
Abstract
Mechanisms that lead to the death of hair cells are reviewed. Exposure to noise, the use of ototoxic drugs that damage the cochlea and old age are accompanied by hair cell death. Outer hair cells are often more susceptible than inner hair cells, partly because of an intrinsically greater susceptibility; high frequency cells are also more vulnerable. A common factor in hair cell loss following age-related changes and exposure to ototoxic drugs or high noise levels is the generation of reactive oxygen species, which can trigger intrinsic apoptosis (the mitochondrial pathway). However, hair cell death is sometimes produced via an extracellular signal pathway triggering extrinsic apoptosis. Necrosis and necroptosis also play a role and, in various situations in which cochlear damage occurs, a balance exists between these possible routes of cell death, with no one mechanism being exclusively activated. Finally, the numerous studies on these mechanisms of hair cell death have led to the identification of many potential therapeutic agents, some of which have been used to attempt to treat people exposed to damaging events, although clinical trials are not yet conclusive. Continued work in this area is likely to lead to clinical treatments that could be used to prevent or ameliorate hearing loss.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hair cells are the sensory receptors of the hearing organ (the cochlea) and balance system (vestibular apparatus) in all vertebrates, and of the pressure-detecting lateral line in fish and amphibians. Each hair cell is equipped with a mechanically sensitive hair bundle composed of stereocilia at its apex. The hair cell responds to physical deflection of this stereociliary bundle via a transducer apparatus, which lies within the bundle and which, on stimulation, allows the influx of cations (K+ and Ca2+) into the hair cell, depolarising it (for a review, see Hackney and Furness 2013). At the base of the hair cell are two types of innervation: afferent fibres that form a variety of types of post-synaptic endings, depending on hair cell type and location, and efferent fibres that are pre-synaptic to the hair cell or the afferent fibres (for a review, see Spoendlin 1985). On depolarisation of the hair cell, neurotransmitter is released onto the afferent endings and action potentials are transmitted along the neurites of the neuron. The efferent nerve endings appear primarily to modulate the activity of the hair cells or afferent fibres. Afferent and efferent fibres travel together in cochlear, vestibular or lateral line nerves (depending on the organ of origin) that convey the information along the appropriate pathway and ultimately to or from the brain.
As hair cells play such a central role within these sensory systems, hair cell loss causes loss of sensory function in each of them. Hair cell loss is most often studied in the auditory system as this is a major cause of hearing impairment in the human population, itself the commonest sensory deficit experienced by both children (Deltenre and Van Maldergem 2013) and adults (Li-Korotky 2012). The environmental causes of hair cell loss are multi-factorial, as are the internal mechanisms that produce it. Hearing loss occurs as a result of genetic abnormality, infection and disease, exposure to chronic or acute episodes of high noise intensity and administration of a number of therapeutic drugs required in cases in which the severity of the condition being treated overrules the need to preserve hearing. It also occurs with age, with the possible involvement of confounding factors such as noise exposure or infection at any time in the life course, and might also be dependent on age-related hearing loss genes (ARHL). Each of these forms of hearing loss has its own features and not all target the hair cell. However, a substantial number do and, in the commonest conditions, hair cell loss is progressive. Although in birds and other vertebrates, hair cells can be replaced by natural regeneration via supporting cell division or transdifferentiation (Wan et al. 2013), in mammals, they do not undergo natural replacement to any large extent. Thus, the hair cell loss is permanent. This review will consider the molecular mechanisms underlying cochlear hair cell loss in mammals.
Organisation of the auditory sensory epithelium and hair cells
In order to understand hair cell loss, it is first necessary to review the structure of the hair cells, where they are located, and their function. In mammals, the hearing organ is the spiral-shaped cochlea, consisting of a membraneous tube spiralling around a central pillar of bone, broader at the base and narrower at the apex (Fig. 1a). Hair cells and surrounding supporting cells are situated in the organ of Corti, a sensory epithelium that sits on a fibrous membrane, the basilar membrane, which runs along this tube longitudinally (Fig. 1b). The basilar membrane forms the lower border of the cochlear duct, a triangular-shaped chamber with its upper boundary formed by Reissner’s membrane and its side wall composed of the stria vascularis and spiral ligament (Fig. 1c). The cochlear duct contains the scala media, a chamber bounded by the cells lining the cochlear duct. Above and below the scala media are two other parallel chambers, the scala vestibuli and the scala tympani. An important feature of these chambers is that, whereas the upper and lower chambers contain a fairly typical extracellular fluid (called perilymph) that has a high sodium ion concentration, the middle chamber is unusual in that it contains a high potassium ion concentration (endolymph). This ionic compositional difference is maintained by homeostatic tissues in the lateral wall and by tight junctions between the apices of all the cells that line the scala media, including the hair cells and their supporting cells, which together form the upper surface of the organ of Corti, called the reticular lamina (Fig. 1d). The stria vascularis, a three-layered tissue lining the lateral wall of the scala media gives rise to a “battery”, developed by a combination of potassium-handling mechanisms and Kir4.1 potassium channels that help power the transduction process, called the endolymphatic potential (for a review, see Wangemann 2006). The maintenance of these ionic differences is relevant not only to the sensory function of the hair cells, but also to the preservation of the cells, as will be seen later.

a Diagram of a human cochlea showing the spiral shape and the location of the cross section shown in b. b The spirally wound duct of the cochlea is here cut across five times and is subdivided by two transverse membranes, the basilar membrane (BM) and Reissner’s membrane (RM). The hair cell epithelium, the organ of Corti (OC) with its overlying tectorial membrane (TM), lies on the basilar membrane. The innervation that supplies the hair cells emanates from the spiral ganglion (SG), the neurons of which also project into the central modiolus (M) and from there to the auditory brain stem (arrow). The area in the red box is equivalent to the image shown in c. c Cross section of the cochlear duct of a guinea pig showing the structures described in b. d Detail of c illustrating the two types of hair cell (inner and outer), the supporting cells that surround them and the TM. Bars 50 μm (c), 25 μm (d)
Acoustic vibrations enter the cochlea at the oval window and form waves of pressure in the fluids that cause the basilar membrane to vibrate, forming a travelling wave along it from the base of the cochlea to its apex. For a pure tone entering the ear, the travelling wave reaches a peak at a specific point along the cochlear duct depending on the frequency of the tone. Complex sounds with multiple frequencies produce multiple peaks with high frequencies peaking towards the base of the spiral and decreasing frequencies peaking further and further towards the apex along a tonopic axis. The basilar membrane vibration in turn moves the organ of Corti up and down and also causes the overlying tectorial membrane to shear in a radial direction across the top of the organ of Corti; this ultimately directs the stimulus energy of the vibration to the stereociliary bundle of the hair cell (Fig. 1d).
Two varieties of hair cell are present, the inner and the outer hair cell. These two types are morphologically, anatomically and functionally distinct and interact differently with the tectorial membrane (for a review, see Fettiplace and Hackney 2006).
Inner hair cells are flask-shaped with a central nucleus and apical hair bundle. Viewed from above, this bundle contains stereocilia in an almost linear arrangement. The stereocilia form rows that increase in height across the bundle like a staircase. Typically, in mice, inner hair cells possess about 50 of these stereocilia. The inner hair cells themselves form a single row running along the basilar membrane-organ of Corti and about 3400 of them are found in a human cochlea (Rask-Andersen et al. 2012). The main function of inner hair cells is to detect the vibration of the basilar membrane and to code for its amplitude in the auditory nerve fibres, between 10 and 20 of which communicate with the base of each inner hair cell. This coding relates to the intensity (or loudness) of the sound, a louder sound producing a greater response in the inner hair cell. The radial motion of the tectorial membrane seems to generate fluid movements across the bundle that elicits transduction. The mechanical activity of the basilar membrane/organ of Corti complex, however, is complicated, as other factors, which will not be considered here, also affect the characteristics of the mechanical response. The frequency of the sound is coded for primarily by the location of the hair cell, with apical hair cells responding to lower frequencies, the response systematically changing to higher frequencies as the location moves more basally, i.e. the coding frequency is determined by virtue of the position of the hair cells along the spiral. The frequency eliciting the best response is specific to the nerve fibres that uniquely innervate each hair cell. Some temporal coding might also occur as for lower frequencies, auditory nerve fibre firing can be phase-locked to the stimulus frequency whereby action potentials occur synchronously with the peak of each cycle of the acoustic wave. Temporal coding is likely to be important in sound localisation as it allows the precise timing of stimuli coming into the two ears to be compared (Joris 2003).
Outer hair cells are more cylindrical than inner hair cells, with a basal nucleus and apical hair bundle, this time arranged as a W-shape (Fig. 2a). The approximately 100 stereocilia per cell (in mice) are organised in rows increasing in height as before but the tips of the tallest row are embedded in the lower surface of the tectorial membrane and so are driven directly by tectorial membrane movement. About three times as many outer hair cells are present as inner hair cells and, hence, about 12,000 occur in a human cochlea but their contribution to auditory nerve activity is weak, if there is any at all. Instead, their main function is to enhance the motion of the basilar membrane. In response to the stimulus delivered to the hair bundle, the outer hair cell transduces like an inner hair cell but the stimulus is converted into contraction-elongation movements that increase the vertical displacement of the basilar membrane. This amplifies the movement to its greatest extent at the frequency that generates the peak of motion, and the amplified signal is detected by the inner hair cell (for a review, see Fettiplace and Hackney 2006).

Scanning electron micrographs of the upper surface of the organ of Corti (reticular lamina). a Normal structure of the reticular lamina: the hair bundles of the hair cells are seen organised in precise rows: the W shapes of the three rows of outer hair cells (OHC) and the more linear bundles of the one row of inner hair cells (IHC) separated by supporting cell apices. b The reticular lamina from a low frequency region of a guinea pig cochlea damaged by kanamycin, a strongly ototoxic aminoglycoside antibiotic. Some outer hair cells are missing, those which have degenerated having been replaced by scars derived from the expanded apices of supporting cells. c The reticular lamina from a high frequency region of a guinea pig cochlea damaged by kanamycin in the same experiment as in b. Very few outer hair cell bundles are left, the outer hair cells of the high frequency region being more susceptible than those of the low frequency region. Inner hair cells (IHC) seem relatively intact at this level of the cell but the cell bodies below the reticular lamina cannot be seen and thus their condition is uncertain. Bar 10 μm
Hair cell loss
Loss of hair cells (Fig. 2b, c) is characteristic of a number of different types of auditory damage or genetic defect and, indeed, of ageing, although in the last-mentioned case there is the potential for cumulative effects other than just those of ageing alone leading to hair cell loss. Noise insult tends to produce hair cell loss in varied locations, related to the type of noise stimulus. Studies of animals by using overexposure to pure tone acoustic stimuli tend to show maximum damage in particular regions along the organ of Corti (e.g. Fredelius et al. 1987); the regions involved are presumably dependent on the frequencies and intensities of sound in the damaging noise, although variability is apparent in the individual responses. However, a typical pattern of hair cell loss in ageing or ototoxicity starts with the outer hair cells at the base of the cochlea, progressively moving apically and including the inner hair cells at a later stage (see Jiang et al. 1993; Mahendrasingam et al. 2011), although there are many exceptions to this. This suggests an increased vulnerability of the higher frequency hair cells.
Some studies have attempted to understand the tonotopic pattern of hair cell loss, and changes are evident in the expression of a variety of proteins along the cochlea possibly affecting the vulnerability of high-frequency hair cells compared with low-frequency hair cells. Jenisen-Smith et al. (2012) suggest that basal outer hair cells are metabolically more susceptible to environmental changes. Another mechanism that has been suggested is failure of calcium homeostasis. In mammals, mutations in the gene encoding plasma membrane calcium ATPase2 (PMCA2) cause deafness (Mammano 2011). Changes in PMCA2 expression along the cochlea have been postulated as being responsible for the greater sensitivity of basal hair cells, although a systematic quantitative study has found no evidence for a substantial difference in PMCA2 expression along the tonotopic axis (Chen et al. 2012).
The evidence of changes in other calcium-regulating proteins nevertheless makes differential susceptibility to calcium toxicity plausible. Calcium loading has been reported in noise-induced hearing loss (Jacob et al. 2013). In another study, the calcium-sensing cochlear protein, otoferlin, has been reported to be present in low-frequency but not high-frequency hair cells (Engel et al. 2006). It has been concluded that otoferlin and calcium-activated potassium channels (BK channels) play important roles in cell survival under noise-damaging conditions. Evidence from studies of non-mammalian systems indicates that the failure of calcium homeostasis is a factor in the death of hair cells (Esterberg et al. 2014).
The vulnerability of the basal outer hair cells to other damaging agents has also been investigated. One suggestion is the involvement of transient receptor potential vanilloid 1 and 4 channels in the susceptibility of high-frequency hair cells to ototoxic antibiotics (Lee et al. 2013). These channels are thought to provide a route of entry of aminoglycosides into the hair cell. However, aminoglycosides can also enter through the transduction channels of the hair cells (Alharazneh et al. 2011). Thus, a combination of characteristics is likely to make the high-frequency hair cells more vulnerable than the low-frequency ones.
Finally, reports that basal outer hair cells are more susceptible to free radicals (Sha et al. 2001) are also worth noting. This could provide a common factor in the typical pattern of hair cell loss. The increased vulnerability has been attributed to a lower expression of glutathione, an anti-oxidant, in basal hair cells.
Processes of hair cell degeneration
Numerous causes of hair cell loss have been reported, ranging from direct physical damage and death to factors that trigger a more defined response, depending on the type and magnitude of damage. Clearly, a significant traumatic event that leads to a break in the reticular lamina and physical damage to the cells themselves does not allow protective mechanisms to be engaged. This is the case when high-intensity sound (130 dB) causes extreme mechanical movements and tearing of the delicate epithelium and leads to the mixing of the endolymph and perilymph (Rauchegger and Spoendlin 1981). Subsequent to this, chronic cell loss can occur by mechanisms that attempt to prevent or limit other damaging effects (Fetoni et al. 2014), as discussed below. In the case of the tearing of the reticular lamina and fluid mixing, the optimal distribution of ions (i.e. primarily potassium and sodium) is disturbed. The hair cells normally sit with their apical surfaces (including the hair bundle) bathed in endolymph and, hence, exposed to high potassium and low sodium ions, with their basolateral surfaces being bathed in perilymph and, hence, exposed to low potassium and high sodium levels. Evidence suggests that elevated potassium is toxic to the hair cell if it builds up to excess (Nouvian et al. 2003). Failure to eliminate potassium from the perilymph could result in this toxic build up.
With less damaging agents, hair cells go through a number of stages before finally becoming lost. Moreover, the loss of the hair cell might be expected to leave a hole in the organ of Corti through which the two distinct fluids, endolymph and perilymph, would mix. However, evidence has been presented that, at least if time permits, the organ of Corti is able to remodel, as the hair cells degenerate so that a scar is formed derived from adjacent supporting cells (Fig. 2b, c).
An example in which this has been studied in detail is the response to ototoxic antibiotics in guinea pigs. As noted previously, the outer hair cells show the greatest susceptibility (Fig. 2). The basal part of the outer hair cell below the reticular lamina tends to degenerate first, whereas the apex retains its tight junctions. Subsequently, the apices are extruded at the same time that the supporting cells swell up and maintain a seal across the hole that would otherwise be formed. The extrusion occurs upwards from the reticular lamina (Forge 1985). This has also been observed in vestibular system epithelia after the trans-tympanic injection of gentamicin (Quint et al. 1998) and in naturally ageing cochleae (D.N. Furness and C.M. Hackney, unpublished observations; Fig. 3). Whether this is always true remains to be established, as holes have been noted occasionally after kanamycin damage in guinea pigs (Furness and Hackney 1986) and the situation for other species might also be different. Nevertheless, morphological features of hair cell loss and supporting-cell expansion indicate that the organ of Corti has evolved mechanisms to prevent or limit the spread of damage.

Mode of hair cell degeneration in guinea pigs following kanamycin damage (a) and old age (b). In both cases, the apices of the outer hair cells appear to be extruded from the surface of the reticular lamina without leaving a gap, because of the expansion of supporting cells. The hair bundles at this stage are still present, even though the apices are no longer attached to the rest of the cell body. Bar 5 μm
When hair cell degeneration has been triggered, two main pathways lead to death, as with other tissues of the body. These are: (1) catastrophic cell death (necrosis) in which the cell contents are released into the environment and (2) programmed cell death (apoptosis) in which a trigger sets in motion a careful and proscribed sequence of energy-dependent events that lead to cellular degeneration without loss of the contents of the cell. A “third pathway” has also been described after noise damage (Bohne et al. 2007). Recently, a form of controlled necrosis, necroptosis, has also been suggested to occur (Park et al. 2012). In general, necrosis is bad for tissues, leading to inflammation and damage to other cells nearby because of the release of intracellular enzymes, whereas apoptosis leads to the destruction of a cell with fewer collateral effects.
Mechanisms of cell death
As a prelude to discussing further the mechanisms leading to death of the hair cell, it is worth briefly reviewing the general features of the two main types of cell death, namely necrosis (necroptosis) and apoptosis. However, the study of cell-death mechanisms is in itself a substantial topic and here only a brief mention of the major molecular players can be given.
Necrosis and necroptosis
Necrosis is characterised by swollen intracellular organelles, pyknosis (condensation of the nucleus), breakdown of the cell membranes, and release of the cell contents (Fig. 4; Nagańska and Matyja 2001). The release of cell contents and the failure also to clear up apoptotic cells provokes or contributes to further inflammatory changes and the death of other cells (Szondy et al. 2014). A form of programmed necrosis (necroptosis) has also been described (for a review, see Yuan and Kroemer 2010). Necroptosis can be triggered by factors such as receptor interacting protein (RIP) kinases.

a Representation of the morphological stages in the two main modes of cell death: necrosis (left) and apoptosis (right). In necrosis, the nucleus condenses and the cell starts to fragment, ultimately releasing its contents into the surrounding environment. In apoptosis, the nucleus condenses, the cell membrane becomes convoluted and apoptotic bodies form containing cell organelles and nuclear material. The apoptotic bodies are phagocytosed by macrophages, hence preventing the release of cell contents into extracellular space. b Simplified representation of the extrinsic and intrinsic pathways involved in apoptosis and some of the key players in each. Note that external signals trigger the extrinsic pathway; the one shown here is FasL but numerous others exist. The intrinsic pathway can be activated by intracellular triggers, such as reactive oxygen species (ROS) amongst others, and also by the extrinsic pathway
Apoptosis
In apoptosis, the cell membranes become convoluted but do not disintegrate. The nucleus becomes pyknotic and starts to break up into smaller parts, with the chromatin becoming condensed (for a review, see Strasser et al. 2011). The cytoplasm also condenses and the cell then forms blebs that are phagocytosed by surrounding macrophages (Fig. 4).
Various methods can detect apoptosis in tissues, such as: looking for apoptotic proteins by using immunohistochemistry; labelling the DNA and looking for morphological changes, for example, by using Hoechst 33342 (Hu et al. 2000); TUNEL staining combined with morphological evaluation (Nishizaki et al. 1999). TUNEL detects DNA fragmentation and severe DNA damage. It relies on the presence of nicks in the DNA that can be localised by terminal deoxynucleotidyl transferase, an enzyme that catalyzes the addition of deoxyuridine triphosphates that are conjugated to a marker. However, such DNA damage might also occur during autolysis (self-destruction caused by the intracellular release of a cell’s own enzymes) and not necessarily only as a consequence of the classical apoptotic pathway.
Apoptosis takes place under genetic control and involves a bewildering array of triggers and signalling pathways. Common triggers include reactive oxygen species (ROS) and extracellular ligands. A role for nitric oxide has also been suggested (see Wang et al. 2010). A variety of genes are involved, such as aspartate-specific cysteine proteases (caspases), apoptotic protease activating factor 1 (APAF-1) a cofactor for caspase 9, the B-cell lymphoma 2 (BCl-2) family including an interacting mediator of cell death (BIM) and hepatic leukaemia factor (HLF) and c-Jun N-terminal kinases (JNK), amongst others. The BCl-2 family includes both pro- and anti-apoptotic genes. Caspases cleave various proteins, with secondary effects such as DNA cleavage leading to chromatin condensation and the stimulation of cytoskeletal proteins such as actin to induce bleb formation and shape changes. They are themselves activated from pro-caspases by other caspases (Strasser et al. 2011).
Apoptosis has also been subdivided into an extrinsic (or death receptor) and an intrinsic (or mitochondrial) pathway (Fig. 3). These pathways differ in that the extrinsic pathway acts through a signalling pathway involving cell-death receptors on the surface, whereas the intrinsic pathway is triggered by cytokine deprivation, intracellular damage or oncogene expression. The two pathways are not mutually exclusive and can interact.
Death-receptor pathways are initiated via receptor oligomerisation induced by their ligands (Guicciardi and Gores 2009), an event that in turn results in the recruitment of specific adaptor proteins and the activation of caspases-8 and −10. An example of the extrinsic pathway (Fig. 4) is that activated by Fas ligand (FasL), which activates antigen 1 (Fas, also known as CD95 and APO-1) followed by FAS adaptor protein (FADD) and then caspase-8. Other extrinsic receptors are tumour necrosis factor-alpha (TNF-α), TNF-related apoptosis-inducing ligand (TRAIL) and death receptors (DR), each of which has its own intracellular mediators leading to caspase activation. TNF-α can also activate JNK and this can lead to the inhibition of Bcl-2, itself a pro-survival factor, by phosphorylation. In the absence of caspase activation, stimulation of death receptors can also lead to necroptosis (for a recent review of death receptor pathways, see Lavrik 2014).
The intrinsic pathway is stimulated by intracellular stressors, including ROS, but can also be triggered by activation through the extrinsic pathway. For example, both ROS (Wu and Bratton 2013) and some of the extracellular signals noted above lead to Bcl-2 family activation, two members of which, the BCl-2 antagonists BCl-2 associated X protein (BAX) and BCl-2 killer protein (BAK), create pores in mitochondria. The pores allow cytochrome c and other materials to be released into the cytoplasm to act as apoptosis-promoting factors. Cytochrome c release can lead to the generation of further ROS, producing a feedback loop increasing the damage to mitochondria and inducing further cytochrome c release. Cytochrome c triggers APAF-1 mediated activation of caspase-9. The intrinsic and extrinsic pathways are similar in that caspase-8 and −9 both initiate effector caspases −3, −6 and −7 leading to apoptosis by proteolysis and DNA cleavage (via the caspase-activated DNAase, CAD). Activated caspase 8 can also cleave Bid, a normally pro-survival member of the Bcl-2 family, forming truncated Bid, which leads to apoptosis (Strasser et al. 2011). Another important protein in the intrinsic pathway is p53, a tumour suppressor. When DNA damage occurs and its repair fails, p53 is phosphorylated and this leads to the activation of BAX triggering the mitochondrial pathway (Niemenen et al. 2013).
Activation of the death receptor ligands does not always lead to apoptosis. For example, TNF-α receptor activation can have anti-apoptotic effects. It can initiate the nuclear factor (NF-κB) pathway and the mitogen-activated protein kinase (MAPK) pathways, which promote pro-survival genes (Marques-Fernandes et al. 2013). NF-κB is a protein complex that controls the transcription of DNA and is associated with cellular responses to stimuli such as stress, cytokines and free radicals.
Apoptosis can also be triggered by caspase-independent pathways. One such trigger is endonuclease G (endoG), which is a DNAase that is released from mitochondria and cleaves DNA in chromatin in the nucleus (Li et al. 2001). Another is apoptosis-inducing factor (AIF), which plays a similar role to endoG (Candé et al. 2002).
Apoptosis is also regulated by micro-RNAs (miRNAs). These are non-coding RNAs that act to determine protein expression during transcription and post-transcriptionally. Many are associated with cancer (for a review, see Jovanovic and Hengartner 2006). For example, the inhibition of several miRNAs (miR-1d, 7, 148, 204, 210, 216 and 296) has been found to lead to increased caspase-3 activity, with one miRNA (miR-214) leading to decreased activity (Cheng et al. 2005). More recently, Xiong et al. (2010) and Roshan et al. (2014) have demonstrated the involvement of miR-29 in apoptosis. miR-29 appears to target two anti-apoptotic molecules, Bcl-2 and myeloid cell leukaemia 1 (Mcl-1), which promotes cell survival by acting on Bcl-2; silencing these proteins mimics the apoptotic-promoting effect of miR-29, whereas their over-expression reduces apoptosis. miR-29 over-expression also stimulates the mitochondrial pathway. This miRNA appears to influence voltage-dependent anion channel 1 (VDAC1), a mitochondrial outer-membrane ion channel, which is up-regulated several‐fold in several brain regions following miR-29 knockdown in mice. Partial recovery of apoptosis can be obtained by the down-regulation of VDAC1 in miR-29 knockdown cells (Roshan et al. 2014).
Another apoptotic regulator is miR-34, which is involved with p53 signalling. miR-34 can induce apoptosis and is itself regulated by p53. This pathway is known to be involved in the suppression of tumourigenesis. miR-34 also appears to be involved in a feedback loop stimulating the up-regulation of p53 (He et al. 2007).
Necrosis and apoptosis of hair cells
Necrosis and apoptosis have been demonstrated in hair cells by a number of studies looking at different forms of auditory damage. In many cases, these two processes occur side-by-side. For example, after kanamycin-induced hair cell damage, cells displaying features either of necrosis or apoptosis can be observed (Jiang et al. 2006). However, determining whether these structural features reflect classical mechanisms is more difficult.
Apoptotic hair cells have been detected by using Hoechst staining after noise damage (Hu et al. 2000) and by using TUNEL during ageing (Usami et al. 1997), although as noted above, the latter might also reflect autolysis (Nishizaki et al. 1999). A number of other studies have shown the cochlear expression of apoptosis-promoting genes, such as caspases and p53 following noise trauma and during ageing (see Hu et al. 2009; Tadros et al. 2008; Fetoni et al. 2014). Attempts to visualise TUNEL staining and other markers for apoptosis after ototoxicity (kanamycin damage) suggest that the classic apoptotic pathways are not always involved (Jiang et al. 2006). Other studies, however, do implicate classic apoptotic pathways in ototoxicity. Certainly, after aminoglycoside toxicity, hair cells with apoptotic features can be observed (Fig. 5). Ototoxic drugs might, therefore, trigger a wider variety of pathways for cell death in the cochlea.

High-frequency inner hair cells in a normal guinea pig cochlea (a) and after kanamycin-induced damage (b). In b, the cell shows signs of undergoing apoptosis. Compared with a, the nucleus (N) is showing condensation and becoming misshapen, the cell membrane is becoming convoluted and irregular and the cytoplasm is condensing. Bar 5 μm
One of the common factors involved in the apoptotic death of hair cells and a potential trigger of cell death is the generation of ROS. ROS are involved in cell signalling pathways and therefore have specific functions in cells (Morgan and Liu 2011). These energetic particles are released during a variety of cell processes and can be created by the increased activity of mitochondria. However, in relatively low doses, oxidative stressors such as ROS can cause mitochondrial injury in hair cells (Baker and Staeker 2012). The increased production of ROS in turn further affects the mitochondria. Indeed, they are known to cause damage to mitochondrial DNA. Noise exposure increases mitochondrial activity and so promotes further formation of ROS. Aminoglycoside antibiotics (Sha and Schacht 1999) and cisplatin (Ravi et al. 1995) both appear to induce the formation of ROS. Another common feature of noise damage and aminoglycoside ototoxicity is the involvement of the JNK pathways (Wang et al. 2003). More details of the contribution of these are found in the individual sections below.
The involvement of ROS in many cases of cochlear damage primarily implicates the intrinsic pathway in hair cell death, as ROS appear to interact with this pathway in multiple ways (Wu and Bratton 2013). ROS alter the function of Bcl-2 family members, regulating the kinases that phosphorylate them. This includes MAPK, JNK and extracellular signal-regulated kinase (ERK; Torres 2003). The production of apoptosis regulator miR-29a is altered by oxidative stress in an organ of Corti cell line exposed to the oxidative stressor tertbutyl hydroperoxide (t-BHP). Exposure to low concentrations appears to down-regulate miR-29a but higher concentrations up-regulate it (Wang et al. 2010). Possibly, with sufficient oxidative stress, this miRNA is increased, initiating apoptosis.
Because the generation of ROS seems to be a major factor in inducing hair cell apoptosis, this raises the question of whether both extrinsic and intrinsic pathways are involved in hair cell loss. Clear FasL expression has not been detected in hair cells, even after experimentally induced labyrinthitis (Bodmer et al. 2003a). The same group has also shown that, in mutants with an absence of Fas receptor function, hair cell loss after aminoglycoside toxicity is no different from that in control mice (Bodmer et al. 2003b). Moreover, no TNFα receptors or other receptors associated with extrinsic pathways have been reported on hair cells. On the other hand, both caspase-8 and TNF receptor type 1-associated death domain protein (TRADD) are found in outer hair cells after a combination of ethacryinic acid (a loop diuretic) and cisplatin ototoxicity (Ding et al. 2007; see also Devrajan et al. 2002). RIPs are expressed by hair cells and play a role in outer hair cell necrosis/necroptosis (Zheng et al. 2014). Increasing emphasis will probably be laid on cochlear studies of necroptosis, as this is a rapidly growing area in its own right (Jouan-Lanhouet et al. 2014).
In agreement with the process of apoptosis in other tissues, macrophages would be expected to be present in order to phagocytose apoptotic bodies and cellular debris from necrosis within the cochlea. Indeed, another common feature of noise-induced damage and aminoglycoside toxicity is the recruitment of macrophages (see Fredelius and Rask-Andersen 1990; Ladrech et al. 2007). Their role lies in the clearing up of the cellular debris produced by cell death.
Cell death in response to processes that cause cochlear damage
Age-related hearing loss
Oxidative stress and mitochondrial dysfunction are clearly involved in ARHL, as in other forms of age-related tissue degeneration (Fujimoto and Yamasoba 2014). Mice that lack the gene for superoxide dismutase 1, an antioxidant that defends cells against oxidative stress, show premature hair cell loss and ARHL (McFadden et al. 1999). Oxidative stress causes Bak to be expressed in the cochlea, hence increasing mitochondrial damage, whereas deletion of the Bak gene prevents early hair cell loss in C57BL/6 J mice (Someya et al. 2009), a strain that shows accelerated ARHL. Evidence obtained from other systems has shown that BAK and BAX work together (Westphal et al. 2011) and might form a pore in the mitochondrial membranes allowing the release of cytochrome c (Bleiken et al. 2013).
mi-RNAs are also likely to play a role in hair cell degeneration in ageing. In a study of mi-RNAs involved in age-related hair cell loss, Zhang et al. (2013) noted that miRNAs that exhibited approximately two-fold up-regulation included members of the miR-29 family and miR-34 family, which are known regulators of pro-apoptotic pathways. In contrast, miRNAs that were down-regulated by about two-fold were members of the miR-181 family and miR-183 family, which are known to be important for proliferation and differentiation, respectively. The shift of miRNA expression favouring apoptosis occurs before hearing threshold elevation and hair cell loss are observed suggesting that they are part of the process leading to the degeneration.
Ototoxicity
Ototoxicity is a general term describing the damaging effect of a variety of drugs on the hearing organ. Drugs that have these effects include anti-cancer agents, such as cisplatin and carboplatin, and aminoglycoside antibiotics, such as streptomycin and gentamicin. Many of these drugs are still in clinical use, despite having the side effect of ototoxicity.
Ototoxic drugs appear to target the hair cells, sometimes selectively. For example, aminoglycoside antibiotics have been used as an experimental tool to generate selective outer hair cell loss (Ryan and Dallos 1975; Harrison and Evans 1979; Fig. 2), although they clearly also affect the inner hair cells, but to a lesser extent (Fig. 5). Aminoglycoside antibiotics can also produce, in common with many other agents that affect hearing, a base-to-apex gradient in their effect on hair cells, with basal hair cells being intrinsically more sensitive than apical ones (Richardson and Russell 1991).
Studies of kanamycin ototoxicity initially implied that apoptosis was the main route of hair cell death (Nakagawa et al. 1998). However, as became apparent subsequently, the modes of hair cell loss produced by ototoxic drugs are varied, even for a single drug. Thus, in the study by Jiang et al. (2006), both apoptotic and necrotic features were observed as a result of kanamycin-induced hair cell loss but a lack of the usual apoptotic markers was noted. This led the authors to conclude that the pathway did not include classic apoptosis. Nevertheless, in another study in which the kanamycin and a loop diuretic (bumetanide) were administered sequentially in mice, outer hair cells were shown to display TUNEL and activated caspase-3 labelling, implying that they mostly died via an apoptotic process (Taylor et al. 2008). Inner hair cells were less susceptible and the effects were delayed but they also showed features of autophagy, apoptosis and necrosis.
Cisplatin also induces apoptotic hair cell loss (Devarajan et al. 2002; Garcia-Berrocal et al. 2007; Ryback et al. 2007; De Freitas et al. 2009), stimulating the expression of a number of apoptotic factors (Jamesdaniel et al. 2008). These include pro-survival activating transcription factor 2 (ATF2), pro-apoptotic serine-threonine protein kinase and RIP. Both extrinsic and intrinsic pathways have been suggested to be involved (Devarajan et al. 2002).
A common feature of ototoxic agents is the production of ROS (Huang et al. 2000; Rizzi and Hirose 2007). ROS have thus been implicated in hair cell death in ototoxicity, including aminoglycoside- and cisplatin-induced hair cell loss (Ryback et al. 2007; Op de Beeck et al. 2011). Cisplatin ototoxicity induces the production of ROS and free radicals and these act on the hair cell membrane to generate 4-hydroxynonenal, a mediator of apoptosis (Huang et al. 2000).
The involvement of miRNA in ototoxicity-induced apoptosis has yet to be thoroughly investigated. One report suggests that miR-34 is up-regulated after kanamycin-induced ototoxicity but this does not appear to be associated with hair cells (Yu et al. 2010).
Acoustic trauma
In noise-induced hair cell loss, as with the other conditions mentioned previously, evidence indicates that ROS play a major role (Ohinata et al. 2000; Fetoni et al. 2013). Thus, the intrinsic pathway is involved. EndoG and AIF have both been detected in the cochlea after noise-induced damage (Yamashita et al. 2004) suggesting that caspase-independent cell death is also a possible mechanism in hair cell loss. AIF and endoG both translocate from the mitochondria into the nucleus in hair cells, the former into both apoptotic and necrotic hair cells, as shown by Han et al. (2006). In the same study, caspase-3 activation has also been detected in hair cells. Thus, caspase-dependent, caspase-independent and necrotic cell death all occur together after noise damage. Activation of the TNFα pathway in noise damage in the cochlea has also been reported, although the data do not show whether this pathway is expressed in hair cells (Hu et al. 2009).
The necrotic process after noise damage appears to involve two pathways, according to morphological evidence presented by Bohne et al. (2007). In this study, the authors suggest that one form of necrotic cell death is typified by outer cells undergoing a swollen phase (oncosis) followed by death (necrosis) and the other (their “third pathway”) by outer hair cells losing their basolateral membrane but retaining cytoplasm in a cylindrical shape. The latter might be a feature specific to cochlea, as Bohne et al. (2007) suggest that it could be a result of exposure to increased levels of potassium from nearby degenerated outer hair cells. However, outer hair cells also have a complex sub-plasma membrane cortical lattice that could potentially hold the cytoplasm in place after loss of the membrane (see Bannister et al. 1988). This morphological feature of a retained shape could simply reflect the possibility that the lattice does not always break down early during hair cell degeneration and that its presence physically constrains the remaining cytoplasm into a cylinder.
As with ototoxic drugs, evidence therefore exists for both apoptosis and necrosis following noise-induced damage. However, the balance between necrosis and apoptosis is unclear. One study has demonstrated that high noise levels can produce membrane damage (Hu and Zheng 2008). However, rather than leading to necrosis, this appears to trigger apoptosis. In a study of permanent-noise-induced outer hair cell loss, Zheng et al. (2014) have shown the necrosis triggering factors RIP1 and RIP3 are up-regulated, together with the apoptosis signalling molecule, caspase-8. Inhibition of caspases reduces the number of apoptotic nuclei but increases the expression of RIP1 and RIP3 and the number of necrotic nuclei. Inhibition of necrosis has the opposite effect decreasing the number of necrotic nuclei and increasing the number of apoptotic nuclei, although no corresponding increase in caspase-8 expression has been detected. Instead, caspase-9 is up-regulated. These results support the suggestion that a balance occurs between the two methods of cell death in noise-induced hair cell loss but that this can vary depending on conditions. Indeed, the intensity of the noise might be a factor in determining this balance (for a review, see Op de Beeck et al. 2011).
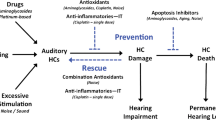
Preventing apoptosis and necrosis as a therapy for ameliorating hair cell loss
Many studies have detailed a plethora of tests of various drugs and agents as potential therapeutic interventions in ototoxicity, ARHL and noise damage. These broadly fall into two categories: those aimed at interrupting the signalling mechanisms that lead to apoptosis and necrosis and those aimed at inhibiting specific parts of the cell-death pathways.
Many strategies centre on the reduction of ROS, the intracellular trigger for intrinsic apoptosis. Blocking the generation of ROS by using anti-oxidant drugs such as N-acetylcysteine (NAC) and salicylate might be protective against their damaging effects (Kopke et al. 2000; Duan et al. 2004). MitoQ, another anti-oxidant that accumulates in mitochondria, is also protective against gentamicin (Ojano-Dirain et al. 2014). In organ cultures exposed to gentamicin, ROS were reduced by melatonin, tacrolimus and dexamethasone, which also caused a reduction in the expression of several factors implicated in apoptosis (Bas et al. 2012). These agents were otoprotective in vivo. Another anti-apoptotic agent, TAT-FNK (a combination of an artificial protein based on Bcl-xL called FNK and of TAT [trans-activation response element], a regulator of viral transcription; see Kashio et al. 2007), inhibited aminoglycoside-induced ototoxicity when released from a gel placed against the round window in guinea pigs (Kashio et al. 2012). The damage was induced by a kanamycin sulphate and ethacrynic acid mixture, and the anti-apoptotic agent was introduced prior to administration of the ototoxic agents.
Inhibition of each of the cell death pathways has been tried with agents that block various stages. Neomycin-induced hair cell loss can be attenuated in organ of Corti explants by inhibiting the expression of dimethylated histone H3K4 by using lysine-specific demethylase 1. This treatment can be recapitulated in vivo and is accompanied by reduced caspase-3 expression (He et al. 2014). Amelioration of the ototoxic and apoptotic promoting effects of cisplatin has also been found by using inhibitors. Examples of this are histone deacetylase inhibitors such as trichostatin A (Wang et al. 2013). Experiments on organ cultures derived from 3-day old mice showed a reduction in cisplatin-induced hair cell loss on pre-treatment with trichostatin A. Microarray analysis revealed that this treatment prior to cisplatin exposure reduced the expression of apoptotic genes associated with the TNF and p53 pathways, compared with cisplatin alone. These were TNFRSF1a, a TNF receptor, and p53 itself.
The effects of aminoglycosides can also be attenuated by necrosis and necroptosis inhibitors (Park et al. 2012). NecroX treatment caused a reduction in hair cell loss in mouse organ of Corti cultures and decreased apoptotic nuclei on TUNEL staining. Similar results were found in zebrafish lateral line (Song et al. 2014). Inhibition of p53 by pre-treatment with pifithrin alpha or KX1-004 also has protective effects on noise damage by reducing apoptosis (Fetoni et al. 2014).
Whereas the numerous studies on otoprotection are pointing the way, an effective clinical treatment has not yet been found, although some clinical trials with NAC have been undertaken (Kopke et al. 2007). In a clinical study of head and neck cancer patients taking cisplatin, no overall benefit was found but a small number of individuals showed some protection by transtypmanic NAC (Yoo et al. 2014). In another study of noise-induced hearing loss, some benefit was found in that NAC reduced the size of temporary threshold shifts (Lin et al. 2010). These and other clinical studies imply a future for agents that interfere with the mechanisms of programmed cell death.
Concluding remarks
In this review, the mechanisms that lead to the death of hair cells have been evaluated. The commonest factor in the three main kinds of cochlear damage is the generation of ROS, which can trigger intrinsic apoptosis. However, in some cases, hair cell death is produced via an extracellular signalling pathway leading to extrinsic apoptosis. Necrosis and necroptosis also play a role and, for a given damaging event, a balance exists between these possible routes of cell death, with no one mechanism being exclusively activated. Further, we should bear in mind that, whereas this review focuses on hair cell death, other targets are present within the cochlea, such as the neural elements in the spiral ganglion and the homeostatic tissues of the lateral wall, in which cell death leading to hearing loss can occur. Finally, the many studies of these mechanisms of hair cell death have led to the identification of numerous potential therapeutic agents, of which some have been used to attempt to treat people suffering cochlear damage. Thus, the continued work in this area is likely ultimately to lead to clinical treatments to prevent or ameliorate hearing loss.
References
Alharazneh A, Luk L, Huth M, Monfared A, Steyger PS, Cheng AG, Ricci AJ (2011) Functional hair cell mechanotransducer channels are required for aminoglycoside ototoxicity. PLoS One 6:e22347
Baker K, Staecker H (2012) Low dose oxidative stress induces mitochondrial damage in hair cells. Anat Rec (Hoboken) 295:1868–1876
Bannister LH, Dodson HC, Astbury AR, Douek EE (1988) The cortical lattice: a highly ordered system of subsurface filaments in guinea pig cochlear outer hair cells. Prog Brain Res 74:213–219
Bas E, Van De Water TR, Gupta C, Dinh J, Vu L, Martínez-Soriano F, Láinez JM, Marco J (2012) Efficacy of three drugs for protecting against gentamicin-induced hair cell and hearing losses. Br J Pharmacol 166:1888–1904
Bleicken S, Landeta O, Landajuela A, Basañez G, García-Sáez AJ (2013) Proapoptotic Bax and Bak proteins form stable protein-permeable pores of tunable size. J Biol Chem 288:33241–33252
Bodmer D, Brors D, Bodmer M, Pak K, Ryan AF (2003a) Fas ligand expression in the organ of Corti. Audiol Neurootol 8:243–249
Bodmer D, Brors D, Pak K, Bodmer M, Ryan AF (2003b) Gentamicin-induced hair cell death is not dependent on the apoptosis receptor Fas. Laryngoscope 113:452–455
Bohne BA, Harding GW, Lee SC (2007) Death pathways in noise-damaged outer hair cells. Hear Res 223:61–70
Candé C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G (2002) Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie 84:215–222
Chen Q, Mahendrasingam S, Tickle JA, Hackney CM, Furness DN, Fettiplace R (2012) The development, distribution and density of the plasma membrane calcium ATPase 2 calcium pump in rat cochlear hair cells. Eur J Neurosci 36:2302–2310
Cheng AM, Byrom MW, Shelton J, Ford LP (2005) Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 33:1290–1297
De Freitas MR, Figueiredo AA, Brito GA, Leitao RF, Carvalho Junior JV, Gomes Junior RM, Ribeiro Rde A (2009) The role of apoptosis in cisplatin-induced ototoxicity in rats. Braz J Otorhinolaryngol 75(5):745–752
Deltenre P, Van Maldergem L (2013) Hearing loss and deafness in the pediatric population: causes, diagnosis, and rehabilitation. Handb Clin Neurol 113:1527–1538
Devarajan P, Savoca M, Castaneda MP, Park MS, Esteban-Cruciani N, Kalinec G, Kalinec F (2002) Cisplatin-induced apoptosis in auditory cells: role of death receptor and mitochondrial pathways. Hear Res 174:45–54
Ding D, Jiang H, Wang P, Salvi R (2007) Cell death after co-administration of cisplatin and ethacrynic acid. Hear Res 226:129–139
Duan M, Qiu J, Laurell G, Olofsson A, Counter SA, Borg E (2004) Dose and time-dependent protection of the antioxidant N-L-acetylcysteine against impulse noise trauma. Hear Res 192:1–9
Engel J, Braig C, Rüttiger L, Kuhn S, Zimmermann U, Blin N, Sausbier M, Kalbacher H, Münkner S, Rohbock K, Ruth P, Winter H, Knipper M (2006) Two classes of outer hair cells along the tonotopic axis of the cochlea. Neuroscience 143:837–849
Esterberg R, Hailey DW, Rubel EW, Raible DW (2014) ER-mitochondrial calcium flow underlies vulnerability of mechanosensory hair cells to damage. J Neurosci 34:9703–9719
Fetoni AR, De Bartolo P, Eramo SL, Rolesi R, Paciello F, Bergamini C, Fato R, Paludetti G, Petrosini L, Troiani D (2013) Noise-induced hearing loss (NIHL) as a target of oxidative stress-mediated damage: cochlear and cortical responses after an increase in antioxidant defense. J Neurosci 33:4011–4023
Fetoni AR, Bielefeld EC, Paludetti G, Nicotera T, Henderson D (2014) A putative role of p53 pathway against impulse noise induced damage as demonstrated by protection with pifithrin-alpha and a Src inhibitor. Neurosci Res 81–82:30–37
Fettiplace R, Hackney CM (2006) The sensory and motor roles of auditory hair cells. Nat Rev Neurosci 7:19–29
Forge A (1985) Outer hair cell loss and supporting cell expansion following chronic gentamicin treatment. Hear Res 19:171–182
Fredelius L, Rask-Andersen H (1990) The role of macrophages in the disposal of degeneration products within the organ of Corti after acoustic overstimulation. Acta Otolaryngol 109:76–82
Fredelius L, Johansson B, Bagger-Sjöbäck D, Wersäll J (1987) Qualitative and quantitative changes in the guinea pig organ of Corti after pure tone acoustic overstimulation. Hear Res 30:157–167
Fujimoto C, Yamasoba T (2014) Oxidative stresses and mitochondrial dysfunction in age-related hearing loss. Oxidative Med Cell Longev 2014:582849
Furness DN, Hackney CM (1986) Morphological changes to the stereociliary bundles in the guinea pig cochlea after kanamycin treatment. Br J Audiol 20:253–259
García-Berrocal JR, Nevado J, Ramírez-Camacho R, Sanz R, González-García JA, Sánchez-Rodríguez C, Cantos B, España P, Verdaguer JM, Trinidad Cabezas A (2007) The anticancer drug cisplatin induces an intrinsic apoptotic pathway inside the inner ear. Br J Pharmacol 152:1012–1020
Guicciardi ME, Gores GJ (2009) Life and death by death receptors. FASEB J 23:1625–1637
Hackney CM, Furness DN (2013) The composition and role of cross links in mechanoelectrical transduction in vertebrate sensory hair cells. J Cell Sci 126:1721–1731
Han W, Shi X, Nuttall AL (2006) AIF and endoG translocation in noise exposure induced hair cell death. Hear Res 211:85-95
Harrison RV, Evans EF (1979) Cochlear fibre responses in guinea pigs with well defined cochlear lesions. Scand Audiol Suppl 9:83–92
He L, He X, Lim LP, Stanchina E de, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ (2007) A microRNA component of the p53 tumour suppressor network. Nature 13:1130–1134
He Y, Yu H, Cai C, Sun S, Chai R, Li H (2014) Inhibition of H3K4me2 demethylation protects auditory hair cells from neomycin-induced apoptosis. Mol Neurobiol (in press)
Hu BH, Zheng GL (2008) Membrane disruption: an early event of hair cell apoptosis induced by exposure to intense noise. Brain Res 1239:107-118
Hu BH, Guo W, Wang PY, Henderson D, Jiang SC (2000) Intense noise-induced apoptosis in hair cells of guinea pig cochleae. Acta Otolaryngol 120:19–24
Hu BH, Cai Q, Manohar S, Jiang H, Ding D, Coling DE, Zheng G, Salvi R (2009) Differential expression of apoptosis-related genes in the cochlea of noise-exposed rats. Neuroscience 161:915–925
Huang T, Cheng AG, Stupak H, Liu W, Kim A, Staecker H, Lefebvre PP, Malgrange B, Kopke R, Moonen G, Van De Water TR (2000) Oxidative stress-induced apoptosis of cochlear sensory cells: otoprotective strategies. Int J Dev Neurosci 18:259–720
Jacob S, Johansson C, Fridberger A (2013) Noise-induced alterations in cochlear mechanics, electromotility, and cochlear amplification. Pflugers Arch 465:907–917
Jamesdaniel S, Ding D, Kermany MH, Davidson BA, Knight PR 3rd, Salvi R, Coling DE (2008) Proteomic analysis of the balance between survival and cell death responses in cisplatin-mediated ototoxicity. J Proteome Res 7:3516–3524
Jensen-Smith HC, Hallworth R, Nichols MG (2012) Gentamicin rapidly inhibits mitochondrial metabolism in high-frequency cochlear outer hair cells. PLoS One 7:e38471
Jiang D, Furness DN, Hackney CM, Lopez DE (1993) Microslicing of the resin-embedded cochlea in comparison with the surface preparation technique for analysis of hair cell number and morphology. Br J Audiol 27:195–203
Jiang H, Sha SH, Forge A, Schacht J (2006) Caspase-independent pathways of hair cell death induced by kanamycin in vivo. Cell Death Differ 13:20–30
Joris PX (2003) Interaural time sensitivity dominated by cochlea-induced envelope patterns. J Neurosci 23:6345–6350
Jouan-Lanhouet S, Riquet F, Duprez L, Vanden Berghe T, Takahashi N, Vandenabeele P (2014) Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol 35:2-13
Jovanovic M, Hengartner MO (2006) miRNAs and apoptosis: RNAs to die for. Oncogene 25:6176–6187
Kashio A, Sakamoto T, Suzukawa K, Asoh S, Ohta S, Yamasoba T (2007) A protein derived from the fusion of TAT peptide and FNK, a Bcl-x(L) derivative, prevents cochlear hair cell death from aminoglycoside ototoxicity in vivo. J Neurosci Res 85:1403–1412
Kashio A, Sakamoto T, Kakigi A, Suzuki M, Suzukawa K, Kondo K, Sato Y, Asoh S, Ohta S, Yamasoba T (2012) Topical application of the antiapoptotic TAT-FNK protein prevents aminoglycoside-induced ototoxicity. Gene Ther 19:1141–1149
Kopke RD, Weisskopf PA, Boone JL, Jackson RL, Wester DC, Hoffer ME, Lambert DC, Charon CC, Ding DL, McBride D (2000) Reduction of noise-induced hearing loss using L-NAC and salicylate in the chinchilla. Hear Res 149:138–146
Kopke RD, Jackson RL, Coleman JK, Liu J, Bielefeld EC, Balough BJ (2007) NAC for noise: from the bench top to the clinic. Hear Res 226:114–125
Ladrech S, Wang J, Simonneau L, Puel JL, Lenoir M (2007) Macrophage contribution to the response of the rat organ of Corti to amikacin. J Neurosci Res 85:1970–1979
Lavrik IN (2014) Systems biology of death receptor networks: live and let die. Cell Death Dis 5:e1259
Lee JH, Park C, Kim SJ, Kim HJ, Oh GS, Shen A, So HS, Park R (2013) Different uptake of gentamicin through TRPV1 and TRPV4 channels determines cochlear hair cell vulnerability. Exp Mol Med 45:e12
Li LY, Luo X, Wang X (2001) Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412:95-99
Li-Korotky HS (2012) Age-related hearing loss: quality of care for quality of life. Gerontologist 52:265–271
Lin CY, Wu JL, Shih TS, Tsai PJ, Sun YM, Ma MC, Guo YL (2010) N-Acetyl-cysteine against noise-induced temporary threshold shift in male workers. Hear Res 269:42–47
Mahendrasingam S, Macdonald JA, Furness DN (2011) Relative time course of degeneration of different cochlear structures in the CD/1 mouse model of accelerated aging. J Assoc Res Otolaryngol 12:437–453
Mammano F (2011) Ca2+ homeostasis defects and hereditary hearing loss. Biofactors 37:182–188
Marques-Fernandez F, Planells-Ferrer L, Gozzelino R, Galenkamp KMO, Reix S, Llecha-Cano N, Lopez-Soriano J, Yuste VJ, Moubarak RS, Comella JX (2013) TNFα induces survival through the FLIP-L-dependent activation of the MAPK/ERK pathway. Cell Death Dis 4:e493
McFadden SL, Ding D, Reaume AG, Flood DG, Salvi RJ (1999) Age-related cochlear hair cell loss is enhanced in mice lacking copper/zinc superoxide dismutase. Neurobiol Aging 20:1–8
Morgan MJ, Liu Z (2011) Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res 21:103–115
Nagańska E, Matyja E (2001) Ultrastructural characteristics of necrotic and apoptotic mode of neuronal cell death in a model of anoxia in vitro. Folia Neuropathol 39:129–139
Nakagawa T, Yamane H, Takayama M, Sunami K, Nakai Y (1998) Apoptosis of guinea pig cochlear hair cells following chronic aminoglycoside treatment. Eur Arch Otorhinolaryngol 255:127–131
Nieminen AI, Eskelinen VM, Haikala HM, Tervonen TA, Yan Y, Partanen JI, Klefström J (2013) Myc-induced AMPK-phospho p53 pathway activates Bak to sensitize mitochondrial apoptosis. Proc Natl Acad Sci U S A 110:E1839–E1848
Nishizaki K, Yoshino T, Orita Y, Nomiya S, Masuda Y (1999) TUNEL staining of inner ear structures may reflect autolysis, not apoptosis. Hear Res 130:131–136
Nouvian R, Ruel J, Wang J, Guitton MJ, Pujol R, Puel JL (2003) Degeneration of sensory outer hair cells following pharmacological blockade of cochlear KCNQ channels in the adult guinea pig. Eur J Neurosci 17:2553–2562
Ohinata Y, Miller JM, Altschuler RA, Schacht J (2000) Intense noise induces formation of vasoactive lipid peroxidation products in the cochlea. Brain Res 878:163–173
Ojano-Dirain CP, Antonelli PJ, Le Prell CG (2014) Mitochondria-targeted antioxidant MitoQ reduces gentamicin-induced ototoxicity. Otol Neurotol 35:533–539
Op de Beeck K, Schacht J, Van Camp G (2011) Apoptosis in acquired and genetic hearing impairment: the programmed death of the hair cell. Hear Res 281:18–27
Park MK, Lee BD, Chae SW, Chi J, Kwon SK, Song JJ (2012) Protective effect of NecroX, a novel necroptosis inhibitor, on gentamicin-induced ototoxicity. Int J Pediatr Otorhinolaryngol 76:1265–1269
Quint E, Furness DN, Hackney CM (1998) The effect of explantation and neomycin on hair cells and supporting cells in organotypic cultures of the adult guinea-pig utricle. Hear Res 118:157–167
Rask-Andersen H, Liu W, Erixon E, Kinnefors A, Pfaller K, Schrott-Fischer A, Glueckert R (2012) Human cochlea: anatomical characteristics and their relevance for cochlear implantation. Anat Rec 295:1791–1811
Rauchegger H, Spoendlin H (1981) Damage of the basilar membrane by acoustic stimulation. Arch Otorhinolaryngol 232:117–122
Ravi R, Somani SM, Rybak LP (1995) Mechanism of cisplatin ototoxicity: antioxidant system. Pharmacol Toxicol 76:386–394
Richardson GP, Russell IJ (1991) Cochlear cultures as a model system for studying aminoglycoside induced ototoxicity. Hear Res 53:293–311
Rizzi MD, Hirose K (2007) Aminoglycoside ototoxicity. Curr Opin Otolaryngol Head Neck Surg 15:352–357
Roshan R, Shridhar S, Sarangdhar MA, Banik A, Chawla M, Garg M, Singh VP, Pillai B (2014) Brain-specific knockdown of miR-29 results in neuronal cell death and ataxia in mice. RNA 20:1287–1297
Ryan A, Dallos P (1975) Effect of absence of cochlear outer hair cells on behavioural auditory threshold. Nature 253:44–46
Rybak LP, Whitworth CA, Mukherjea D, Ramkumar V (2007) Mechanisms of cisplatin-induced ototoxicity and prevention. Hear Res 226:157–167
Sha SH, Schacht J (1999) Stimulation of free radical formation by aminoglycoside antibiotics. Hear Res 128:112–118
Sha SH, Taylor R, Forge A, Schacht J (2001) Differential vulnerability of basal and apical hair cells is based on intrinsic susceptibility to free radicals. Hear Res 155:1–8
Someya S, Xu J, Kondo K, Ding D, Salvi RJ, Yamasoba T, Rabinovitch PS, Weindruch R, Leeuwenburgh C, Tanokura M, Prolla TA (2009) Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc Natl Acad Sci U S A 106:19432–19437
Song JJ, Chang J, Choi J, Im GJ, Chae SW, Lee SH, Kwon SY, Jung HH, Chung AY, Park HC, Choi J (2014) Protective role of NecroX-5 against neomycin-induced hair cell damage in zebrafish. Arch Toxicol 88:435-441
Spoendlin H (1985) Anatomy of cochlear innervation. Am J Otolaryngol 6:453-467
Strasser A, Cory S, Adams JM (2011) Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J 30:3667–3683
Szondy Z, Garabuczi E, Joós G, Tsay GJ, Sarang Z (2014) Impaired clearance of apoptotic cells in chronic inflammatory diseases: therapeutic implications. Front Immunol 5:354
Tadros SF, D'Souza M, Zhu X, Frisina RD (2008) Apoptosis-related genes change their expression with age and hearing loss in the mouse cochlea. Apoptosis 13:1303–1321
Taylor RR, Nevill G, Forge A (2008) Rapid hair cell loss: a mouse model for cochlear lesions. J Assoc Res Otolaryngol 9:44–64
Torres M (2003) Mitogen-activated protein kinase pathways in redox signaling. Front Biosci 8:369–391
Usami S, Takumi Y, Fujita S, Shinkawa H, Hosokawa M (1997) Cell death in the inner ear associated with aging is apoptosis? Brain Res 747:147–150
Wan G, Corfas G, Stone JS (2013) Inner ear supporting cells: rethinking the silent majority. Semin Cell Dev Biol 24:448–459
Wang J, Van De Water TR, Bonny C, Ribaupierre F de, Puel JL, Zine A (2003) A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J Neurosci 23:8596–8607
Wang P, Zhang P, Huang J, Li M, Chen X (2013) Trichostatin A protects against cisplatin-induced ototoxicity by regulating expression of genes related to apoptosis and synaptic function. Neurotoxicology 37:51–62
Wang Y, Chen C, Loake GJ, Chu C (2010) Nitric oxide: promoter or suppressor of programmed cell death? Protein Cell 1:133–142
Wangemann P (2006) Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol (Lond) 576:11–21
Westphal D, Dewson G, Czabotar PE, Kluck RM (2011) Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta 1813:521–531
Wu CC, Bratton SB (2013) Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid Redox Signal 19:546-558
Xiong Y, Fang JH, Yun JP, Yang J, Zhang Y, Jia WH, Zhuang SM (2010) Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology 51:836–845
Yamashita D, Miller JM, Jiang HY, Minami SB, Schacht J (2004) AIF and EndoG in noise-induced hearing loss. Neuroreport 15:2719–2722
Yoo J, Hamilton SJ, Angel D, Fung K, Franklin J, Parnes LS, Lewis D, Venkatesan V, Winquist E (2014) Cisplatin otoprotection using transtympanic L-N-acetylcysteine: a pilot randomized study in head and neck cancer patients. Laryngoscope 124:E87–E94
Yu L, Tang H, Jiang XH, Tsang LL, Chung YW, Chan HC (2010) Involvement of calpain-I and microRNA34 in kanamycin-induced apoptosis of inner ear cells. Cell Biol Int 34:1219–1225
Yuan J, Kroemer G (2010) Alternative cell death mechanisms in development and beyond. Genes Dev 24:2592–2602
Zhang Q, Liu H, McGee J, Walsh EJ, Soukup GA, He DZ (2013) Identifying microRNAs involved in degeneration of the organ of Corti during age-related hearing loss. PLoS One 8:e62786
Zheng HW, Chen J, Sha SH (2014) Receptor-interacting protein kinases modulate noise-induced sensory hair cell death. Cell Death Dis 5:e1262
Acknowledgments
Thanks are due to Professor C.M. Hackney CBiol FSB for reading and commenting on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Furness, D.N. Molecular basis of hair cell loss. Cell Tissue Res 361, 387–399 (2015). https://doi.org/10.1007/s00441-015-2113-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-015-2113-z