds to 3′UTR of RUNX3 and post-transcriptionally repressed protein expression. a Putative binding site of miR-301a in the RUNX3 3′UTR region (detected by RNAhybrid). b miR-301a down-regulated luciferase activities controlled by wild-type RUNX3 3′UTR, but did not affect luciferase activity controlled by mutant RUNX3 3′UTR. c Anti-miR-301a up-regulated luciferase activities controlled by wild-type RUNX3 3′UTR, but did not affect luciferase activity controlled by mutant RUNX3 3′UTR. d RUNX3 protein in SGC-7901 cells was detected by Western blot at 48 h post-transfection with miR-control, miR-301a, anti-miR-control, anti-miR-301a (100 nM). GAPDH was used as an internal loading control. e RUNX3 protein was detected in SGC-7901/vector or SGC-7901/miR-301a cells from subcutaneous tumor in nude mice. f qRT-PCR was performed to analyze the mRNA expression of Bim and MMP9 in SGC-7901 cells of transfection of miR-301a mimics or inhibitors. g The mRNA expression of SKA2 was analyzed in SGC-7901 cells with transfection of miR-301a mimics or inhibitors using qRT-PCR. *p < 0.05, **p < 0.01
miR-301a downregulates RUNX3 expression post-transcriptionally
To identify how miR-301a regulates RUNX3, Western blot and qRT-PCR analysis were performed. Forty-eight hours after transfection, we examined the RUNX3 protein level in SGC-7901 cells using Western blot analysis. The RUNX3 protein level was greatly suppressed in SGC-7901/miR-301a, whereas it was upregulated in SGC-7901/anti-miR-301a (Fig. 6d). Despite the effect of miR-301a on the RUNX3 protein level, no effect on the RUNX3 mRNA level was detected by qRT-PCR analysis (Online Resource S6). In addition, we examined expression of the RUNX3 protein in SGC-7901/retro-miR-301a or SGC-7901/vector derived from subcutaneous tumors of nude mice by western blot analysis, the RUNX3 protein level in SGC-7901/retro-miR-301a demonstrated downregulation compared with that in SGC-7901/vector (Fig. 6e). These results strongly suggest that miR-301a negatively regulates RUNX3 expression post-transcriptionally.
Overexpression of miR-301a downregulates Bim and upregulates MMP9 and SKA2
During CDDP-induced apoptosis, one of the major targets for transcription regulation by RUNX3 is the apoptotic activator Bim [19]. Reduced expression of Bim was observed 72 h after transfection of SGC-7901 cells with miR-301a mimics with the addition of CDDP compared with control (p = 0.025; Fig. 6f). The opposite effect on Bim gene expression was observed when SGC-7901 cells were transfected with miR-301a inhibitors (p = 0.019). Moreover, as another major target of RUNX3, MMP9 plays important roles in invasion and metastasis [20]. We found that overexpressed miR-301a upregulates mRNA expression level of MMP9 (p = 0.014). In contrast, knockdown of miR-301a in SGC-7901 cells resulted in significant reduction of MMP9 (p = 0.018). These results suggests that transfection of miR-301a mimics or inhibitors indirectly regulates the expression of Bim and MMP9. In a previous work, it was reported that miR-301a feedback regulated the expression of its host gene SKA2 in lung cancer A549 cells [21]. Next, we verified whether miR-301a regulated SKA2 in gastric cancer. As shown in Fig. 6g, knockdown of miR-301a significantly reduced the expression level of SKA2 (p = 0.024), but no significant change of SKA2 expression was observed in SGC-7901 cells of miR-301a overexpression (p = 0.157).
miR-301a-induced proliferation and invasion depends on the expression of RUNX3
To validate that miR-301a promotes cellular proliferation and invasion via the expression of RUNX3, RUNX3 was greatly downregulated by siRNA and miR-301a or negative control (NC) was co-transfected (Online Resource S7). As expected, NC + si-RUNX3 transfected SGC-7901 cells grew faster than miR-301a + NC transfected cells at the third day following transfection (p = 0.027). However, there was no significant difference between the NC + si-RUNX3 group and the miR-301a + si-RUNX3 group. It suggested that when downstream RUNX3 was depleted, the proliferation-promoting effect of miR-301a was largely compromised (Fig. 7a). Similar results were observed in the cellular invasion assay (Fig. 7b, c).

miR-301a-induced proliferation and invasion depends on the expression of RUNX3. a Proliferation of SGC-7901 cells detected by WST assay. b Average number of invasive cells per field from three independent experiments. c Representative fields of SGC-7901 cells in the invasion assay. *p < 0.05, **p < 0.01
Discussion
Accumulating evidence has indicated that aberrant expression of miRNAs may be a common mechanism involved in the development of various cancers [22–24]. Investigation of cancer-specific miRNAs and their targets is necessary for further elucidation of their role in the pathogenesis of tumors, and may be important for the design of novel therapeutic targets [10, 25, 26].
Overexpression of miR-301a has been previously reported in various types of human cancers, including hepatocellular carcinoma, pancreatic tumors, small cell lung cancer, and breast cancer [15–17, 27], indicating that miR-301a potentially plays an important role during the development of cancer. Indeed, it has been reported that miR-301a acts as an NF-κB activator in pancreatic cancer cells [28] and promotes proliferation and invasion in breast cancer. Furthermore, overexpression of miR-301a has been reported to be strongly associated with the recurrence of lymph nodal or distant metastasis and associated with worse disease-free survival (DFS) in lymph node negative (LNN) invasive ductal breast cancer [27]. In this study, we detected miR-301a expression in nine gastric cancer cells and the tumor tissues and its adjacent non-tumor tissues by qRT-PCR. We found that miR-301a was overexpressed in gastric cell lines and tissues. We also found that miR-301a expression demonstrated a strong association with tumor differentiation (p = 0.009), but did not show an association with lymph node metastasis. And as a target gene of miR-301a, RUNX3 play an important role in gastric epithelial cells transdifferentiating into intestinal-type cells and carcinogenesis [29]. Thus, because miR-301a is frequently overexpressed in gastric cancer, it could potentially be a biomarker for predicting clinical behaviors of gastric cancer.
The miR-301a and its host gene SKA2 play important roles in the cell mitotic index. Depletion of either miR-301a or SKA2 leads to a strong increase in the mitotic index [18]. Considering depletion of miR-301a reducing SKA2 expression [21] and depletion of SKA2 arresting the mitosis in a M-phase-like state [18], we speculated that miR-301a regulated the mitotic index through SKA2 via M phase delay. This speculation was partially confirmed in our work that the inhibition of miR-301a arrested SGC-7901 cells in G2/M phase for cytometry, which contributed to the miR-301a knockdown-induced growth inhibition. As a centromere protein, enhanced SKA2 expression did not affect mitosis in the M phase. For this reason, when miR-301a was overexpressed, as we observed, SGC-7901 cells in G2/M phase remained constant. We found that miR-301a expression was not constant, but fluctuates with the cell cycle. It implied that miR-301a might take part in the cell cycle through alteration of miR-301a expression levels. The specific mechanism needs further study.
One miRNA potentially targets different mRNAs. The miR-301a has been reported to target MEOX2 in lung cancer [21], and to target FOXF2, BBC3, PTEN, and COL2A1 in breast cancer [27]. The expression level of miR-301a exhibits an association with RUNX3 protein level in eight gastric cancer cell lines (p = 0.001) [30]. Interestingly, a discrepancy is shown between RUNX3 mRNA and protein level in the above work. The absence of mRNA–protein correlation suggests that the relation between mRNA and protein is not strictly linear, but has a more complex and intrinsic dependence. Different regulation mechanisms (such as post-transcriptional regulation of genes, synthesis and degradation rates), affect the amount of the two molecules differentially. We further demonstrated that RUNX3 is a potential target gene of miR-301a according to bioinformatics algorithms. RUNX3 is a crucial tumor suppressor in gastric cancer, and appears to be principally prominent in epithelial cells of the adult gastrointestinal tract and in hematopoietic cells. The RUNX3 is frequently downregulated in human gastric cancer [31]. The gastric mucosa of RUNX3-null mice develop gastric epithelial hyperplasia because of increased cell proliferation and suppressed apoptosis [32]. It has been shown that targeted restoration of RUNX3 expression in gastric cancer cells inhibits tumor growth and prevents metastasis [31]. In humans, RUNX3 controls the growth and differentiation of gastric epithelial cells [33]. Our study demonstrated that miR-301a potentially functions as an oncogene; based on our experimental evidence, we also demonstrated that miR-301a-induced proliferation and invasion depends on the expression of RUNX3 in the manner of directly post-transcriptional downregulation in gastric cancer. Therefore, we consider that RUNX3 should be at least one of target genes of miR-301a.
In conclusion, our findings indicate that miR-301a is up-regulated in gastric cancer and affects the proliferation and metastatic potential of gastric cancer cells, partially through the regulation of RUNX3. Thus, the identification of the role of miR-301a as an oncogene through targeting RUNX3 in gastric cancer helps us to further elucidate the potential molecular mechanisms of gastric cancer development, and could potentially have diagnostic as well as therapeutic value in the future.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90.
Stock M, Otto F. Gene deregulation in gastric cancer. Gene. 2005;360:1–19.
Yasui W, Yokozaki H, Fujimoto J, Naka K, Kuniyasu H, Tahara E. Genetic and epigenetic alterations in multistep carcinogenesis of the stomach. J Gastroenterol. 2000;35(Suppl 12):111–5.
Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Pasquinelli AE, Hunter S, Bracht J. MicroRNAs: a developing story. Curr Opin Genet Dev. 2005;15:200–5.
Ahmed FE. Role of miRNA in carcinogenesis and biomarker selection: a methodological view. Expert Rev Mol Diagn. 2007;7:569–603.
Michael MZ, O’Connor SM, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–91.
Cummins JM, He Y, Leary RJ, Pagliarini R, Diaz LA Jr, Sjoblom T, et al. The colorectal microRNAome. Proc Natl Acad Sci USA. 2006;103:3687–92.
Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA. 2004;101:11755–60.
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–70.
Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–98.
Pallante P, Visone R, Ferracin M, Ferraro A, Berlingieri MT, Troncone G, et al. MicroRNA deregulation in human thyroid papillary carcinomas. Endocr Relat Cancer. 2006;13:497–508.
Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, Okanoue T, et al. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006;25:2537–45.
Jiang J, Gusev Y, Aderca I, Mettler TA, Nagorney DM, Brackett DJ, et al. Association of MicroRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clin Cancer Res. 2008;14:419–27.
Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, Frankel WL, et al. Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer. 2007;120:1046–54.
Miko E, Czimmerer Z, Csanky E, Boros G, Buslig J, Dezso B, et al. Differentially expressed microRNAs in small cell lung cancer. Exp Lung Res. 2009;35:646–64.
Hanisch A, Sillje HH, Nigg EA. Timely anaphase onset requires a novel spindle and kinetochore complex comprising Ska1 and Ska2. EMBO J. 2006;25:5504–15.
Yano T, Ito K, Fukamachi H, Chi XZ, Wee H. The RUNX3 tumor suppressor upregulates Bim in gastric epithelial cells undergoing transforming growth factor beta-induced apoptosis. Mol Cell Biol. 2006;26:4474–88.
Chen Y, Wei X, Guo C, Jin H, Han Z, Han Y, et al. Runx3 suppresses gastric cancer metastasis through inactivation of MMP9 by upregulation of TIMP-1. Int J Cancer. 2011;129:1586–98.
Cao G, Huang B, Liu Z, Zhang J, Xu H, Xia W, et al. Intronic miR-301 feedback regulates its host gene, ska2, in A549 cells by targeting MEOX2 to affect ERK/CREB pathways. Biochem Biophys Res Commun. 2010;396:978–82.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–9.
Metzler M, Wilda M, Busch K, Viehmann S, Borkhardt A. High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosom Cancer. 2004;39:167–9.
Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005;102:3627–32.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8.
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257–61.
Shi W, Gerster K, Alajez NM, Tsang J, Waldron L, Pintilie M, et al. MicroRNA-301 mediates proliferation and invasion in human breast cancer. Cancer Res. 2011;71:2926–37.
Lu Z, Li Y, Takwi A, Li B, Zhang J, Conklin DJ, et al. miR-301a as an NF-kappaB activator in pancreatic cancer cells. EMBO J. 2011;30:57–67.
Fukamachi H, Mimata A, Tanaka I, Ito K, Ito Y, Yuasa Y. In vitro differentiation of Runx3−/− p53−/− gastric epithelial cells into intestinal type cells. Cancer Sci. 2008;99:671–6.
Lai KW, Koh KX, Loh M, Tada K, Subramaniam MM, Lim XY, et al. MicroRNA-130b regulates the tumour suppressor RUNX3 in gastric cancer. Eur J Cancer. 2010;46:1456–63.
Wei D, Gong W, Oh SC, Li Q, Kim WD, Wang L, et al. Loss of RUNX3 expression significantly affects the clinical outcome of gastric cancer patients and its restoration causes drastic suppression of tumor growth and metastasis. Cancer Res. 2005;65:4809–16.
Li QL, Ito K, Sakakura C, Fukamachi H, Inoue K, Chi XZ, et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109:113–24.
Fukamachi H, Ito K, Ito Y. Runx3−/− gastric epithelial cells differentiate into intestinal type cells. Biochem Biophys Res Commun. 2004;321:58–64.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (No. 30872476, No. 30900670, No. 81101585, No. 91229106 and No. 81172324), and Science and Technology Commission of Shanghai Municipality (No. 12XD1403700, and No. 12PJ1406300).
Conflict of interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
535_2012_733_MOESM2_ESM.tif
S2 The miR-301a expression in SGC-7901 cells was effectively altered by transient transfection of miR-301a (miR-301a mimics) as detected by RT-PCR. U6 snRNA was used for normalization. (TIFF 429 kb)
535_2012_733_MOESM3_ESM.tif
S3 The miR-301a expression in SGC-7901 cells was effectively altered by transient transfection of anti-miR-301a (miR-301a inhibitors) as detected by RT-PCR. (TIFF 434 kb)
535_2012_733_MOESM6_ESM.tif
S6 RUNX3 mRNA expression in SGC-7901 cells was analyzed by qRT-PCR 48 h post-transfection with miR-301a precursor and control precursor (100 nM). The results was shown as fold change relative to the control precursor-transfected SGC-7901 cells. (TIFF 541 kb)
Rights and permissions
About this article
Cite this article
Wang, M., Li, C., Yu, B. et al. Overexpressed miR-301a promotes cell proliferation and invasion by targeting RUNX3 in gastric cancer. J Gastroenterol 48, 1023–1033 (2013). https://doi.org/10.1007/s00535-012-0733-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-012-0733-6