
Abstract
Key message
A set of five genic-SSR markers ( p < 0.1) are significantly associated ( r 2 = 19.92%) with six critical pomological traits. This information will facilitate marker-assisted genetic enhancement in mango.
Abstract
India is considered the home of mango cultivation and is the highest contributor to the global mango production. However, India has low productivity because of the lack of understanding of the genomic complexity of regular bearing habit, fruit quality, and yield-related traits. In the present investigation, 17 critical pomological traits in mango were mapped through genome-wide association mapping using the generalized linear model (GLM) and mixed linear model (MLM) approaches. Significant phenotypic variability in the selected traits was observed in 60 mango varieties, which indicated the suitability of these varieties for association studies. Genotyping of the varieties was performed using 87 polymorphic genic-simple sequence repeats (SSR) markers. Population structure analysis performed using the genotyping data revealed three distinct sub-populations that corroborated with the geographical origin of the varieties. Subsequently, the association analysis performed using the GLM identified 23 genic-SSR markers associated with 13 different pomological traits. However, only five genic-SSR markers were associated with six different pomological traits in MLM approach. The A and C alleles of the MSSR 190 locus were significantly associated with fruit diameter, pulp: stone ratio and fruit weight with significant phenotypic variance, and the B allele of the MSSR 146 locus was associated with fruit length and fruit width in both the GLM and MLM approaches with a high amount of phenotypic variance. The ranges of phenotypic variation explained by the associated loci were 13.18–42.39 and 13.31–27.34% in the GLM and MLM approaches, respectively. Extensive phenotypic and genotypic characterization of mango varieties, population structure, and markers associated with critical traits provided in this study will facilitate marker-assisted improvement programs in mango.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mango (Mangifera indica L.) is the favorite fruit of India because of its sweetness, richness of taste, enormous variability, large production volume, and variety of end usage (Singh 1960). Mango, belonging to family Anacardiaceae, and genus Mangifera, is an amphidiploid or allotetraploid tree (2n = 40). It has a small genome size of approximately 450 Mbp (Arumuganathan and Earle 1991). It originated in the northern foothills of India-Myanmar region. It includes 73 genera and approximately 830 species (Singh 1996; Yamanaka et al. 2006). India is the largest producer of mango in the world, with an annual production of 18 million tons from an area of 2.50 million hectares (NHB 2014), contributing approximately 56% of the total world production. Despite sustained research efforts for increasing the production and productivity over the past four decades, the productivity (t/ha) of mango orchards in India remains low at 7.20 with that of China 9.35, Thailand 8.20, Pakistan 10.62, Mexico 9.33 and Brazil 15.83 (Yadav and Pandey 2016). In addition, most of the produced fruit does not fulfill the quality requirement of consumer and meet export quality standards. Several factors are associated with low productivity and poor fruit quality. Among these factors, the alternate fruit-bearing habit of mango is of immense concern, because it causes considerable economic losses to growers because of poor yield or failure of the entire crop during the “off year” and low price during “on year” caused by a glut in the market (Monselise and Goldschmidt 1982). The sub-standard fruit quality [in terms of peel color, dwarfness, total soluble solids (TSS), ascorbic acid, fiberlessness, shape, size and pulp:stone ratio] for table and export purposes is another major problem in mango production. In the past, breeding approaches have been used by several researchers to improve yield and quality parameters such as alternate bearing (Roy 1953; Singh 1961; Luckwill 1970; Shamy and Eissa 1990; Davenport 2007; Ramírez et al. 2010; Nakagawa et al. 2012), fruit peel color (Sharma et al. 1983; Sharma 1987; Iyer and Subramanyam 1987; Pandey and Singh 1993, 1999; Pandey 1994; Prabhuram 1998) and fruit shape and size (Nayak 2010; Lavi et al. 1989). The main hurdles limiting the genetic studies and breeding programs of mango are a long juvenile period, large tree size, single-seededness, high fruit drop, sex ratio, self-incompatibility, and high requirement for cross-pollination (Nayak 2010). Therefore, efforts to enhance genomic resources and genetic understanding in mango are needed.
For genetic and genomics studies, microsatellite markers or simple sequence repeat (SSR) markers, have become the markers of choice because of their robustness, co-dominance, locus-specific nature, and abundance in genome. Consequently, they have been extensively used in genetic diversity analysis studies in mango (Surapaneni et al. 2013; Dillon et al. 2014; Begum et al. 2014; Gitahi et al. 2014; Ravishankar et al. 2015; Dinesh et al. 2015; Panwar et al. 2015). The SSR markers have been successfully used for genetic mapping, gene tagging, and genetic diversity assessment in numerous fruit crops including mango (Dinesh et al. 2015; Liang et al. 2015; Chaluvadi et al. 2014; Li et al. 2014; Rana et al. 2015). Identification of linked markers underlying major economic traits, such as disease resistance or stress tolerance in mango is extremely necessary. Furthermore, regular bearing, fruit quality, and yield-associated traits are extremely essential in mango. Thus far, however, sufficient efforts have not been made towards the identification of genomic loci governing crucial phenotypic traits in mango. Genic-SSRs are a type of gene-specific marker derived from the transcribed regions of the genome. They specifically target the functional region of the genome. They have potential for linkage to loci that mainly contribute to phenotypes. Such an endeavor will facilitate marker-assisted selection (MAS) and planning future improvement programs by selecting parents with favorable alleles. A promising approach for plant breeders, association analysis eliminates the main drawback of classical linkage analysis, namely prolonged and expensive development of specific mapping populations. Furthermore, the association analysis approach has the potential to assess a large number of alleles with higher mapping resolution (Yu et al. 2006; Gupta et al. 2014) than other approaches.
The identification of genomic regions responsible for alternate bearing, fruit quality, and yield-related traits is the first step for the identification of candidate genes. At present, few studies on the identification of genomic loci for the selected traits, such as biennial bearing habit in different fruit crops in olive (Sadok et al. 2013; Yanik et al. 2013; Dündar et al. 2013), apple (Guitton et al. 2012), cranberry (Schlautman et al. 2015), and citrus (Garcia et al. 2000), are available. Genomic SSR markers have been used to dissect fruit quality and yield attributes in apple (Espley et al. 2007; Morimoto et al. 2013), raspberry (McCallum et al. 2010), pear (Quilot et al. 2004), pomegranate (HarelBeja et al. 2015), peach (Verde et al. 2002), apricot (Salazar et al. 2013), sweet cherry (Ganopouls et al. 2011), and papaya (Blas et al. 2012).
Apart from the use of genic-markers, several analytical advances are available for the identification of loci governing traits. These advances include the stratification of population structure and statistical models such as generalized linear model (GLM) and mixed linear model (MLM) approaches. A unified, mixed-model approach for association mapping combined with a population structure analysis is a dependable and robust system for identifying reliable quantitative trait loci (QTL) (Yu and Buckler 2006; Zhao et al. 2007). This study focuses on analyzing the population structure of some varieties and identifying linked molecular markers for alternate bearing, fruit quality, and yield-related traits for mango. The proposed research work on association studies would considerably facilitate the understanding of the genetic basis of the component traits involved in alternate bearing, fruit quality, yield, and its components will enable the development of functional markers for these economic traits and will accelerate the mango breeding programs.
Materials and methods
Selection of varieties and genomic DNA extraction
A total of 60 mango varieties were collected from the scientifically maintained mango germplasm block of Division of Fruits and Horticultural Technology, ICAR-Indian Agricultural Research Institute, New Delhi, India (Table 1), where uniform treatment was given to all the varieties based on scientific recommendations and operations, such as spraying, intercultural operation, water and nutrient scheduling, insect pest management, pruning or training, harvesting, and weeding. These mango varieties were collected from different regions of India. Except for a few recently bred hybrids, all other varieties represent land races, which originated as chance seedlings and have been conserved in situ by vegetative propagation. These varieties were selected for screening and evaluation of component traits to identify the loci governing alternate bearing, fruit quality, and yield. The young, light-green tender leaves (5 g) of each variety were used for DNA extraction using the CTAB method described by Murray and Thompson (1980) with minor modifications. To overcome the co-precipitation of polysaccharides, the DNA precipitation step included NaCl (5 M) treatment (Porebski et al. 1997), and polyvinyl pyrrolidone (PVP, 2%) was used for the removal of phenolic compounds. Purified DNA was quantified using nanodrop (Thermo Scientific, USA). The integrity was assessed using agarose (0.8%) gel electrophoresis.
Phenotypic evaluation
To assess the phenotypic variability, correlation, and heritability as detailed below, a total of ten randomly selected fruits were used for bio-metric observation in each variety and replication. Data of 17 major quantitative traits with three replications (a single tree was selected to represent one replication) were analyzed using a randomized block design suggested by Singh and Chaudhury (1977). Uniform cultural practices were applied to experimental materials. The fruits were harvested at the horticultural maturity stage and for uniform ripening, the mango fruits immersed in ethrel solution (700 ppm; 1.8 mL/L) prepared in lukewarm water (52 ± 1 °C) for 5 min. The addition of carbendazim (0.5 g/L) to this solution provided protection from fungal diseases after harvest. For estimation of the alternate bearing index (ABI) and other traits, 5-year and 2-year average data, respectively, were considered and analyzed.
Fruit yield
Fruit yield was recorded by counting the number of fruits at the time of harvesting. The total yield in kg per tree was calculated by multiplying the number of fruits per tree with average fruit weight.
Alternate bearing index
A 5-year (2011, 2012, 2013, 2014, and 2015) yield per tree data were used and calculated as per formula given by Monselise and Goldschmidt (1982). ABI = {1/n − 1}{|(a 2 − a 1)|/(a 2 + a 1) + |(a 3 − a 2)|/(a 3 + a 2)}; where n is the number of years, and a 1, a 2, …, a( n − 1), an is the yield of corresponding years.
Peel color
The peel color of each variety was measured at ripening using a calibrated Hunter Lab UltraScan PRO colorimeter attached with an Easy Match QC software (Hunter Associate Laboratory Inc., Reston, USA), and the values were expressed as L* (lightness; 0 = black, 100 = white), a* (−a = greenness, +a = redness), and b* (−b = blueness, +b = yellowness). The values of C for chromaticity (color intensity), and H for hue were calculated using the following equations.
Fruit length
The fruit length was measured from the base of the fruit to the top of the groove at the calyx end using digital vernier calipers and expressed in millimeters.
Fruit diameter
The fruit diameter was measured at the maximum width of the fruit using digital vernier calipers and expressed in millimeters.
Estimation of fruit shape index
The fruit shape index (FSI) was calculated as the ratio of fruit length and diameter measured by vernier calipers.
The FSI value was determined as follows:
Pulp:stone ratio
This ratio was calculated by dividing the pulp weight by fresh stone weight.
Total soluble solids
Total soluble solids (oBrix): the total soluble solids (TSS) content of the berry was determined with the help of hand refractometer at room temperature 26 ± 2 °C (AOAC 1994).
Titratable acidity
Titratable acidity was determined using 10-g aliquots of strawberry fruits poured into 50 mL of distilled water and titrated against 0.1 N NaOH to an end-point of pH 8.1, where a light pink color developed. Titratable acidity was expressed as the percentage of citric acid and was calculated using the method given in AOAC (1994).
TSS/acid
This ratio was calculated by dividing TSS and titratable acidity values.
Ascorbic acid
Ascorbic acid content of fresh fruit was determined spectrophotometrically by metaphosphoric acid extraction of 2,6-dichlorophenol indophenol dye as described by Robinson and Stotz (1945) using a NanoDrop spectrophotometer (Model Thermo 8000, USA) at a wavelength of 500 nm. The results were expressed as mg ascorbic acid × 100/g fresh weight.
SSR genotyping
A total of 100 new genic-SSR loci were designed and synthesized from the leaf transcriptome sequencing data of mango (M. indica L.) cv. Amrapali (Table S1) (Singh et al. 2014; Mahato et al. 2015). The selected markers were chosen randomly from different contigs of mango leaf transcriptome sequences (unpublished data) hypothesizing their genome-wide distribution because although mango genome sequencing is in progress, no reliable high-density linkage map is available for this crop. Both PCR reaction master mix composition and temperature conditions were empirically standardized for newly synthesized genic-SSR markers. Each 10 µL reaction mixture contained 3 μL genomic DNA (10 ng/μL), 1.0 μL 10× PCR buffer (10 mM Tris HCl, 50 mM KCl, pH 8.3), 0.5 μL dNTP mix (0.2 mM each of dATP, dGTP, dTTP, and dCTP), 0.5 μL (10 pmol) of each of the two primers, 0.15 μL Taq DNA polymerase (5 U/μL) (Vivantis Technologies, USA), and 4.35 μL MilliQ water. The PCR reaction was carried out as follows: 1 cycle of 3 min at 94 °C for initial denaturation, then 36 cycles with a denaturation step at 94 °C for 30 s, an annealing step for 1 min at the respective annealing temperatures of each primer in the range of 48.3–53 °C, an initial extension at 72 °C for 1 min, and a last cycle for final extension at 72 °C for 10 min. A 3% metaphor agarose gel was used to resolve the amplified PCR products. Amplified results were confirmed by three replicate assays. Nonspecific fragments (errors due to stuttering) and null alleles amplified by genic-SSR markers were analyzed using Microchecker 2.2.3 (van Oosterhout et al. 2004). The clear and reproducible alleles amplified by each genic-SSR marker were scored based on their fragment size (bp).
Statistical analysis
Variations in 17 pomological traits studied among 60 mango varieties were observed in three independent replicates (consisting one tree in each replication) for two consequent years (2013 and 2014), except for ABI, which was calculated on the basis of 5-year yield data (2011–2015) under field conditions. The data were analyzed using the ANOVA procedure of SAS 9.2 (SAS Institute, Cary, NC, USA) to derive their summary statistics including mean, range, standard deviation, variance, and coefficient of variation. Furthermore, the Shapiro–Wilk normality test, measuring the data distribution of each trait, was carried out using the univariate procedure in the SAS 9.2 software. The positive and negative correlations among the 17 pomological traits in 60 varieties were measured using Pearson correlation coefficient at 1 and 5% levels of significance in SAS 9.2. Genotypic (\(\sigma_{\text{g}}^{2}\)) and phenotypic variances (\(\sigma_{\text{ph}}^{2}\)) were obtained from the analysis of variance table according to Comstock and Robinson (1952) using the following equations: (where r: replication, s: season MS1: mean square for variety, MS2: mean square for cultivar × season).
Broad-sense heritability was estimated as per standard methods described by Lush (1949), Johnson et al. (1955) and Hanson et al. (1956).
where, \(h_{\text{b}}^{2}\) is the heritability, \(\sigma_{\text{g}}^{ 2}\) is the genotypic variance, \(\sigma_{\text{p}}^{ 2}\) is the phenotypic variance.
In the current study, 100 novel genic-SSRs were selected and used for genotyping. Among those markers, 87 polymorphic markers were used for further population structure and marker–trait analysis. The remaining 13 genic-SSRs were monomorphic and were not included in the analysis.
For population structure, we assumed an admixed model with a uniform prior probability and independent allele frequency of the number of populations, K. All the runs with 100,000 MCMC replicates after a burn-in of 100,000 replicates were conducted for K = 2–10. Five independent runs were performed for each value of K to generate our estimate of the true number of sub-populations (Pritchard et al. 2000). The ‘Structure harvester’ program (http://taylor0.biology.ucla.edu) was used to determine the final K value (K = 3 was optimum for this analysis), based on both the LnP (D) and Evanno’s ΔK (Evanno et al. 2005; Earl et al. 2012). Markers exhibiting a probability value (p value) less than 0.01 thresholds were considered significantly associated with the particular phenotypic trait. The association of each alleles of the locus with the trait of interest was tested using two approaches, namely the GLM and MLM (Yu et al. 2006) approaches, wherein the population structure and kinship in the model was implemented using the TASSEL v2.0.1 software (Bradbury et al. 2007). The MLM model exhibited the least variation in observed p values from expected p values in the quartile–quartile plot compared with the variation of the Q (population structure) or K (kinship) model only.
Results
Phenotypic evaluation
Experimental field data were distributed normally, except for traits such as L*, a*, fruit length, fruit width, and FSI (Table S2; Fig. S1). Significant differences among individuals were observed through ANOVA for all measured traits (Table S3; Table 2). Among the traits analyzed, the ABI varied from 0.03 to 0.93 (average of 0.42 ± 0.03); however, the pulp:stone ratio ranged from 0.45 to 9.37 (3.28 ± 0.23) (Table 3). The yield range was 13.50–88.00 and the fruit weight varied in the range 71.62–327.50 (average 30.84 ± 1.79 kg) and 163.16 ± 8.04 g), respectively. The a*, pulp:stone ratio, ABI, and fruit weight exhibited greater CVs, 1.11, 0.53, 0.63, and 0.38, respectively, and a minimum in hue value (0.07). The broad-sense heritability ranged from 75.47 (TSS) to 93.23% (fruit weight) and all the selected traits had an H 2 higher than 75%, suggesting that all these traits are stably inherited (Table 3) (Fig. 1).

Trends of alternate bearing index over 5-year period (2011–2015) among 60 mango varieties. Vertical bar represents mean ± standard error
The correlations among the yield and quality traits are presented in Table 4. The b* value was found positively and significantly correlated with L* and a*. Similarly, the significantly positive correlation was also found between chromaticity, L*, a*, and b*. The yield exhibited a positive and significant correlation with the fruit weight, fruit diameter, and fruit width. The fruit length had a significant and positive correlation with fruit weight, however, the fruit diameter exhibited a positive and significant association with fruit weight and fruit width, whereas a negative and significant association with fruit length. The FSI showed a significantly positive correlation with the fruit length, and a significantly negative association with fruit diameter. In the case of sugar:acid ratio, a highly positive significant correlation was observed with the TSS, however, a negative significantly correlation was found with titratable acidity. The ascorbic acid exhibited a positive significant correlation with titratable acidity and had exhibited highly negative significant correlation with yield per tree. These wide phenotypic trait variations among 60 mango varieties indicated that the constituted association panel was suitable for association mapping for selected traits.
Population structure analysis
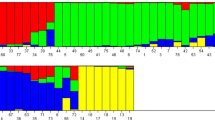
Of the 100 genome-wide genic-SSR loci, 87 loci were found polymorphic and 13 loci were monomorphic in nature and these polymorphic markers were further used in determining the population structure and marker–trait association analysis (Table S4). The Bayesian model-based program STRUCTURE 2.2 was used to infer the population structure. Admixture model-based simulations were carried out by varying K from 2 to 10 with ten runs for each K, using all 60 varieties which revealed evident knees at K = 3. The average LnP(D) (log-likelihood) value increased continuously with the increase in K from 2 to 10. However, its most apparent inflection was obtained at one of the best replicates of K = 3. The results of population numbers (K) were further confirmed using ∆K estimation. A sharp peak with the maximum value of ∆K was obtained at K = 3, thereby confirming the classification of 60 mangoes varieties into three distinct population groups (Fig. 2). Using this approach, 60 varieties were assigned to the corresponding A–C subpopulations, representing 23.33% (14), 66.66% (40), and 10.00% (6) of the varieties used for analysis (Table 1). The three sub-populations (A–C) had fixation index (Fst) equal to 0.15, 0.22, and 0.16, respectively, supporting the existence of moderate population structures. The overall Fst value estimated within the subpopulations was 0.176 (Table 5).

Model based population structure plot for each variety with K = 3, using Structure software with 87 polymorphic genic-SSR loci. Color codes are as follows: Population A red, Population B green, and Population C blue. The single vertical line represents an individual variety and different segments of each vertical line show extent of admixture in an individual (color figure online)
Furthermore, the genetic distances among these three sub-populations by Fst value were also measured which showed the variable level of genetic differentiation between inferred populations. The Fst value ranged from 0.133 to 0.63 with an average 0.381, revealing smallest genetic distances between sub-pop B and C the largest between sub-population A and C (Table 6). There are mixed proportions of accessions with admixture with parental varieties in sub-populations defined by the structure, which might have grouped in different defined clusters. In the sub-population A, the percentage of mixed individuals was found to be 57.14%, likewise 25% for B and 50% for C having q K values of less than 80% for all three populations.
Association analysis
Marker–allele–trait association was performed for 17 pomological traits using GLM and MLM models using the TASSEL software. By performing genome scanning, a total of 23 genic-SSR markers were associated with 13 different pomological traits and contributed 13.18–42.39% of the phenotypic variation in the GLM approach. In combination, with all 23 markers explained an average of 21.20% of the trait variation (Table 7). However, only five SSR markers were associated with six different pomological traits and contributed 13.31–27.34% of the phenotypic variation (Table 8) in the MLM approach. In combination, all five markers explained an average of 19.92% of the trait variation.
In the GLM approach, a significant correlation was revealed by the B allele of MSSR 146, MSSR 153, MSSR 156, MSSR 173, and MSSR 130 markers for fruit width. The A/B allele of MSSR 186, A/B allele of MSSR 147, B allele of MSSR156, B allele of MSSR 137, B allele of MSSR 179, and B allele of MSSR 146 were markers for fruit length. The C allele of MSSR 190, C allele of MSSR 189, B allele of MSSR 130, and B allele of MSSR 156 were markers for fruit diameter. The C allele of MSSR 155 and A allele of MSSR 187 were markers for acidity. The B allele of MSSR 160 for TSS. The B allele of MSSR 174, MSSR 178, and MSSR 146 were markers for ABI. The A allele of MSSR 173, A/B alleles of MSSR 102, A allele of MSSR 163, B allele of MSSR 146, A/C alleles of MSSR 190, and MSSR 189 were markers for fruit weight. The A allele of MSSR 190 and A/C alleles of MSSR 189 were markers for pulp:stone ratio. The A allele of MSSR 140, B allele of MSSR 179, and MSSR 174 were markers for yield/tree. The A/B allele of MSSR 106, and B allele of MSSR 146 were markers for hue. The B allele of MSSR 187, MSSR 117, and B/C allele of MSSR 155 were markers for TSS/acid ratio. The A/B allele of MSSR 137 was a marker for FSI (Table 7). Of the total 23 associated genic-SSR markers, 11 were found to be associated with more than one pomological trait. The MSSR 146 marker showed significant p value for multiple traits such as fruit width, ABI, fruit length, hue value, and fruit weight with considerable phenotypic variance. Similarly, the MSSR 190 was associated with the fruit diameter, pulp:stone ratio, and fruit weight. The MSSR 189 exhibited association with fruit diameter, pulp:stone ratio, and fruit weight; however, MSSR 155 was associated with fruit length and TSS/acid ratio. Similarly, MSSR 156 was associated with fruit length and fruit width, MSSR 130 with fruit diameter and fruit width, and MSSR 179 with fruit length and yield per tree. Meanwhile, MSSR 173 was associated with fruit weight and fruit width, MSSR 137 with FSI and fruit length, MSSR 147 with fruit length and yield, and MSSR 187 with acidity and TSS/acidity. These markers can be regarded as multi-trait associations.
However, in the MLM approach, a total of five SSR loci with different alleles showed a significant association with six pomological traits. The B allele of MSSR 146 marker was significantly associated with two traits, namely fruit length and fruit width with 27 and 24% of the phenotypic variance, respectively. The B allele of MSSR 160 with TSS, A/B allele of MSSR 106 with hue, A allele of MSSR 140 with yield per tree, and B allele of MSSR 174 with ABI. Of five significantly associated markers, one (B allele of MSSR 146) was associated with more than one trait, including fruit length and fruit width with considerable phenotypic variance (Table 8). The Manhattan plots of MSSR 146, in which the genomic coordinates are displayed along the X-axis and the negative logarithm of the association p value for each genic-SSR are displayed on the Y-axis, clarify that each dot on the Manhattan plot depicts a genic-SSRs. The Manhattan plot displaying the significant association of MSSR 146 with fruit width in both the approaches is depicted in Figs. 3 and 4.

Manhattan plots for fruit width trait using GLM

Manhattan plots for fruit width trait using MLM
Discussion
Phenotypic evaluation
Association analysis is a powerful tool to establish allele-trait associations and provides estimates of phenotypic variations exhibited by each locus. In the current study, significant phenotypic variations were observed for some pomological traits. The higher coefficients of variation for a*, pulp:stone, ABI, and fruit weight, suggests high potential for selection in a mango improvement program. Selection applied to one character can affect other characters because of the genetic correlation between traits (Cheng et al. 2004). The high heritability was observed for fruit characteristics such as fruit weight (93.23%), L* (90.30%), fruit width (90.40%), pulp:stone ratio (82.23%), fruit diameter (81.23%), and fruit length (81.06), thereby suggesting that selection for this character would be more effective because of a close correspondence between the genotype and phenotype (Singh 1991; Vasugi et al. 2013). The fruit weight and regular fruiting behavior are two determinative factors for high and consistent yield in mango. Because these traits are directly linked to each other, the major components of yield would lead to the selection of varieties with high fruit yield.
The alternate bearing trait showed highly significant negative correlation with yield per tree, which emphasized the hypothesis of the low number of fruit per tree is a sign of alternate bearing habit of varieties. In ber , Islam et al. (2010) and Saran et al. (2007) also reported similar findings among different pomological characteristics. The chromatic association (L*, a*, b*, hue, and chroma) is critical for the consumer; mango fruits with a red blush on the yellow peel are mostly preferred. Except for a*, all the color value exhibited a negative association with yield. The positively associated value of chromaticity parameter a*, which indicates the high blush color affecting the cosmetic appearance. Mango fruits having a sufficient red blush on the peel are preferred in the international trade (Sethi et al. 2011). It is desirable to give due consideration to the pulp:stone ratio, while selecting mango cultivars for higher productivity. Higher pulp:stone ratio is desirable while selecting mango cultivars for pickle making. Among the quality traits, namely L*, a*, b*, hue value, chromaticity, total soluble solids, titratable acidity, and ascorbic acid are the desirable attributes for fruit flavor, however, these traits do not affect the production. Mango cultivars having excellent flavor and taste may be selected without compromising total productivity of the crop. The major correlation analysis findings indicated that fruit weight, fruit diameter, stone length, and stone diameter were the major attributes contributing to fruit yield. Hence, selection of large-sized fruits will increase pulp recovery, which is suitable for the processing industry. Similarly, a highly positive correlations among fruit traits (fruit weight, fruit length, fruit diameter, stone weight, and stone diameter) were also reported by Majumder et al. (2013), Prasad (1987), Bhowmick and Banik (2008), Barholia and Yadav (2014), Karibasappa et al. (1999), Rai et al. (2001) and Yadav et al. (2003) for various mango cultivars. Knowledge about the correlation studies between various fruit quality, yield, and yield-related traits would certainly provide an idea, which could be utilized for the selection of desirable traits in future breeding programs and could enable possible the selection of all correlated traits concurrently by evaluating only one trait during the selection (León et al. 2004). These wider phenotypic trait variations among 60 mango varieties indicated that the constituted association panel was suitable for association mapping for selected traits.
Population structure analysis
Usually, mango has a moderate genetic base with moderate to high variability. However, because of the inclusion of different varieties, including native and exotic collection of germplasm and improved genotypes, a wide variability was observed in this study. A wider variation in the morphological traits was also detected in mango by Majumder et al. (2013), Gitahi et al. (2014), Begum et al. (2014), and Kheshin et al. (2016) also reported considerable variations in the composition of the mango population. Population stratification contributes to false positive results (Cardon and Bell 2001; Yu and Buckler 2006). The population structure with respect to geographical origin contributes toward pseudo-associations (Maskri et al. 2012). Therefore, adequate methods need to be implemented to control the effects of population structure to avoid the high rate of Type-I error (Agrama et al. 2007). In this study, a Bayesian model-based population structure analysis indicated the occurrence of three sub-populations largely according to the major geographic regions of their origin or genetic makeup.
The varieties having prefix Pusa (Pusa Surya, Pusa Arunima, Pusa Shresth, Pusa Lalima, Pusa Peetamber) grouped with Mallika and Amrapali and the most popular varieties of Northern India. These varieties were bred and collected from the same geographical locality, namely New Delhi, India and shared common parents, which indicates their genetic closeness. In addition, to other popular varieties from Uttar Pradesh (India) as Langra, Chausa, and Lucknow Safeda were grouped together indicating geographically and genetically closeness. In fact, the Indian mango gene pool comprises primarily the germplasm collected and bred in North India, and hence, such results are obvious. The overlapping of structures by a number of varieties indicated that such varieties showed common ancestry. Similarly West Indian, East Indian, South Indian, and Brazilian varieties clustered together, which indicates that these varieties probably had a complex breeding history with inter, intra-crossing, introgression, and out crossing among germplasms from diverse spatial and genetic backgrounds, followed by strong selection pressure for desirable traits. The varieties belonging to Florida, USA, clustered together the genetic relatedness among selected varieties was expected because they were related to each other by their pedigree. “Haden”, a seedling of Indian variety Mulgoba gave rise to several Floridian mango varieties like “Tommy Atkins”, “Edward”, and “Eldon”. Florida (USA) served as a secondary geographical diversity center for several varieties as Pusa Surya (Eldon), Sensation, Smith, Elard, and Edward (Lopez-Velenzuela et al. 1997) and these varieties were grouped together owing to their common geographical origin (Schnell et al. 1995; Olano et al. 2005). Separation based on their geographical location was evident among the selected mango varieties. However, some of the varieties could not be differentiated according to geographical patterns and hence admixtures in the populations can be attributed primarily due to cross-pollination, gene flow, particularly orchards situated in close proximity, chance seedling, and exclusive vegetative propagation. The information generated in this study population structure and ancestral background would facilitate germplasm conservation and management strategy and selection of suitable parents in mango breeding programs.
Marker–trait association analysis
In general, the GLM approach detects higher numbers of significant marker–trait associations than does the MLM approach (Neumann et al. 2011), however, GLM-based analysis considers only the Q matrix generated during the population structure analysis, whereas the MLM approach simultaneously accounts for both, population structure as well as kinship statistics (genetic relatedness among individuals). Hence, the MLM approaches are more reliable. Only 22.27% of the markers associated with different traits in the GLM approach also exhibited significant marker–trait association in the MLM. Furthermore, the extent of phenotypic variation in the MLM was comparatively less than that in the GLM, which proves that it was a more stringent approach for eliminating the spurious associations than was GLM. Studies conducted in rapeseed, Arabidopsis, bread wheat, and soybean (Cai et al. 2014; Yu et al. 2006; Zhao et al. 2007; Kumar et al. 2014) demonstrated the effectiveness of the MLM approach over the GLM. Because population structure can cause spurious associations (Kang et al. 2008) the MLM approach, incorporates kinship in the association analysis allowing an improved control of type I and type II error rates over GLM due to relatedness and population structure (Yu et al. 2006; Sa 2016).
In the current study, we observed that alleles of various loci were significantly associated with multiple traits. A typical example was the B allele of the MSSR 146 locus, which was highly associated with fruit length and fruit width in the GLM and MLM approaches with a high amount of phenotypic variance and fruit weight and ABI in the GLM approach only. Because fruit length, fruit width, fruit weight, and ABI are significantly correlated with fruit yield per tree, the associated alleles of marker MSSR 146 may be used to select fruit yield per tree. Meanwhile, B allele of MSSR 160 was significantly associated with TSS and the A and B allele of MSSR 106 was associated with hue in both the GLM and MLM approaches. The plants that bear fruits and have higher TSS and hue values are expected to provide higher fruit quality with an attractive peel color and can meet the international standards of export of mango. Similarly, the A allele of MSSR 140 and the B allele of MSSR 174 were associated with yield per tree and bearing habit. The similar type of association analysis for SSR and ISSR markers in sweet cherry cultivars revealed 114 SSR alleles apart from the three ISSR markers correlated with fruit harvest time and soluble solids and four ISSR markers with fruit skin color (Ganopouls et al. 2011). In olive, significant associations were determined between five SSR markers and oleic, stearic, linolenic acids, and linoleic of olive oil (Ipek et al. 2015), similarly using the MLM approach, a total of 16 significant associations for chemical traits and 12 for physical traits were identified in almond (Font i Forcada et al. 2015). In peach (Cao et al. 2012), estimated the genome-wide marker–trait associations for ten complex traits, such as red pigment in the pulp, pulp color around the stone, flesh texture, pulp adhesion, pulp firmness, fruit weight, ripening time, chilling, flowering time, requirement, and fruit development period. In this study, among the particular alleles of a total of 23 associated genic-SSR markers, 11 were found to be associated with more than one pomological traits in GLM, and the single allele of one locus in MLM with two traits indicates the precise evaluation of traits.
The alleles of different genic-SSR loci, which exhibit overlapping association with diverse traits, signify the biological correlation among them. Therefore, it was presumed that the loci governing fruit length, fruit width, fruit weight, alternate bearing habit, and yield per tree are probably located closest to each other on the same chromosome. In this investigation locus, the A and C alleles of MSSR 190 were significantly associated with fruit diameter, pulp:stone ratio, and fruit weight with significant phenotypic variance in the GLM and the B allele of MSSR 146 was found to be associated with the fruit length and fruit width in the MLM approaches with high phenotypic variance. Therefore, the MSSR 190 and MSSR 146 markers can be used in breeding programs for yield improvement in mango. In the Manhattan plots, MSSR 146 exhibited the strongest associations by exhibiting the smallest p values (e.g., SSR 146 for fruit width) and the highest negative logarithms value in both the approaches. These markers were considered for correlating pomological traits that have not been reported. Alternatively, they confirmed the suspected correlation with pomological traits, but these correlations require further investigation.
Conclusion
Considering the perennial nature of mango and its inherent genetic hurdles, the alleles that are significantly associated with five genic-SSR markers will prove extremely useful to identify promising candidate loci governing complex traits such as alternate bearing, fruit quality, and yield. This association analysis may be sufficient for marker-assisted selection and mining alleles related to major traits in mango germplasm collection, which will further facilitate early identification of desired cultivars while simultaneously saving significant time and resources. Knowledge regarding these associated loci would make valuable contributions to mango breeding programs particularly in the absence of genetic information such as linkage map and QTL. The identification of new SSR markers for effective MAS and fine resolution mapping of the genes or QTL underlying a trait.
Author contribution statement
SL, AKS, SKS, and NKS proposed, initiated, and led the project, interpreted scientific information, and participated in manuscript preparation. BPS and NS participated in DNA extraction, genotyping, statistical analysis, and manuscript preparation. MS maintained and provided the mango varieties and participated in manuscript preparation.
Abbreviations
- AMOVA:
-
Analysis of molecular variance
- ABI:
-
Alternate bearing index
- SSR:
-
Simple sequence repeat
- GLM:
-
Generalized linear model
- MLM:
-
Mixed linear model
References
Agrama HA, Eizenga GC, Yan W (2007) Association mapping of yield and its components in rice. Mol Breed 19:341–356
AOAC (1994) Official methods of analysis, 16th edn. Association of Official Analytical Chemists, Arlington
Arumuganathan K, Earle ED (1991) Nuclear DNA content of some important plant species. Plant Mol Biol 9:208–218
Barholia AK, Yadav S (2014) Fruit quality characters and their relation with weight per fruit in mango over years. Orient J Chem 30(3):34–39
Begum H, Reddy MT, Malathi S, Reddy BP, Narshimulu G, Nagaraju J, Siddiq EA (2014) Morphological and microsatellite analysis of intra-varietal heterogeneity in ‘Beneshan’ mango (Mangifera indica L.). Int J Agric Food Res 3(2):16–33
Bhowmick N, Banik BC (2008) Genetic variability and correlation studies for fruit physico-chemical properties of some mango cultivars grown under new alluvial zone of west Bengal. Asian J Hortic 3(2):346–349
Blas A, Qingyi Y, Olivia V, Robert P, Paul M, Ray M (2012) Genetic mapping of quantitative trait loci controlling fruit size and shape in papaya. URCE Mol Breed 29(2):457–462
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Cai D, Xiao Y, Yang W, Ye W, Wang B, Younas M, Wu J, Liu K (2014) Association mapping of six yield-related traits in rapeseed (Brassica napus L.). Theor Appl Genet 127:85–96
Cao K, Wang L, Zhu L, Fang G, Chen WC, Jing L (2012) Genetic diversity, linkage disequilibrium, and association mapping analyses of peach (Prunus persica L.) Landraces in China. Tree Genet Genomes 8:975–990
Cardon RL, Bell JI (2001) Association study designs for complex disease. Nat Rev Genet 2:91–99
Chaluvadi SR, Khanam S, Mohammed AMA, Jeffrey LB (2014) Genetic diversity and population structure of native and introduced date palm (Phoenix dactylifera) germplasm in the United Arab Emirates. Trop Plant Biol 7(1):30–41
Cheng CH, Seal AG, Boldingh HL, Marsh KB, MacRae EA, Murphy SJ, Ferguson AR (2004) Inheritance of taste characters and fruit size and number in a diploid Actinidia chinensis (Kiwifruit) population. Euphytica 138:185–195
Comstock RR, Robinson HF (1952) Genetic parameters, their estimation and significance. In: Proceedings of the 6th international Grassland congress, vol 1. National Publishing Company, Washington, DC, pp 248–291
Davenport TL (2007) Reproductive physiology of mango. Braz J Plant Physiol 9(4):363–376
Dillon NL, David JI, Bally ISE, Wright CL, Devitt LC, Dietzgen RG (2014) Expressed sequence tag-simple sequence repeat (EST-SSR) marker resources for diversity analysis of mango (Mangifera indica L.). Diversity 6:72–87
Dinesh MR, Ravishankar KV, Sthapit B, Parthasarathy VA, Sandya BS, Nischita P, Lavanya B (2015) Genetic diversity studies in certain indigenous mango (Mangifera indica L.) varieties. Indian J Plant Genet Res 28(1):153–160
Dündar E, Suakar O, Unver T, Dagdelen A (2013) Isolation and expression analysis of cDNAs that are associated with alternate bearing in Olea europaea L. cv. Ayvalik. BMC Genom 14:219–226
Earl DA, Von H, Bridgett M (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Espley RV, Hellens RP, Puterill J, Kutty-Amma S, Allan AC (2007) Red colouration in apple fruit is due to the activity of a MYB transcription factor, MdMYB10. Plant J 49:414–427
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620
Font i Forcada C, Oraguzie N, Reyes-Chin-Wo S, Espiau MT, Socias i Company R, Fernández i Martí A (2015) Identification of genetic loci associated with quality traits in almond via association mapping. PLoS One 10(6):12765
Ganopouls IV, Konstantinos K, Ioannis C, Irene K, Athanasios ST (2011) Genetic diversity, structure and fruit trait associations in Greek sweet cherry cultivars using microsatellite based (SSR/ISSR) and morpho-physiological markers. Euphytica 181(2):237–251
García MR, Asíns MJ, Carbonell EA (2000) QTL analysis of yield and seed number in citrus. Theor Appl Genet 101(3):487–493
Gitahi R, Kasili R, Kyallo M, Kehlenbeck K (2014) Morphological and genetic diversity of local mango accessions from the Eastern region of Kenya. In: Bridging the gap between increasing knowledge and decreasing resources Tropentag, 17–19 September 2014, Prague
Guitton B, Segura V, Kelner JJ, Foster T, Gardiner S, Chagné D, Costes E (2012) Genetic analysis and QTL detection for biennial bearing in apple. Acta Hortic (ISHS) 929:65–72
Gupta S, Kumari K, Muthamilarasan M, Parida SK, Prasad M (2014) Population structure and association mapping of yield contributing agronomic traits in foxtail millet. Plant Cell Rep 33:881–893
Hanson CH, Robinson HF, Comstock RS (1956) Biometrical studies of yield in segregating populations of Korean Lezpedeza. Agron J 48:268–272
HarelBeja R, Sherman A, Rubinstein M, Eshed R, BarYaakov I, Trainin T, Ophir R, Holland D (2015) A novel genetic map of pomegranate based on transcript markers enriched with QTLs for fruit quality traits. Tree Genet Genomes 11(5):1–18
Ipek M, Ipek A, Seker M, Gul MK (2015) Association of SSR markers with contents of fatty acids in olive oil and genetic diversity analysis of an olive core collection. Genet Mol Res 14(1):2241–2252
Islam MN, Hossain MM, Rahman MM, Uddin MS, Rahman MM (2010) Heritability, correlation and path coefficient analysis in twenty ber genotypes. Acad J Plant Sci 3(2):92–98
Iyer CPA, Subramanyan MD (1987) Improvement of mango by selection and hybridization. In: Annual report. Indian Institute of Horticulture Research, Bangalore, p 16
Johnson HW, Robinson HF, Comstock RE (1955) Estimates of genetic and environmental variability in soybean. Agron J 47:314–318
Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E (2008) Efficient control of population structure in model organism association mapping. Genetics 178(3):1709–1723. doi:10.1534/genetics.107.080101
Karibasappa GS, Nalawadi UG, Sulikeri GS (1999) Characterization of mango germplasm in North Karnataka, India. Indian J Plant Genet Res 12:330–340
Kheshin MAE, Hossam AS, Allatif AMA (2016) Morphological and molecular analysis of genetic diversity among some ‘Sukkary’ mango (Mangifera indica L.) genotypes. J Hortic Sci Ornam Plants 8(1):1–10
Kumar B, Talukdar A, Bala I, Verma K, Lal SK, Sapra RL, Namita B, Chander S, Tiwari R (2014) Population structure and association mapping studies for important agronomic traits in soybean. J Genet. doi:10.1007/s12041-014-0454-0
Lavi U, Tomer E, Gazit S (1989) Inheritance of agriculturally important traits in mango. Euphytica 4(12):56–61
León L, Martín LM, Rallo L (2004) Phenotypic correlations among agronomic traits in olive progenies. J Am Soc Hortic Sci 129:271–276
Li M, Zhong Z, Xingjun M, Jingjing Z (2014) Genetic diversity and population structure of Siberian apricot (Prunus sibirica L.) in China. Int J Mol Sci 15:377–400
Liang W, Dondini L, Franceschi PD, Paris R, Sansavini S, Tartarini S (2015) Genetic diversity, population structure and construction of a core collection of apple cultivars from Italian germplasm. Plant Mol Biol Rep 33:458–473
Lopez-Velenzuela JA, Martinez O, Paredes-Lopez O (1997) Geographic differentiation and embryo type identification in Mangifera indica L. cultivars using RAPD markers. Hortic Sci 32:1105–1108
Luckwill LC (1970) The control of growth and fruitfulness of apple trees. In: Luckwill LC, Cutting CV (eds) Physiology of tree crops. Academic Press, London, pp 237–254
Lush JL (1949) Heritability of quantitative characters in farm animals. Proceedings of inter-cropping. Cong Genet Hered (Suppl):356–357
Mahato AK, Sharma N, Singh AK, Singh A, Singh SK, Prakash J, Srivastava M, Gaikwad K, Sharma TR, Singh NK (2015) Identification of genic SSR and SNP by leaf transcriptome sequencing of mango (Mangifera indica L.) varieties Neelam and Dushehari. In: Plant and animal genome XXIII conference, 10–14 Jan 2015, San Diego
Majumder DAN, Hassan MA. Rahim L, Kabir MA (2013) Genetic diversity in mango (Mangifera indica L.) through multivariate analysis. Bangladesh J Agric Res 38(2):343–353
Maskri YA, Sajjad M, Khan SH (2012) Association mapping: a step forward to discovering new alleles for crop improvement. Int J Agric Biol 14:153–160
McCallum S, Woodhead M, Christine AH, Kassim A, Paterson A, Graham J (2010) Genetic and environmental effects influencing fruit colour and QTL analysis in raspberry. Theor Appl Genet 121(4):611–627
Monselise S, Goldschmidt EE (1982) Alternate bearing in fruit trees. Hortic Rev 4:128–173
Morimoto T, Hiramatsu Y, Banno K (2013) A major QTL controlling earliness of fruit maturity linked to the red leaf/red flesh trait in apple cv. ‘Maypole’. J Jpn Soc Hortic Sci 82(2):97–105
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acid Res 8:4321–4325
Nakagawa M, Honsho C, Kanzaki S, Shimizuc K, Utsunomiyaa N (2012) Isolation and expression analysis of FLOWERING LOCUS T-like and gibberellins metabolism genes in biennial-bearing mango trees. Sci Hortic 139:108–117
Nayak D (2010) Characterization of mango hybrids for fruit quality traits. Ph.D. thesis submitted to PG School, IARI, New Delhi
Neumann K, Kobiljski B, Dencic S, Varshney RK, Borner A (2011) Genome-wide association mapping: a case study in bread wheat (Triticum aestivum L.). Mol Breed 27:37–58
NHB (2014) Indian horticulture database 2013. National Horticulture Board, Gurgaon
Olano CT, Schnell JR, Quintanilla WE, Campbell RJ (2005) Pedigree analysis of Florida mango cultivars. Proc Fla State Hortic Soc 118:192–197
Pandey SN (1994) Breeding for improved shelf-life in mango (Mangifera indica L.). In: National seminar on strategies for packaging and storage of fresh and processed fruits and vegetables in 21st century (abstract 90). IARI, New Delhi
Pandey SN, Singh OP (1993) Breeding mango for colour. In: Golden jubilee symposium on horticultural research: changing scenario (abstract 95). Horticultural Society of India, Bengaluru
Pandey SN, Singh OP (1999) Evolving red-peeled mango (Mangifera indica L.) hybrid for sustainable export (abstract). In: 4th agricultural science congress, pp 205–206
Panwar S, Singh KP, Namita N, Sonah H, Deshmukh R, Sharma TR (2015) Identification and characterization of microsatellites in ESTs of Rosa species: insight in development of SSR markers. Indian J Agric Sci 85(3):429–433
Porebski S, Bailey LG, Baum BR (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep 15:8–15
Prabhuram R (1998) Breeding studies with special reference to red peel colour in mango (Mangifera indica L.). Ph.D. thesis. Division of Fruits and Horticultural Technology, IARI, New Delhi
Prasad A (1987) Correlation studies on growth, behaviour and fruit characters with yield components in mango. Indian J Hortic 44:183–193
Pritchard JK, Matthew S, Peter D (2000) Inference of population structure using multi-locus genotype data. Genetics 155:945–959
Quilot B, Wu BH, Kervella J, Génard M, Foulongne M, Moreau K (2004) QTL analysis of quality traits in an advanced backcross between Prunus persica cultivars and the wild relative species P. Davidiana. Theor Appl Genet 109(4):884897
Rai M, Reddy NN, Prasad VSRK (2001) Genetic variation and its relationship with yield components in mango cultivars grown under eastern Indian conditions. Indian J Hortic 58:314–320
Ramírez F, Davenport TL, Fischer G (2010) The number of leaves required for floral induction and translocation of the florigenic promoter in mango (Mangifera indica L.) in a tropical climate. Sci Hortic 123:443–453
Rana JC, Rakesh KC, Sharma V, Rana M, Verma N, Verma B, Sharma TR (2015) Genetic diversity and structure of Pyrus accessions of Indian Himalayan region based on morphological and SSR markers. Tree Genet Genomes 11:821. doi:10.1007/s11295-014-0821-2
Ravishankar KV, Bommisetty P, Bajpai A, Srivastava N, Mani BH, Vasugi C, Rajan S, Dinesh MR (2015) Genetic diversity and population structure analysis of mango (Mangifera indica) cultivars assessed by microsatellite markers. Trees 29:775–783
Robinson WB, Stotz E (1945) The indophenolxylene extraction method for ascorbic acid and modifications for interfering substances. J Biol Chem 16:217–225
Roy RS (1953) Study of irregular bearing of mango (Mangifera indica L.). Indian J Hortic 10:157–160
Sa KJ, Ko WR, Kim OG, Ma SJ, Choi HJ, Lee JK (2016) Association analysis and population structure of flowering-related traits in super sweet corn inbred lines with simple sequence repeat markers. Genes Genomics 38 (891). doi:10.1007/s13258-016-0434-2
Sadok IB, Celton JM, Essalouh L, Aabidine AZ, Garcia G, Martinez S, Grati-Kamoun N, Rebai A, Costes E, Khadari B (2013) QTL mapping of flowering and fruiting traits in olive. PLoS One 8(5):e62831. doi:10.1371/journal.pone.0062831
Salazar JA, Ruiz D, Egea J, Martnez GP (2013) Transmission of fruit quality traits in apricot (Prunus armeniaca L.) and analysis of linked quantitative trait loci (QTLs) using simple sequence repeat (SSR) markers. Plant Mol Biol Rep 31(6):506–510
Saran PL, Godara AK, Lal G, Yadav IS (2007) Correlation and path coefficient analysis in bergenotypes for yield and yield contributing traits. Indian J Hortic 64(4):459–460
Schlautman B, Covarrubias-Pazaran G, Garcia LAD, Johnson-Cicalese J, Iorrizo M, Rodriguez-Bonilla L, Bougie T, Bougie T, Wiesman E, Steffan S, Polashock J, Vorsa N, Zalap J (2015) Development of a high-density cranberry SSR linkage map for comparative genetic analysis and trait detection. Mol Breed 35:177. doi:10.1007/s11032-015-0367-5
Schnell RJ, Ronning CM, Knight RJ (1995) Identification of cultivars and validation of genetic relationship in (Mangifera indica L) using RAPD markers. Theor Appl Gent 90(2):269–274
Sethi S, Srivastav M, Samuel DVK, Singh AK, Dubey AK, Singh G (2011) Evaluation of newly developed mango (Mangifera indica) hybrids for their storage behaviour and peel colour. Indian J Agric Sci 81(3):252–255
Shamy HA, Eissa AB (1990) Alternate bearing in some mango cultivars in relation to the chemical composition of trees. Agric Sci (Cairo) 35(2):931–938
Sharma DK (1987) Mango breeding. Acta Hortic 196:61–67
Sharma DK, Majumder PK, Singh RN (1983) Induction of mutation in mango (Mangifera indica L.). J Nuclear Agric Biol 12(1):14–17
Singh LB (1960) The mango (reprint). Leonard Hill, London
Singh RN (1961) Studies in the differentiation and development of fruit buds in mango (Mangifera indica L.). V. Effect of defoliation, decapitation and de-blossoming on fruit-bud differentiation. Indian J Hortic 18:1–11
Singh BD (1991) Principles of plant breeding. Kalyani Publishers, Ludhiana
Singh RN (1996) Mango. ICAR, New Delhi
Singh RK, Chaudhary BD (1977) Biometrical methods in quantitative genetic analysis. Kalyani Publishers, New Delhi, pp 57–58
Singh NK, Mahato AK, Sharma N, Gaikwad K, Srivastava M, Tiwari K, Dogra V, Rawal SC, Rajan S, Singh AK, Sharma TR (2014) A draft genome of the king of fruit, mango (Mangifera indica L.). In: Plant and animal genome XXII conference, San Diego
Surapaneni M, Lakshminarayan RV, Hameedunnisa B, Reddy BP, Neetasri C, Nagaraju J, Anwar SY, Siddiq EA (2013) Population structure and genetic analysis of different utility types of mango (Mangifera indica L.) germplasm of Andhra Pradesh state of India using microsatellite markers. Plant Syst Evol 299(7):1215–1229
Van Oosterhout C, Hutchinson W, Wills D, Shipley P (2004) MICROCHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol 4:535–538
Vasugi C, Sekar K, Dinesh MR, Venugopalan R (2013) Studies on genetic divergence in unique indigenous mango accessions of Western Ghat. Int J Curr Res 5(9):2494–2499
Verde I, Quarta R, Cedrola C, Dettori MY (2002) QTL analysis of agronomic traits in a BC1 peach population. Acta Hortic 592:302–304
Yadav AS, Pandey DS (2016) Geographical perspectives of mango production in India. Imp J Interdiscip Res 2(4):257–264
Yadav VB, Wangchu L, Singh B, Sajeed A, Mitra SK, Ali S (2003) Correlation studies among fruit characters of different clones of mango cv. Langra. Environ Ecol 21:119–122
Yamanaka N, Hasran M, He-Xu D, Tsunematsu H, Idris S, Ban T (2006) Genetic relationships and diversity of four Mangifera species revealed through AFLP analysis. Genet Resour Crop Evol 53:949–954
Yanik H, Turktas M, Dundar E, Hernandez P, Dorado G, Turgay U (2013) Genome-wide identification of alternate bearing-associated microRNAs (miRNAs) in olive (Olea europaea L.). BMC Plant Biol 13:10. doi:10.1186/1471-2229-13-10
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160
Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C, Tang C, Toomajian C, Zheng H, Dean C, Marjoram P, Nordborg M (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet 3(1):e4. doi:10.1371/journal.pgen.0030004
Acknowledgements
The authors thank the Head, Division of Fruits and Horticultural Technology, Indian Council of Agricultural Research-IARI, Director, Indian Council of Agricultural Research-IARI, New Delhi, Dr. N.K. Singh, National professor and B.P. Pal chair, advisory committee, Dr. Rupesh Deshmukh, University Laval, Quebec, Canada and ICAR, New Delhi for providing resources for this study. We also would like to thank our anonymous reviewers for the great effort they have put into reviewing our paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Data archiving statement
The authors declare that all the work described in this manuscript followed the standard Tree-structure and function journal policy. All the primers used in the current study have been derived from leaf RNA-seq of mango variety Amrapali, and to date, no information pertain to this manuscript is submitted to any public databases.
Additional information
Communicated by L. Kalcsits.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lal, S., Singh, A.K., Singh, S.K. et al. Association analysis for pomological traits in mango (Mangifera indica L.) by genic-SSR markers. Trees 31, 1391–1409 (2017). https://doi.org/10.1007/s00468-017-1554-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00468-017-1554-2