
Abstract
Background
Although most gastrointestinal stromal tumours (GIST) carry oncogenic mutations in KIT exons 9, 11, 13 and 17, or in platelet-derived growth factor receptor alpha (PDGFRA) exons 12, 14 and 18, around 10% of GIST are free of these mutations. Genotyping and accurate detection of KIT/PDGFRA mutations in GIST are becoming increasingly useful for clinicians in the management of the disease.
Method
To evaluate and improve laboratory practice in GIST mutation detection, we developed a mutational screening quality control program. Eleven laboratories were enrolled in this program and 50 DNA samples were analysed, each of them by four different laboratories, giving 200 mutational reports.
Results
In total, eight mutations were not detected by at least one laboratory. One false positive result was reported in one sample. Thus, the mean global rate of error with clinical implication based on 200 reports was 4.5%. Concerning specific polymorphisms detection, the rate varied from 0 to 100%, depending on the laboratory. The way mutations were reported was very heterogeneous, and some errors were detected.
Conclusion
This study demonstrated that such a program was necessary for laboratories to improve the quality of the analysis, because an error rate of 4.5% may have clinical consequences for the patient.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastrointestinal stromal tumours (GIST) are mesenchymal tumours mainly arising along the intestinal tract, mostly in the stomach and in the small intestine. The tumours are characterized by exclusive mutation in KIT (70–80%) or platelet-derived growth factor receptor alpha (PDGFRA) (~10%) [1–4]. The remaining GIST, referred to as wild-type (WT-GIST), do not carry KIT or PDGFRA mutations. Mutations are found in exons 9, 11, 13 and 17 of KIT and in exons 12, 14 and 18 of PDGFRA [5–10].
Within KIT exon 9, one major mutation is a duplication of 6 nucleotides (c.1504_1509dup) [2], although a few other rare mutations have been reported [11, 12]. The KIT exon 11 mutations are quite heterogeneous, encompassing mainly in-frame deletions of variable sizes, basic amino acid substitutions, the duplications in the 3′ part of the exon, or more complex deletions-insertions [13–15]. Within KIT exons 13 and 17, mainly point mutations have been described [16, 17]. The majority of mutations in KIT are found in exon 11 (80%) and exon 9 (5–18%). The mutations in KIT exons 13 and 17 are rare, but they can be detected frequently in imatinib-refractory GIST in addition to the primary mutation [16, 18, 19]. Mutations in PDGFRA concern predominately GIST of gastric origin [10]. Most of these mutations are located in exon 18 (deletions and point mutations) [7], whereas mutations in exons 12 and 14 are less frequent [12]. Notably, the most frequent mutation in exon 18 of PDGFRA, p.Asp842Val, confers primary resistance to imatinib mesylate treatment [20]. Some mutations detected in the homozygous state were reported to have an adverse prognostic impact in GIST patients, predisposing them to liver metastasis [21, 22].
Imatinib mesylate was one of the first targeted anti-cancer therapies developed. Several clinical trials have proved that the response to imatinib in GIST correlates with the tumour genotype, with the best response observed in tumours with KIT exon 11 mutations [23, 24]; these observations are of the utmost importance. Data have also shown that patients with exon 9 KIT mutations fare better in terms of progression-free survival on a higher dose level, i.e. 800 mg daily, which is therefore recommended treatment in this subgroup [25]. The inefficacy of the standard dose of the drug for GIST bearing a common mutation in KIT exon 9 or WT-GISTs, the resistance induced by some mutations and the potential adverse side effects of the drug, render the tumour KIT/PDGFRA mutational status necessary to propose an alternative therapy to the patients [25, 26].
The most widely available materials come from fixed tumour tissues and the fixative most commonly used is formalin. Unfortunately, DNA extracted from fixed tissues is not of the best quality [27], and the DNA is often broken and chemically modified [28]. In these cases, polymerase chain reaction (PCR) amplification may be difficult and specific PCR primers must be chosen that yield short PCR amplification products. Moreover, sequencing artefacts due to the fixative are routinely detected and may be misinterpreted as mutations [29]. Special care must be taken when analysing mutations with DNA extracted from fixed paraffin-embedded tumours.
Pathology laboratories are increasingly asking for GIST mutational status and here we proposed a quality control program for mutation detection. The objective of this program was to allow laboratories to evaluate their own practice in GIST mutation detection and eventually to improve the quality of the service.
Materials and methods
Centres
Eleven laboratories (centres 1–11) participated in this study. Seven laboratories were affiliated to the Conticanet structure (Connective Tissue Cancers Network to Integrate European Experience in Adults and Children) (centres 5–11) spread across France (n = 3), the UK (n = 1), Belgium (n = 1) and Italy (n = 2). The laboratories not affiliated were from France (n = 3) and Poland (n = 1).
Material
Each Conticanet laboratory submitted 7 DNA samples (except one laboratory that submitted 8 samples), each comprising at least 5 μg of DNA that had been previously analysed for KIT and PDGFRA mutations, to the investigator coordinating laboratory. Samples were chosen in a blind manner and the selection of one every ten samples included in the routine process was proposed. A total of 50 DNA samples were collected; 9 extracted from frozen tumours and 41 from formalin-fixed tumours (Table 1). DNA was extracted using commercial kits (centre 5, Wizard DNA clean-up system, Promega, Madison, WI, USA; centres 6, 8, 9, 10, QIAamp DNA tissue kit, Hilden, Germany; centre 7, RecoverAll Nucleic Acid, Applied biosystems, Foster City, CA, USA; centre 11, High Pure PCR template preparation kit, Roche, Mannheim, Germany). Each centre submitting DNA completed a document with the following information for each sample: first three letters of the patient name, tumour identification number, information about the tumour (primary tumour, relapse, metastasis, tumour under imatinib treatment), information about the type of preservation (frozen or fixed tumours, and type of fixative), DNA concentration and mutational analysis report (type of mutation or polymorphism, and the zygosity status). On reception at the investigator centre, each DNA sample was quantified, and divided into 4 aliquots with a minimum DNA quantity of 1 μg each.
An aliquot from each DNA sample was sent to 3 other centres, meaning that each DNA sample was analysed by four different groups (the laboratory sending the DNA to the investigator and the three centres performing the quality control analyses). Overall, this resulted in 50 DNA samples analysed by four centres, giving 200 mutational reports. Each Conticanet centre analysed 20 samples (140 exons) and other centres screened 15 specimens (105 exons) for a total of 1400 exons analysed.
Each laboratory performing the test had to fill in a document reporting the mutation detected with the zygosity status and the polymorphisms detected.
Methods
Each centre analysed the sample for KIT (exons 9, 11, 13 and 17) and PDGFRA (exons 12, 14 and 18) mutations according to its routine process. The reference sequences used in this study are NM_000222.2 for KIT and NM_006206.4 for PDGFRA.
Most centres analysed the mutations by direct sequencing or by a pre-screening method followed by sequencing, specifically centres 2, 4, 7, 8, 9 and 10 used direct sequencing; centres 1 and 3 used length analysis of the PCR product (LAPP) for KIT exons 9 and 11, then direct sequencing for the other exons; centre 6 used length analysis of the PCR product for exons 9, denaturing high-performance liquid chromatography (DHPLC) screening and sequencing for the other exons; centres 5 and 11 used DHPLC screening and sequencing for all the exons.
Results
Obtaining the results
All results were sent back within the allocated time of 4.5 months. The time for delivering the results varied from 1 to 4.5 months (mean 3.7 months).
Efficiency of the analysis
Nine centres out of 11 (82%) analysed all samples entirely. Centres 3 and 4 analysed 44% and 49% of the 105 exons, respectively. Overall, the 11 centres evaluated mutations for 1287 exons out of 1400. Yet, laboratories were unable to amplify 1.4% of the exons (18/1287). Seventy-three percent of the centres (n = 8) amplified all the exons.
Mutation analysis
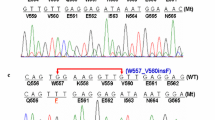
Fifty samples were analysed 4 times, giving 200 reports. Correct results were defined when at least two groups detected the same mutation. When there was no consensus for the correct mutation, the sample was analysed by the reference centre (Department of Human Genetics, University of Leuven, Leuven, Belgium). For two samples (4%), the reference centre was asked to check for the KIT exon 13 and 17 (sample 1) and the KIT exon 11 (sample 5). Mutations in the KIT exon 13 (c.1988G>T) and exon 17 (c.2446G>C) were detected for sample 1. The KIT exon 11 was detected to be a wild type for sample 5 (Table 2).
Overall, at least one group did not report the detectable mutation in 8 out of 200 analysis (Table 3), giving 4% false negative results. The rate of false negative results for mutation detection varied according to the laboratory and ranged from 0 to 15% (mean 4.5%, Table 4). As presented in Table 3, the false negative results concerned mainly some mutations for KIT exon 11. One false positive mutation (2%) was detected (sample 5), i.e. centre 6 detected a false positive mutation in KIT exon 11 (c.1656_1676 del). In addition, centre 3 detected no deletion nor insertion after length analysis of the PCR product. The two other centres (centres 1 and 2) were unfortunately unable to amplify a PCR product. The reference centre confirmed the absence of mutation for KIT exon 11 (Table 2).
Homozygous mutations
One sample out of 50 had a homozygous/hemizygous mutation in KIT exon 11 (c.1669_1725del). The homozygous status of the mutation was reported by two groups. The third group did not report on this status and the remaining group detected no mutation.
Polymorphism detection
Because of the variable location of the primers according to laboratory, some polymorphisms were not detected by some groups. We reduced the analysis to the two most frequent polymorphisms [single-nucleotide polymorphism (SNP) rs2228230 C/T, NM_006206.4:c.2472C>T, PDGFRA exon 18 and SNP rs55789615 C/T, NM_000222.2:c.2394C>T, KIT exon 17] located in the exonic sequence of the two genes. Three laboratories did not check for the polymorphisms (centres 3, 4, 7). Seventeen polymorphisms were present over the 50 samples. At least one laboratory failed to detect almost half of the polymorphisms present (8/17), and one laboratory detected 2 false positive polymorphisms (error rate 20%). Four laboratories failed to detect at least one of the polymorphisms (Table 5). One centre had a false positive rate of polymorphism detection of 40% (2 polymorphisms detected out of 5 analysed). In fact, this laboratory detected the polymorphisms NM_006206.4:c.2472C>T for 2 samples, although it was not detected by three other groups analysing these samples. Samples were analysed once more for PDGFRA exon 18 sequence and no polymorphism was detected.
Reporting of the mutation
The nucleotide and protein mutational report was requested. On the standard operational procedure sent to each participant, it was suggested to describe the mutation according to the guidelines for sequence variation of the Human Genome Variation Society (HGVS; http://www.hgvs.org/mutnomen). For five samples, the laboratories detected the mutation at the correct exon, but the mutation nomenclature was wrong. One laboratory was responsible for four wrong results (Table 6). For seven cases, there was discordance between the DNA and the protein sequence formula (Table 7). The mutation annotation was very heterogeneous and was not always annotated according to the HGVS recommendations. Some examples of this are given in Tables 6 and 7.
Discussion
Mutation analysis of KIT and PDGFRA in GIST is important for the confirmation of diagnosis in doubtful cases (as exemplified by CD117-immunonegative GIST) and for the optimal treatment of patients with imatinib mesylate [30]. Most tumours carrying mutations for KIT and some PDGFRA mutations are sensitive to the drug at a dose of 400 mg/day [31]. However, when a tumour carries the KIT exon 9 mutation (c.1504_1509dup), 800 mg/day might be more effective in obtaining an objective clinical response [24]. On the other end of the scale, the PDGFRA p.Asp842Val mutation confers primary insensitivity to imatinib [20]. The accuracy of mutation detection in GIST is an important challenge for laboratories. The mutational analysis may be difficult because of the number of exons to screen (7 exons), the high number of different type of mutations existing, especially in exon 11 of KIT, and the paraffin-embedded fixation material that limits DNA quality. To date, no consensus has been defined for GIST mutation detection. Here we investigated a mutation detection quality control program in GIST at the European level within the framework of Conticanet.
Overall, this program was successful because 82% of the centres gave a complete report for the GIST mutation detection. Overall, the discrepancy rate was 4.5% including eight false negative results and one false positive result. The high rate of false negative results that we observed concerned mainly exon 11 of KIT, and for 6 out of 8 samples the wrong result reported a wild-type GIST. The undetectable mutations were located in exons 13 and 17 (1 sample), exon 9 (1 sample) and exon 11 of KIT (5 samples) and exon 14 of PDGFRA (1 sample). The absence of detection of KIT exon 11 deletion by one group (number 7) for three samples was explained by the location of the primers. In this group, in order to improve the PCR amplification of highly degraded samples, the KIT exon 11 was amplified by two sets of primers. In all three instances, the mutations consisted of large deletions (36 and 57 bp), comprising the regions where the entire or at least the 3′ end of one primer was located and only the wild-type allele was amplified. All three deletions were detected retrospectively upon usage of a new set of primers covering the whole exon 11. Regarding the two other cases, where deletion of KIT exon 11 was falsely not detected, no explanation was found. In fact, one centre (number 2) was able to detect KIT exon 11 deletions for some other samples, while direct sequencing was used for mutation detection. The size of the PCR product cannot explain this discrepancy, because the mutation gave a shorter PCR product than the wild-type allele. For this group and for one other sample, the same deletion was detected (sample 21). The second group (group 1) that was not able to detect the KIT exon 11 deletion used the LAPP method for detecting some deletions or duplications. The mutation not detected concerned a small deletion of 3 nucleotides, and the sensitivity of the technique could explain this problem. Nevertheless, a small deletion of 3 nucleotides in KIT exon 11 for sample 47 was detected by the same group. Sample 31 showed a duplication in KIT exon 9 and the mutation was not detected by direct sequencing, although the analogous mutation was detected by the same group for another sample. The PCR reaction was performed again by this group changing the primer pairs and the KIT exon 9 duplication was detected. As reported by Lasota et al. [5] for KIT exon 11 duplication located at the 3′ end of the exon, a modification of the primers’ sequence could increase the rate of detection of mutation in exon 9 of KIT in fixed paraffin-embedded GIST. Centre 11 screened the mutation for a primary imatinib-naïve GIST (sample 36). This sample bore a mutation in exons 12 and 14 of PDGFRA. Although the three other groups detected these two mutations by direct sequencing, group 11 did not detect the mutation in exon 14 by means of pre-screening with DHPLC. The DHPLC technique is a very sensitive method of mutation detection [32], but the analysis of the abnormal profile may be difficult in particular with this unusual mutation. Moreover, this tumour was a primary imatinib-naïve GIST and a second mutation in the sample was not expected. Less attention to a DHPLC profile might explain the absence of this mutation detection. A surprising result concerned sample 1 for which only two groups were able to analyse all the exons. The DNA was extracted from a GIST after imatinib treatment. Each group detected two mutations, but each of them detected a different secondary mutation (one in exon 13 and one in exon 17), although all three mutations were present in the sample.
One false positive result was observed in this study. Centre 6 reported a deletion in exon 11 of KIT for sample 5, although the other centres did not. In addition, this centre reported the result as being inconsistent despite several analyses. In our hands, a similar observation was made while repeating the experiment. In fact, we tested several dilutions of DNA (data not shown) and surprisingly, according to the quantity of DNA to amplify, the mutation was not detected, detected, or detected in the homozygous state. A preferential allelic amplification seemed to be obtained according to the amount of DNA amplified, and we hypothesise that low DNA quantity allowed the allele bearing the mutation to be amplified. This observation could explain why some centres were not able to detect some deletion or duplication mutation by direct sequencing. We propose that in order to improve the mutation detection at least two dilutions of DNA should be used for DNA extracted from paraffin-embedded GIST. The same observation could explain why one centre reported a heterozygous mutation (sample 29), although two other centres reported homozygous status for this. The detection of two main polymorphisms was evaluated in this study, because they are located in the exonic sequences and can be detected regardless of the primer location. The rate of detection of the polymorphisms was lower compared with the mutation detection. The percentage error ranged from 14 to 100%, depending on the centres. This may be explained by less attention being paid by the laboratories to the polymorphisms detection, for which no clinical implication has been reported to date.
Most of the DNA samples were extracted from fixed paraffin-embedded tumours and DNA is known to be of a poor quality using this strategy. To improve the PCR reaction for degraded DNA, PCR primers are usually designed in order to obtain a short PCR product. Moreover, the DNA may be chemically modified by the fixative making it difficult to perform PCR amplification [27]. In this study, only 3 laboratories were unable to obtain a PCR product for at least one out of 7 exons. Surprisingly, one DNA sample extracted from a frozen tumour failed to be amplified by PCR for exons 9, 11, 13 and 17 of KIT and for exon 18 of PDGFRA by one centre, and for exon 11 of KIT (the only exon screened) by a second group. The analysis was successful for the third group and for the fourth group (only exon 9 of KIT was analysed). The choice of the PCR primers does not seem to explain this absence of PCR amplification, because the same centres analysed some other DNAs with success. The quality of the DNA may not be the reason behind the failure to perform PCR amplification, because one of the DNA samples of concern was extracted from a frozen tumour and was amplified by PCR by the other 3 groups.
Gastrointestinal stromal tumours are considered to be rare, although increasingly more are identified nowadays because of appropriate immunohistochemistry application and mutation screening. For research purposes, tumours should be collected in some database with histological, immunohistological, clinical and mutational data available for investigators. It is important to make mutational reports uniform at the DNA and protein level, and standard recommendations exist for mutation nomenclature (http://www.hgvs.org/mutnomen) [33]. We observed multiple errors in the nomenclature used to report the mutations. In Conticanet, a GIST database (http://www.conticagist.org) was constructed and each mutation report was standardized according to the HGVS recommendation, making some research easier to conduct.
This program was very successful, allowing laboratories to evaluate their own practice and to improve their procedure. One laboratory changed their primers for detection of KIT exon 11 mutation and one other changed all primers because the previous ones produced too many large PCR products for analysis conducted on DNA extracted from paraffin-embedded tissues. The PCR amplification must be optimal for avoiding analysis delay and also to limit the PCR contamination due to repeating PCR amplification of the same DNA.
We suggest the following recommendations for KIT and PDGFRA mutation screening. PCR primers must be located in intronic sequences and generate a PCR product with a maximum size of 300 bp. Yet, according to the PCR product size and to the PCR amplification efficiency, we suggest using primers from centres 5, 6, 8 or 9. The mutation detection error rate for KIT exons 9 and 11 was 0, 4.1 and 4.7% for DHPLC, the LAPP screening method and direct sequencing, respectively. For this reason, we suggest screening for KIT exon 9 and 11 mutations by DHPLC. DHPLC is also a good screening method for resting exons to avoid a laborious direct sequencing. However, sequencing the DHPLC variant from an independent PCR product serves as adequate confirmation of the result for quality control.
Such a quality control program is necessary, because the mean global error rate with clinical implication based on 200 reports was 4.5%. Centralization of mutational analysis in laboratories enrolled in an external quality assurance program and with expertise in the disease may be useful in order to make mutational analysis more widely available and more accurate.
References
Tomillo L, Terracciano LM. An update on molecular genetics of gastrointestinal stromal tumours. J Clin Pathol. 2006;59:557–63.
Hirota S, Nishida T, Isozaki K, Taniguchi M, Nakamura J, Okazaki T, Kitamura Y. Gain of function mutation at the extracellular domain of KIT in gastrointestinal stromal tumours. J Pathol. 2001;193:505–10.
Heinrich MC, Corless CL, Duensing A, MsGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CDM, Fletcher JA. PDGFRA activating mutations in gastrointestinal stromal tumours. Science. 2003;299:708–10.
Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813–25.
Lasota J, Wasag B, Steigen SE, Limon J, Miettinen M. Improved detection of KIT exon 11 duplication in formalin-fixed, paraffin-embedded gastrointestinal stromal tumours. J Mol Diagn. 2007;9:89–94.
Corless CL, McGreevey L, Town A, Schroeder A, Bainbridge T, Harrel P, Fletcher JA, Heinrich MC. KIT gene deletions at the intron 10–exon 11 boundary in GI stromal tumours. J Mol Diagn. 2004;6:366–70.
Wasag B, Debiec-Rychter M, Pauwels P, Stul M, Vrabckx H, Oosterom AV, Hagemeijer A, Sciot R. Differential expression of KIT/PDGFRA mutant isoforms in epithelioid and mixed variants of gastrointestinal stromal tumours depends predominantly on the tumor site. Mod Pathol. 2004;17:889–94.
Chen LL, Holden JA, Choi H, Zhu J, Wu EF, Jones KA, Ward JH, Andbacka RH, Randall RL, Scaife CL, Hunt KK, Prieto VG, Raymond AK, Zhang W, Trent JC, Benjamin RS, Frazier ML. Evolution from heterozygous to homozygous KIT mutation in gastrointestinal stromal tumor correlates with the mechanism of mitotic nondisjunction and significant tumor progression. Mod Pathol. 2008;21:826–36.
Lasota J, Stachura J, Miettinen M. GISTs with PDGFRA exon 14 mutations represent subset of clinically favorable gastric tumours with epithelioid morphology. Lab Invest. 2006;86:94–100.
Lasota J, Dansonka-Mieszhowska A, Sobin LH, Miettenen M. A great majority of GISTs with PDGFRA mutations represent gastric tumours of low or no malignant potential. Lab Invest. 2004;84:874–83.
Hostein I, Longy M, Gastaldello B, Geneste G, Coindre JM. Detection of a new mutation in KIT exon 9 in a gastrointestinal stromal tumour. Int J Cancer. 2006;118:2089–91.
Mazzola P, Spitale A, Banfi S, Mazzucchelli L, Frattini M, Bordoni A. Epidemiology and molecular biology of gastrointestinal stromal tumours (GISTs): a population-based study in the South of Switzerland, 1999–2005. Histol Histopathol. 2008;23:1379–86.
Biasco G, Velo D, Angriman I, Astorino M, Baldan A, Baseggio M, Basso U, Battaglia G, Bertin M, Bertorelle R, Bocus P, Brosolo P, Bulzacchi A, Cannizzaro R, Da Dalt GF, Di Battista M, Errante D, Fedrigo M, Frustaci S, Lionetti I, Massani M, Mencarelli R, Montesco MC, Norberto L, Pantaleo MA, Pasquali C, Pastorelli D, Rossi CR, Ruffolo C, Salvagno L, Saponara MS, Vittadello F, Zaccaria F, Zovato S, Farinati F. Gastrointestinal stromal tumours: report of an audit and review of the literature. Eur J Cancer Prev. 2009;18:106–16.
Conca E, Negri T, Gronchi A, Furnagalli E, Tamborini E, Pavan GM, Fermeglia M, Pierotti MA, Pricl S, Pilotti S. Activate and resist: L576P-KIT in GIST. Mol Cancer Ther. 2009;8:2491–5.
Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathol. 2008;53:245–66.
Chen LL, Trent JC, Wu EF, Fuller GN, Ramdas L, Zhang W, Raymond AK, Prieton VG, Oyedeji CO, Hunt KK, Pollock RE, Feig BW, Hayes KJ, Choi H, Macapinlac HA, Hittelman W, Velasco MA, Patel S, Burgess MA, Benjamin RS, Frazier ML. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumours. Cancer Res. 2004;64:5913–9.
Lasota J, Corless CL, Heinrich MC, Debiec-Rychter M, Sciot R, Wardelmann E, Merkelbach-Bruse S, Schildhaus HU, Steigen SE, Stachura J, Wozniak A, Antonescu C, Daum O, Martin J, Del Muro JG, Miettinen M. Clinicopathologic profile of gastrointestinal stromal tumours (GISTs) with primary KIT exon 13 or exon 17 mutations: a multicenter study on 54 cases. Mod Pathol. 2008;21:476–84.
Wardelmann E, Thomas N, Merkelbach-Bruse S, Pauls K, Speidel N, Buttner R, Bihl H, Leutner CC, Heinicke T, Hohenberger P. Acquired resistance to imatinib in gastrointestinal stromal tumours caused by multiple KIT mutations. Lancet Oncol. 2005;6:249–51.
Antonescu CR, Besmer P, Guo T, Arkun K, Horn G, Koryotowski B, Leversha MA, Jeffrey PD, Desantis D, Singer S, Brennan MF, Maki RG, DeMatteo RP. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–90.
Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, Vranckx H, Wasag B, Prenen H, Roesel J, Hagemeijer A, Van Oosterom A, Marynen P. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumours and activity of the PKC412 inhibitor against imatinib resistant mutants. Gastroenterology. 2005;128:270–9.
Lasota J, vel Dobosz AJ, Wasag B, Wozniak A, Kraszewska E, Michej W, Ptasynski K, Rutkowski P, Sarlomo-Rikala M, Steigen SE, Schneider-Stock R, Stachura J, Chosia M, Ogun G, Ruka W, Siedlecki JA, Miettinen M. Presence of homozygous KIT exon 11 mutations is strongly associated with malignant behavior in gastrointestinal stromal tumours. Lab Invest. 2007;87:1029–41.
Emile JF, Bachet JB, Tabone-Eglinger S, Terrier P, Vignault JM. GIST with homozygous KIT exon 11 mutations. Lab Invest. 2008;88:456–7.
Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247–53.
Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, Ryan CW, von Mehren M, Blanke CD, Rankin C, Benjamin RS, Bramwell VH, Demetri GD, Bertagnolli MM, Fletcher JA. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;20:5360–7.
Prene H, Cools J, Mentens N, Folens C, Sciot R, Schoffski P, Oosterom AV, Marynen P, Debiec-Rychter M. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res. 2006;12:2622–7.
Schittenhelm MM, Shiraga S, Schroeder A, Corbin AS, Griffith D, Lee FY, Bokemeyer C, Deininger AS, Druker BJ, Heinrich MC. Dasatinib (BMS-354825) a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66:473–81.
Srinivasan M, Sedmak D, Jewel S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am J Pathol. 2002;161:1961–71.
Masuda N, Ohnishi T, Kawamoto S, Monden M, Okubo K. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acid Res. 1999;27:4436–43.
Williams C, Ponten F, Moberg C, Söderkvist P, Uhlén M, Pontén J, Sitbon G, Lundeberg J. A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am J Pathol. 1999;155:1467–71.
Blay JY, Bonvalot S, Casali P, Choi H, Debiec-Richter M, Dei Tos AP, Emile JF, Gronchi A, Hogendoorn PC, Joensuu H, Le Cesne A, McClure J, Maurel J, Nupponen N, Ray-Coquard I, Reichardt P, Sciot R, Stroobants S, van Glabbeke M, van Oosterom A, Demetri GD. Consensus meeting for the management of gastrointestinal stromal tumors. Report of the GIST Consensus Conference of 20–21 March 2004, under the auspices of ESMO. Ann Oncol. 2005;16:566–78.
De Giorgi U. KIT mutations and imatinib dose effects in patients with gastrointestinal stroma tumours. J Clin Oncol. 2007;25:1146–7.
Battochio A, Mohammed S, Winthrop D, Lefresne S, Mulder K, Chu Q, O’Hara C, Lai R. Detection of c-KIT and PDGFRA gene mutations in gastrointestinal stromal tumours: comparison of DHPLC and DNA sequencing methods using a single population-based cohort. Am J Clin Pathol. 2010;133:149–55.
Ogino S, Gulley M, den Dunnen JT, Wilson RB. Standard mutation nomenclature in molecular diagnostics. J Mol Diagn. 2007;9:1–6.
Acknowledgments
This work was supported by CONTICANET (Connective Tissue Cancers Network to Integrate European Experience in Adults and Children), Lyon, France. We are grateful to Pippa McKelvie-Sebileau for help with the English manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Hostein, I., Debiec-Rychter, M., Olschwang, S. et al. A quality control program for mutation detection in KIT and PDGFRA in gastrointestinal stromal tumours. J Gastroenterol 46, 586–594 (2011). https://doi.org/10.1007/s00535-011-0375-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-011-0375-0