
Abstract
Magnetic double-shell hybrid microspheres (Fe3O4@SiO2@p(NIPAM-co-GMA)) have been developed as a promising supported substrate for the immobilization of cellulase. Since the surface of the magnetic microspheres not only contains an epoxy group from GMA (glycidyl methacrylate) that can covalently bind to the enzyme, but also has an intelligent temperature response property from NIPAM (N-isopropylacrylamide), the cellulase can be covalently bonded to the magnetic microspheres and have a temperature-sensitive capability. The immobilized cellulase has the recovery ability of cellulase activity after a high-temperature inactivation. The average amount and activity of immobilized enzymes, respectively, was 233 mg g−1, 57.4 U mg−1 under the optimized conditions. The experimental results show that the immobilized cellulase has a wider catalytic temperature range, better temperature and storage stability. The residual activity still remained about 65.6% of the initial activity after the sixth catalysis run, which indicated that the immobilized enzyme had high reproducibility.
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cellulose is the most abundant polysaccharide polymer on the earth, which can be degraded to low-molecular glucose by enzyme hydrolysis cooperated by three kinds of cellulases, including endoglucanases, exoglucanases and β-glucosidases. The resultant glucose can be further used to produce biofuel or other chemicals, so cellulose was expected as the most promising renewable source of energy [1, 2]. Cellulase-based catalytic hydrolysis is an efficient, eco-friendly and mild process, however, the high cost of cellulase and its instability against harsh environment limited its industrial application.
Enzyme immobilization is attractive for the reduction of enzyme cost by recycling and enhancement of enzyme stability. The immobilization of enzyme on various insoluble carriers, such as magnetic nanoparticles (MNPs) [3,4,5,6], mesoporous silica nanoparticles [7], functionalized molybdenum sulfide nanosheets [8], gold-doped magnetic silica nanoparticles [9] and magnetic polymer [10] seems to be a feasible way to solve the above-mentioned problems. The encapsulation of magnetic Fe3O4 by silica shell have the advantages of protecting the magnetic cores from erosion, increasing hydrophilic property and providing active silanol group for subsequent surface modification [11,12,13]. The polymer shell can not only prevent poly-condensation reaction induced by surface hydroxyl groups on the silica shell, but also easily incorporate various functional group such as reactive epoxy group from constitute comonomer (GMA) [14, 15].
Progress towards the prevention of heat-induced inactivation of enzyme using smart polymers as artificial chaperone has been made in recent years [16, 17]. Most proteins are likely to lose their catalytic activity upon heating, which is believed to the result of undesired aggregation. Inspired by natural chaperone, the thermo-sensitive polymers are developed as artificial chaperones to provide hydrophobic binding sites to capture unfolded proteins against irreversible aggregation when heating, and then release refolded protein upon cooling [18]. Thereby the thermal stability of proteins can be enhanced.
Herein, we synthesized the Fe3O4@SiO2@p(NIPAM-co-GMA) double-shell microspheres as carrier for cellulase immobilization, which offer a number of advantages including good biocompatibility, rapid separation form the digested peptides simply using a magnet and enhanced stability. To the best of our knowledge, the use of Fe3O4@SiO2@p(NIPAM-co-GMA) carrier for the cellulase immobilization has not been reported yet. Their binding capacity towards cellulase was examined. The catalysis reactivity of immobilized cellulase for the hydrolysis of carboxymethyl cellulose (CMC) at different pH and temperature were investigated and compared with their free state. Other enzyme property comparison such as thermal stability and storage stability were also conducted in this paper. Beyond that, we also utilize the immobilized cellulase to refold the thermally denatured cellulase.
Experiment
Materials
Ferric chloride (FeCl3·6H2O), ammonium acetate (NH4·Ac), trisodium citrate, ethylene glycol, ethanol, ammonium hydroxide aqueous solution (25 wt %), tetraethyl orthosilicate (TEOS), acetonitrile, carboxymethyl cellulose (CMC), glucose, 3,5-dinitrosalicylic acid (DNS), sodium hydroxide (NaOH), potassium sodium tartrate tetrahydrate (Na–K tartrate), phenol were obtained from Sinopharm Chemical Reagent Co., Ltd. Cellulase (≥ 45 units/mg) from Trichoderma viride and, N-isopropylacrylamide (NIPAM) and γ-methacryloxypropyltrimethoxysilane (MPS) were bought from Aladdin Industrial Corporation. 2,2′-Azobisisobutyronitrile (AIBN) was supplied by Tianjin Guangfu Fine Chemical Research Institute and recrystallized from methanol. N,N′-Methylenebisacrylamide (MBA), was bought from Aladdin Industrial Corporation and recrystallized from acetone. Glycidyl methacrylate (GMA) was bought from Aladdin Industrial Corporation and vacuum distilled. All other reagents were of analytical grade and used without any further treatment. Deionized water was used in all the experiments.
Synthesis of Fe3O4 microspheres
Fe3O4 microspheres were prepared by a modified solvothermal reaction [19]. Typically, 2.700 g of FeCl3·6H2O, 7.708 g of NH4·Ac and 0.8 g of trisodium citrate trihydrate were dissolved in 140 ml of ethylene glycol in a round-bottomed flask. The mixture was stirred vigorously for 1 h to form a homogeneous solution. The mixture was transferred into a Teflon-lined stainless-steel autoclave. The autoclave was heated at 200 °C for 16 h and then cooled to room temperature. The black product was separated from the mixture by using a magnet and was washed with ethanol. The washing and magnetic separation steps were repeated several times. The Fe3O4 microspheres were dried in a vacuum oven until they reached a constant weight.
Synthesis of Fe3O4@SiO2–MPS core–shell microspheres
Fe3O4@SiO2 microspheres were prepared by a sol–gel method through the coating of the silica layer onto the Fe3O4 microspheres [20, 21]. 0.3 g of Fe3O4 microspheres were suspended in the mixture of 40 ml of ethanol, 10 ml of water, and 1.5 ml of ammonium hydroxide aqueous solution (25 wt %). Then, 0.5 ml of TEOS was added to the above mixture under mechanical stirring drop by drop. After 12 h, 0.6 ml of MPS was added to the mixture, and the reaction was allowed to proceed for 24 h at 60 °C. Finally, the product was separated with the help of a magnet, and washed with ethanol for several times. The product Fe3O4@SiO2–MPS was dried in a vacuum oven until a constant weight.
Synthesis of Fe3O4@SiO2@p(NIPAM-co-GMA) double-shell microspheres
Coating a p(NIPAM-co-GMA) layer onto Fe3O4@SiO2–MPS microspheres was performed by distillation–precipitation polymerization (DPP) [22, 23] of NIPAM and GMA in acetonitrile, with MBA as the cross-linker and AIBN as the initiator. 50 mg of Fe3O4 @SiO2–MPS microspheres were dispersed in 40 ml of acetonitrile in a 250-ml single-necked flask and sonicated for 30 min. 150 ml of GMA, 50 mg of NIPAM, 200 mg of MBA and 8 mg of AIBN were added in the flask to initiate the polymerization. The flask was submerged in a heating oil bath and attached to a fractionating column, Liebig condenser, and a receiver. The mixture was heated from ambient temperature to the boiling state within 30 min. The reaction was ended after 20 ml of acetonitrile distilled from the mixture within about 1 h. The obtained Fe3O4@SiO2@p(NIPAM-co-GMA) microspheres were separated and washed using ethanol to remove excess reactants and the polymer microspheres (without a Fe3O4@SiO2–MPS microspheres core). The products were dried in a vacuum oven until a constant weight.
Characterization
The morphology of the resultant microspheres was determined by transmission electron microscopy (TEM, Tecnai G2 F30 S-TWIN, USA). The sample dispersed in ethanol was dropped onto the surface of a carbon-coated copper grid, and then dried under vacuum state at room temperature. The magnetic properties of the resultant microspheres were studied in the dried state with a vibrating sample magnetometer (VSM, LakeShore 7410, USA) at room temperature. X-ray diffraction (XRD) patterns were analyzed using a DX-1000 diffractometer (Shimadzu, XRD-6100, Japan) with Cu Κα radiation at 40 kV and 36 mA. The chemical structure of the microspheres was determined by a Fourier transform infrared (FT-IR) spectra spectrometer (Nicolet Nexus 470, USA) over potassium bromine pellet, and the diffuse reflectance spectra were scanned over the range of 400–4000 cm−1. Thermogravimetric analysis (TGA) data were performed by a simultaneous thermal analyzer (Netzsch STA 499C, Germany) at a heating rate of 10°C/min from 25 to 800 °C under N2 atmosphere.
Immobilization of cellulase on Fe3O4@SiO2@p(NIPAM-co-GMA) double-shell microspheres
The support solution was prepared by suspending Fe3O4@SiO2@p(NIPAM-co-GMA) double-shell microspheres in 10 mL of 0.1 M acetate buffer (pH 4.5) solution. The prepared 5 mg mL−1 cellulase solutions were mixed with support solution in a container, followed by incubator shaker at a temperature of 30 °C, 200 rpm speed for 0.5 h. After incubation, the obtained immobilized cellulase was separated by a magnet. Then, the supernatant is carefully decanted without loss of any conjugate. Next, the immobilized cellulase was re-dispersed into fresh acetate buffer solution to wash and remove the unbound cellulase.
The concentration of residual cellulase in the adsorption solution was detected using UV/VIS spectrophotometer (UV-2450, Shimadzu (China) Co., Ltd.) according the Bradford protein assay method [24]. The loading mass of cellulase can be calculated by the following equation:
In this equation, C0 (mg mL−1) expressed the initial concentration of protein solution; Ct (mg mL−1) expressed the equilibrium concentration of protein solution; V (mL) expressed the volume of protein solution; m (g) was the quality of the support material.
Determination of cellulase enzyme activity
The activity of free cellulase and immobilized cellulase were determined according to the CMC assay [25, 26]. Here, 0.1 ml of cellulase solution at an appropriate dilution was mixed with 0.9 ml of 1% CMC solution (dissolved in 0.1 M acetate buffer, pH 4.5) in a tube to proceed the hydrolysis. The reaction was carried out for 0.5 h at 50 °C for free cellulase and 60 °C for immobilized cellulase. The amount of glucose produced during the reaction was measured as absorbance at 540 nm using the 3,5-dinitrosalicylic acid (DNS) reagent [27]. One unit of cellulase activity was defined as 1 µmol of glucose produced per minute of enzyme assay.
Results and discussion
Preparation and characterization of Fe3O4@SiO2@p(NIPAM-co-GMA) double-shell microspheres
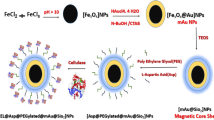
The procedure for the synthesis of double-shell magnetic materials is shown in Fig. 1. Briefly, a modified solvothermal reaction was used to synthesis the Fe3O4 microspheres with diameters of around 250 nm. Then, a layer of SiO2 was coated on the Fe3O4 microspheres surface by the sol–gel method. Then MPS was modified on the surface of microspheres to form the available double bonds. Finally, the polymer layer composed of GMA and NIPAM was synthesized on the microspheres surface by the one-step distillation–precipitation polymerization (DPP), the temperature-sensitive monomer and the epoxy group were introduced onto the surface of microspheres.

Schematic diagram of preparation of Fe3O4@SiO2@p(NIPAM-co-GMA) double-shell microspheres
The acquired magnetite microspheres are shown in the TEM image of Fig. 2. Representative TEM images of Fe3O4 and Fe3O4@SiO2–MPS microspheres are shown in Fig. 2a, b. The Fe3O4 microspheres gave an average diameter of 250 nm and presented a spherical shape with a rough surface. After adding SiO2 layer, the size of the microspheres was increased to around 280 nm. The obtained Fe3O4@SiO2–MPS microspheres possessed a well-defined core–shell structure. Figure 2c shows a typical TEM image of the obtained Fe3O4@SiO2@p(NIPAM-co-GMA) double-shell microspheres with a relative uniform size of ~ 350 nm. The well-defined double-shell structure is distinctly observed.

TEM images of a Fe3O4, b Fe3O4@SiO2–MPS, c Fe3O4@SiO2@p(NIPAM-co-GMA)
The FT-IR spectra of the Fe3O4 microspheres, the Fe3O4@SiO2–MPS microspheres and the Fe3O4@SiO2@p(NIPAM-co-GMA) microspheres are shown in Fig. 3. The peaks at 596 and 3432 cm−1 are attributed to the stretching vibration of Fe–O and the –OH peaks on magnetic microsphere surface. The peak at about 1631 cm−1 was associated with carboxyl groups available from the stabilizer citrate in the surface of Fe3O4 microspheres. After coating on SiO2 layer on Fe3O4 microspheres, the peaks at 1637 and 3432 cm−1 became weak. The strong absorption peak at 796, 1087 and 459 cm−1 was assigned to the symmetrical stretching vibration, antisymmetric stretching vibration and the flexural vibrations of Si–O–Si. The peak at 949 cm−1 was attributed to the flexural vibrations of Si–OH. Thus, SiO2 shell was successfully coated on Fe3O4 microspheres. The absorption peaks at 1400 and 1632 cm−1 were the methyl group characteristic absorption peak and the vinyl group stretching vibration from MPS, which proved that MPS was successfully modified. As shown in Fig. 3c, the two absorption peaks at 1727 and 1386 cm−1 were attributed to the vibration of the ester group in GMA and the isopropyl group in NIPAM, respectively. In addition, absorption peaks at 3365 and 1525 cm−1 appeared as a result of the stretching vibration and the flexural vibrations of the N–H group. The absorption peak at 1659 cm−1 was assigned to the stretching vibration of amido group. These results further proved that the p(NIPAM-co-GMA) shell was successfully coated onto the Fe3O4@SiO2–MPS microspheres through the DDP method.

FT-IR spectra: a Fe3O4, b Fe3O4@SiO2–MPS, c Fe3O4@SiO2@p(NIPAM-co-GMA)
The XRD pattern of the product is shown in Fig. 4. Six characteristic peaks for magnetite marked by their indices (220) (311) (400) (422) (511) (440), matched well with the standard JCPDS data (74–748). No obvious XRD peaks arising from impurities were found, which revealed that magnetite microspheres were successfully synthesized with high purity.

XRD patterns of a Fe3O4, b Fe3O4@SiO2–MPS, c Fe3O4@SiO2@p(NIPAM-co-GMA)
The magnetic properties of the three kinds of microspheres were studied using a vibrating sample magnetometer (VSM), which are shown in Fig. 5. The hysteresis curves show that the three kinds of microspheres have no obvious remanence and coercivity at 298.15 K, indicating that they have superparamagnetic properties. The superparamagnetism is coming from the core Fe3O4 small nanocrystals. As a control, the saturation magnetization (Ms) of the Fe3O4 microspheres was 57 emu·g−1. With the addition of the SiO2 layer and the polymer layer, the Ms values of the microspheres were reduced to 32 and 8 emu·g−1, respectively. In addition, due to the high magnetic properties of the Fe3O4 core, the final product of Fe3O4@SiO2@p(NIPAM-co-GMA) microspheres can be separated from the solution in 30 s with a magnet, as the inside picture shows in Fig. 5.

Magnetization curves of a Fe3O4, b Fe3O4@SiO2–MPS, c Fe3O4@SiO2@p(NIPAM-co-GMA)
The components of composite microspheres are measured by thermogravimetric analysis (TGA), and the results are displayed in Fig. 6. The organic components were decomposited and inorganic components remained with the increase of temperature. The 16.95 wt% loss of Fe3O4 microspheres was attributed to the weight ratio of citrate stabilizer, indicating the Fe3O4 content is 83.05 wt%. After coating by SiO2 layer, the loss of 13.95 wt% was assigned to the physically adsorbed water and small amount of MPS on the silica surface. When the outmost p(NIPAM-co-GMA) layer was introduced, the weight loss of the composite microspheres was about 77.14 wt%, which is much higher than Fe3O4@SiO2–MPS (13.95 wt%). The first weight loss (15%) until 200 °C was due to the evaporation of the physically adsorbed water or solvent, and the second major weight loss (62%) from 200 to 600 °C was due to the decomposition of the polymer component in the second shell layer of the corresponding microspheres.

TGA curves of a Fe3O4, b Fe3O4@SiO2–MPS, c Fe3O4@SiO2@p(NIPAM-co-GMA)
Optimization of immobilization conditions
A certain amounts of cellulase (6, 12, 24, 36, 48, 60, 72, or 84 µg) and magnetic material were mixed for 2 h at 30 °C. The effect of initial enzyme content on immobilization is shown in Fig. 7a. As the initial enzyme content increased, the loading amount of enzyme increased gradually until almost no change. The most likely reason for this is that the binding sites on the material gradually reach saturation. The optimized initial enzyme content is 60 µg.

Effect of a initial enzyme content, b temperature and c reaction time on cellulase immobilization. The concentration of the magnetic material is 0.1 mg mL−1 in 0.1 M acetate buffer (pH 4.5) solution
The effect of temperature on immobilization was investigated on 25–55 °C. It can be seen from Fig. 7b, with the increase of the temperature, the amounts of immobilized enzymes slightly increased. According to the effect of temperature on the enzyme activity, the free enzyme was gradually inactivated as the temperature increased. Combined with the above analysis, we set the temperature of the immobilized enzyme process to 30 °C in order to maximize the enzyme activity and take into account the actual production requirements.
The effect of incubation time was investigated under the temperature of 30 °C, and the results are shown in Fig. 7c. As can be seen from the figure, with the extension of the reaction time, the amount of immobilized enzyme gradually increased and was nearly stable thereafter for up to 30 min. This may be because the extension of the reaction time is conducive to the amino groups of the enzyme molecules and the epoxy of support on the reaction, after a certain period of time the material load to saturation, and then extension of time is useless, so the immobilization time is 30 min. The average amount and activity of immobilized enzymes, respectively, was 233 mg g−1, 57.4 U mg−1 under the above optimized conditions. In addition, the loading amount of immobilized cellulase was comparably higher than previously reported supports for cellulase, for example, 18.4 mg g−1 on magnetoresponsive graphene by covalent [28], 43.4 mg g−1 on clay/PGMA by absorption [29], 43.9 mg g−1 on sol–gel matrix by entrapment [30].
Effect of temperature on cellulase activity
The optimum catalytic temperature of free cellulase and immobilized cellulase was examined by altering the reaction temperatures involved 30, 40, 50, 60, 70, and 80 °C. The results are shown in Fig. 8a. From the graph we can see that the optimum temperature of the free enzyme is 50 °C, and of the immobilized enzyme is 50–60 °C. This shows that the range of catalytic temperature for immobilized cellulase is larger. The enzyme activity has improved stability due to being protected by temperature-sensitive polymer monomer NIPAM under high temperature. A similar shift was reported in many cases of enzyme immobilization [31].

Effect of a temperature, b pH on the activity of free cellulase and immobilized cellulase. The reaction was carried out for 0.5 h at different temperature and pH. Relative activities were calculated by using the highest activity of free cellulase and immobilized cellulase as 100%
Effect of pH on cellulase activity
The optimum pH of immobilized cellulase and free cellulase was determined at different buffers with a pH of 3.6–8.0 at 50 °C, and the results are shown in Fig. 8b. The maximum activity was at 4.8 for immobilized cellulase, which was the same as free cellulase. It seemed obvious that the activity of immobilized cellulase was generally higher over most of the pH values as compared to the free cellulase, which indicated that immobilized cellulase showed preferable pH adaptability.
Thermal stability
The thermal stabilities of free cellulase and immobilized cellulase were investigated by incubation for 2 h at 30, 40, 50, 60, 70, 80 °C, respectively. Figure 9a shows that the thermal stabilities of free cellulase and immobilized cellulase were identical below 50 °C, but the thermal stability of the immobilized cellulase was better than that of the free cellulase above 50 °C. The activity of both immobilized cellulase and free cellulase decreased; however, the former decreased slowly and less than the latter. The free enzyme retained 35.2% of its original activity with heat treatment at 60 °C for 2 h, while the immobilized enzyme retained 85.1% of its original activity. The residual activity of the immobilized cellulase was 32.2% after 2 h test at 80 °C; whereas, the free cellulase had kept only 21.0%. The thermal stability of immobilized cellulase was improved compared with the published data [32]. The promising stability of the immobilized cellulase is very important for practical applications.

a Thermal stability of free cellulase and immobilized cellulase. Relative activities were calculated by using the highest activity of free cellulase and immobilized cellulase as 100%. b Storage stability of free cellulase and immobilized cellulase. The thermal stabilities of free cellulase and immobilized cellulase were investigated by incubation for 2 h at 30, 40, 50, 60, 70, 80 °C. The storage stabilities of free cellulase and immobilized cellulase were investigated for different times in 4 °C. The relative activities were calculated by using the 0 day activity of free cellulase and immobilized cellulase as 100%
Storage stability
The storage stabilities of free cellulase and immobilized cellulase were investigated for different times in cold storage (4 °C). According to Fig. 9b, the remaining activity for free cellulase and immobilized cellulase were 34.7 and 59.6%, respectively, after 40 days. It was obvious that the stability of cellulase was enhanced by immobilization, and this is consistent with the thermal stability data.
Kinetic parameters
The kinetic parameters (Km) of free cellulase were changed after immobilization on the support material. Specifically, the apparent K m values of free cellulase and immobilized cellulase were 0.6224 and 0.5485 mg mL−1, respectively. The lower Km values of immobilized cellulase suggested a higher affinity toward the substrate CMC after immobilization. The change in the affinity of cellulase to its substrate may be caused by constructional changes in the enzyme because of immobilization program or by the lower approachability of substrate to the active site of immobilized cellulase. The Vmax is increased from 3.5 to 8.6 after the immobilization, which is indicated that the immobilized cellulase can provide better stability than the free cellulase.
Reusability study on the immobilized cellulase
The reusability of the immobilized enzyme in the hydrolysis of CMC test was assessed under optimized reaction condition. After each cycle, the immobilized cellulase was removed by a magnet. The immobilized cellulase was then collected and washed simultaneously with distilled water. The activity of the immobilized enzyme after first cycle was defined as the control and recognized as a relative activity of 100%. The reusability assay was repeated six cycles, the results are shown in Fig. 10. The activity of the immobilized cellulase decreased with number of times of reuse compared to its initial value and the immobilized cellulase kept over 65.6% after 6 times reuse, it was improved compared with the published data 50% after 6 times reuse [32]. This resultant activity loss might be caused by several factors, including loss of constituents of the cellulase complex, end-product inhibition, and protein denaturation. Another reason for the loss in the activity could be that some physically adsorbed cellulase might have been present initially, but it fell off during the reusability assay [4].

Influence of repeats on the relative activity of the immobilized cellulase at 0.5 h, pH 4.5, and 60 °C. Relative activities were calculated by using the first time activity of immobilized cellulase as 100%
Refolding of thermally denatured cellulase
The recovery ability of cellulase activity after a high-temperature inactivation was discussed in this part, the free cellulase and immobilized cellulase were put in the extreme temperatures of 80 °C for 5 min, then transfer them to the temperature of 50 or 4 °C for 30 min or 1 h. The results are shown in Table 1.
It is shown that the free enzyme after high-temperature inactivation, and then placed in the low-temperature conditions, the enzyme activity of the free cellulase retained only about 23.4%, However, the immobilized cellulase activity recovered to about 63.0% after being restored at 4 °C for 30 min, reached about 72.2% after 60 min. After being restored at 50 °C for 30 min, the activity of immobilized cellulase is recovered about 50.1%, reached about 56.5% after 60 min. The inactivation of the enzyme activity through heat treatment is due to the thermal deformation of the polypeptide chain of the enzyme [33].
The inactivation of the enzyme at high temperature is often due to the expansion of the polypeptide chain of the enzyme at high temperature, exposing its hydrophobic sites, so that the irreversible adhesion between the peptides, resulting in the denaturation and inactivation of the enzyme. The polymer formed by the temperature-sensitive monomer NIPAM on the magnetic material can change the hydrophilicity at the critical temperature (Fig. 11). We concluded that the monomer is condensed at high temperature, making enzyme deformation restricted under high temperature. As the temperature recovers, NIPAM is restored to the original shape, and there is no aggregation of enzyme molecules polypeptide under high temperature. The temperature-sensitive monomer NIPAM can protect the stability of the molecular structure of the enzyme, which has a strong ability to restore the enzyme activity.

Schematic diagram of inactivation of free enzyme and recovery activation of immobilized enzyme
Conclusion
In conclusion, Fe3O4@SiO2@p(NIPAM-co-GMA) double-shell hybrid microsphere were developed as a promising supported substrate for the immobilization of cellulase. It has a double-shell structure with a diameter of about 350 nm and has good superparamagnetism, which can be rapidly separated from the solution in the presence of an external magnetic field for 30 s. Then, we immobilized cellulase on the magnetic composite microspheres and investigated the effects of different conditions on the loading amount of cellulase. In the analysis of thermal stability, when the temperature reaches 60 °C, the residual activity of free enzyme remained only about 35.1%, while the residual activity of immobilized enzyme remained about 85.2%, which is much higher than that of free enzyme. The remaining relative enzyme activity was 65.6% of initial activity after 6 times of carboxymethyl cellulose catalyzed by immobilized enzyme, which means that the immobilized enzyme had high reusability. In addition, we also studied the renaturation of immobilized cellulase after heat denaturation, and found that the immobilized enzyme could reach 72.2% of the initial activity after heating and cooling, which indicated that it had a certain ability of renaturation.
References
Klemm D, Heublein B, Fink HP, Bohn A (2005) Cellulose: fascinating biopolymer and sustainable raw material. Angew Chem Int Ed Engl 44:3358–3393
Huber GW, Iborra S, Corma A (2006) Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering. Chem Rev 106:4044–4098
Abraham RE, Verma ML, Barrow CJ, Puri M (2014) Suitability of magnetic nanoparticle immobilised cellulases in enhancing enzymatic saccharification of pretreated hemp biomass. Biotechnol Biofuels 7
Khoshnevisan K, Vakhshiteh F, Barkhi M, Baharifar H, Poor-Akbar E, Zari N, Stamatis H, Bordbar AK, Khoshnevisan K, Vakhshiteh F (2017) Immobilization of cellulase enzyme onto magnetic nanoparticles: applications and recent advances. 442, pp 66–73
Heidarizadeh M, Doustkhah E, Rostamnia S, Rezaei PF, Harzevili FD, Zeynizadeh B (2017) Dithiocarbamate to modify magnetic graphene oxide nanocomposite (Fe3O4-GO): a new strategy for covalent enzyme (lipase) immobilization to fabrication a new nanobiocatalyst for enzymatic hydrolysis of PNPD. Int J Biol Macromol 101:696–702
Khoshnevisan K, Barkhi M, Ghasemzadeh A, Tahami HV, Pourmand S (2016) Fabrication of coated/uncoated magnetic nanoparticles to determine their surface properties. Mater Manuf Processes 31:1206–1215
Lee YC, Dutta S, Wu KCW (2014) Integrated, cascading enzyme-/chemocatalytic cellulose conversion using catalysts based on mesoporous silica nanoparticles. Chemsuschem 7:3181–3181
Das R, Mishra H, Srivastava A, Kayastha A (2017) Covalent immobilization of β-amylase onto functionalized molybdenum sulfide nanosheets, its kinetics and stability studies: A gateway to boost enzyme application. Chem Eng J 328:215–227
Cho EJ, Jung S, Kim HJ, Lee YG, Nam KC, Lee HJ, Bae HJ (2012) Co-immobilization of three cellulases on Au-doped magnetic silica nanoparticles for the degradation of cellulose. Chem Commun 48:886–888
Kamat RK, Ma WF, Yang YK, Zhang YT, Wang CC, Kumar CV, Lin Y (2013) Adsorption and hydrolytic activity of the polycatalytic cellulase nanocomplex on cellulose. ACS Appl Mater Interfaces 5:8486–8494
Pan MR, Sun YF, Zheng J, Yang WL (2013) Boronic acid-functionalized core–shell–shell magnetic composite microspheres for the selective enrichment of glycoprotein. ACS Appl Mater Interfaces 5:8351–8358
Yi DK, Selvan ST, Lee SS, Papaefthymiou GC, Kundaliya D, Ying JY (2005) Silica-coated nanocomposites of magnetic nanoparticles and quantum dots. J Am Chem Soc 127:4990–4991
Khoshnevisan K, Barkhi M, Zare D, Davoodi D, Tabatabaei M (2012) Preparation and characterization of CTAB-coated Fe3O4 nanoparticles. Synth React Inorg M 42:644–648
Bayramoglu G, Doz T, Ozalp VC, Arica MY (2017) Improvement stability and performance of invertase via immobilization on to silanized and polymer brush grafted magnetic nanoparticles. Food Chem 221:1442–1450
Ince A, Bayramoglu G, Karagoz B, Altintas B, Bicak N, Arica MY (2012) A method for fabrication of polyaniline coated polymer microspheres and its application for cellulase immobilization. Chem Eng J 189:404–412
Huang F, Wang JZ, Qu AT, Shen LL, Liu JJ, Liu JF, Zhang ZK, An YL, Shi LQ (2014) Maintenance of amyloid beta peptide homeostasis by artificial chaperones based on mixed-shell polymeric micelles. Angew Chem Int Ed Engl 53:8985–8990
Ganguli S, Yoshimoto K, Tomita S, Sakuma H, Matsuoka T, Shiraki K, Nagasaki Y (2009) Regulation of lysozyme activity based on thermotolerant protein/smart polymer complex formation. J Am Chem Soc 131:6549–6553
Liu X, Liu Y, Zhang ZK, Huang F, Tao Q, Ma RJ, An YL, Shi LQ (2013) Temperature-responsive mixed-shell polymeric micelles for the refolding of thermally denatured proteins. Chem Eur J 19:7437–7442
Ma WF, Xu SA, Li JM, Guo J, Lin Y, Wang CC (2011) Hydrophilic dual-responsive magnetite/PMAA core/shell microspheres with high magnetic susceptibility and pH sensitivity via distillation–precipitation polymerization. J Polym Sci Part A: Polym Chem 49:2725–2733
Bourgeatlami E, Lang J (1999) Encapsulation of inorganic particles by dispersion polymerization in polar media. J Colloid Interface Sci 210:281
Stöber W, Fink A, Bohn E (1968) Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci 26:62–69
Ma W, Zhang Y, Li L, Zhang Y, Yu M, Guo J, Lu H, Wang C (2013) Ti4+-immobilized magnetic composite microspheres for highly selective enrichment of phosphopeptides. Adv Funct Mater 23:107–115
Bai F, Xinlin Yang A, Huang W (2004) Synthesis of narrow or monodisperse poly(divinylbenzene) microspheres by distillation–precipitation polymerization. Macromolecules 37:3641–3649
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Khoshnevisan K, Bordbar AK, Zare D, Davoodi D, Noruzi M, Barkhi M, Tabatabaei M (2011) Immobilization of cellulase enzyme on superparamagnetic nanoparticles and determination of its activity and stability. Chem Eng J 171:669–673
IUPAC (2009) Measurement of cellulase activities. Pure Appl Chem 59:257–268
Pardo AG, Forchiassin F (1999) Influence of temperature and pH on cellulase activity and stability in Nectria catalinensis. Rev Argent Microbiol 31:31–35
Gokhale AA, Lu J, Lee I (2013) Immobilization of cellulase on magnetoresponsive graphene nano-supports. J Mol Catal B-Enzym 90:76–86
Bayramoglu G, Senkal BF, Arica MY (2013) Preparation of clay-poly(glycidyl methacrylate) composite support for immobilization of cellulase. Appl Clay Sci 85:88–95
Ungurean M, Paul C, Peter F (2013) Cellulase immobilized by sol–gel entrapment for efficient hydrolysis of cellulose. Bioproc Biosyst Eng 36:1327–1338
Liu X, Liu Y, Zhang Z, Huang F, Tao Q, Ma R, An Y, Shi L (2013) Temperature-responsive mixed-shell polymeric micelles for the refolding of thermally denatured proteins. Chemistry 19:7437–7442
Zang LM, Qiu JH, Wu XL, Zhang WJ, Sakai E, Wei Y (2014) Preparation of magnetic chitosan nanoparticles as support for cellulase immobilization. Ind Eng Chem Res 53:3448–3454
Yan Q, Yuan JY, Yuan WZ, Zhou M, Yin YW, Pan CY (2008) Copolymer logical switches adjusted through core-shell micelles: from temperature response to fluorescence response. Chem Commun 6188–6190
Acknowledgements
This work was supported by the National Natural Science Foundation of China (nos. 21676124, 31470434 and 21576124), China Postdoctoral Science Foundation funded project (no. 2017M610308), and Jiangsu Postdoctoral Science Foundation funded project (no. 1701107B).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Han, J., Rong, J., Wang, Y. et al. Immobilization of cellulase on thermo-sensitive magnetic microspheres: improved stability and reproducibility. Bioprocess Biosyst Eng 41, 1051–1060 (2018). https://doi.org/10.1007/s00449-018-1934-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00449-018-1934-z