
Abstract
Polymorphic glutathione S-transferase (GST) enzymes are involved in the metabolism of xenobiotics. They are of particular interest when studying disease susceptibility and adverse drug responses. The present work deals with the genetic polymorphisms and expression of the five GST classes (alpha, mu, pi, theta and zeta) in human lung and hepatocyte cell lines. We have determined their bioavailability for in vitro approaches. Common genetic polymorphisms of GSTM1 (*0, null), GSTT1 (*0) and GSTP1 (*A/*B, I105V) are detectable. The frequencies of the polymorphisms are within the expected range for a Caucasian population with one exception. The GSTM1*0 allele is 1.5-fold more frequent in lung cell lines. GST mRNAs are frequently but not uniformly distributed among unstimulated in vitro conditions. Lung cell lines show an approximately six-fold higher total GST transcript expression than hepatocyte cell lines. Additional GST transcripts have been identified for GSTT1; they represent alternative new splicing variants that occur in cancerous cell lines and in healthy lung tissue and blood. GST enzyme activity is mainly influenced by GSTP1. The activity promoted by 1-chloro-2,4-dinitrobenzene is significantly correlated to the GSTP1 mRNA expression level (R2=0.77, P<0.001). Individual human cell lines thus express GST isoenzymes in a similar pattern to human tissue. The most common genetic polymorphisms are present among the cell lines and have to be considered for in vitro stimulation approaches in a combinatory pattern.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Soluble glutathione-S-transferase (GST) enzymes (EC 2.5.1.18) belong to the group of phase II metabolizing enzymes that facilitate the biotransformation of a large range of exogenous toxic xenobiotics and endogenous compounds of lipid or DNA oxidation (Falany 1997; Glatt 2000). Eight classes of soluble GST, with as many as five isoenzymes per class, have been described in mammalian cells. Each GST isoenzyme exhibits unique substrate specificity.
Many of the cytosolic/soluble GSTs are expressed in the liver, a finding consistent with their role in detoxification. GSTP and M3 are specifically expressed in the lung, in bronchial and bronchiolar epithelium and in alveolar macrophages (Piipari et al. 2003). However, the tissue-specific expression of GST has not been extensively investigated (Hayes and Strange 2000).
GSTs become physiologically important during protection against oxidative stress. They conjugate glutathione to an electrophilic species that can adduct protein or DNA and generate a reactive oxygen species (Hayes and Strange 2000). By catalyzing the conjugation of electrophiles with glutathione, they inactivate these often cytotoxic and/or genotoxic substances (Mannervik and Danielson 1988). Each of the GSTM1, GSTM3, GSTP1 and GSTT1 genes is involved in the detoxification of peroxidation-derived products formed during chronic infection and inflammation (Flamant et al. 2004). However, in a number of cases, glutathione conjugation can also activate chemicals to mutagenic metabolites (Thier et al. 1993).
GST enzymes are polymorphic. They are of particular interest in the study of disease susceptibility and adverse drug responses. Common polymorphisms among the GSTs have the potential to alter the susceptibility of an individual to carcinogens and toxins and to influence the toxicity and efficacy of drug treatment (Hayes and Strange 2000). Whereas some of these polymorphisms appear innocuous, others have been implicated in altering susceptibility to diseases such as cancer (Hayes and Strange 2000; Harries et al. 1997; Olshan et al. 2000) and Parkinson’s disease (Menegon et al. 1998). For instance, a GSTP1 polymorphism is involved in bronchial hyper-responsiveness and liver disease (Fryer et al. 2000; Henrion-Caude et al. 2002). An amino acid substitution at codon 105 (Ile105Val) results in the existence of two isoenzymes that display different catalytic activities and substrate specificities (Rebbeck 1997; Ji et al. 1999; Autrup 2000; Indulski and Lutz 2000). In combination with a GSTT1 null genotype, the GSTP1 (I105V) genotype has an approximately four-times higher risk of acquiring chronic obstructive pulmonary disease (COPD; Gaspar et al. 2004).
Another member of a GST class, the GSTM3 isoenzyme, displays peroxidase activity (Crawford et al. 2000) and has been associated with different outcomes of inflammatory diseases, such as multiple sclerosis (Mann et al. 2000) and rheumatoid arthritis (Mattey et al. 1999). The GSTM3*B allele variant is significantly more likely to have a better lung function than a GSTM3*A/*A genotype (Flamant et al. 2004).
The common polymorphisms of the GSTs are caused by different genetic variations. Single nucleotide polymorphisms (SNP; GSTP1) and gene deletions (GSTM1, GSTT1) are the most frequent genetic variations. The SNPs may be localized within the protein-coding region (GSTP1, I105V) or in the promoter region of a gene. Promoter polymorphisms are thus additionally responsible for differences in the activity of four GSTs and susceptibility to diseases and the effect of drugs (Guy et al. 2004).
Therefore, variations in GST activity have a major impact on the sensitivity of human cells and tissues to the GST substrate. The present work is concerned with the genetic polymorphisms and expression of the five GST classes alpha, mu, pi, theta and zeta on human cell lines. We have determined their use for in vitro exposure to environmentally relevant xenobiotics. Our studies focus on human cell lines of lung and liver, which represent the target organs with respect to the pathways involved in human respiratory exposure and ingestion and subsequent major detoxification.
Materials and methods
Human probes
Normal tissue samples from patients undergoing surgery for the treatment of lung carcinoma (n=5) were obtained in accordance with protocols of the local committee of medical ethics; all patients gave their written consent. The normal lung specimens were resected as far away as possible from carcinoma lesions. Histological diagnosis and tumour staging were established following the classification proposed by the International Union against Cancer. Samples were cryo-preserved in liquid nitrogen.
The non-adherent growing human small-cell lung cancer (SCLC) cell lines NCI-H82 (Little et al. 1983) and NCI-H69 (Turner et al. 1992), the non-SCLC cell line MR65, the adherent growing non-SCLC cell lines A549 (Lieber et al. 1976), Colo-699, LCLC-103H (Bepler et al. 1988), Oka-C-1 (Asahi et al. 1996), EPLC-272H (Heidtmann et al. 1993), KNS-62 (Takaki 1980), NCI-H322 (Gazdar et al. 1990), NCI-H358 (Brower et al. 1986) and BEN (Ellison et al. 1975) and the hepatocyte cell line HepG2 (Aden et al. 1979) were obtained from the European Collection of Cell Cultures and German Collection of Microorganisms and Cell Cultures.
Human hepatocyte cell lines IHH1 (immortalized human hepatocyte no. 1), HH1 (human hepatoma no. 1), IHFL1 (immortalized human fetal liver cells no. 1) and IHI1 (immortalized human islet cells no. 1) were kindly provided by Jan G. Hengstler from the University of Leipzig. The phenotype of the hepatocytes was controlled by measurement of glucose, lactate and urea in the supernatant and the CYP3A4 and GST activity of the cell homogenate.
Lung cell lines were kept in RPMI1640/10% fetal calf serum (FCS). Hepatocyte cell lines were held in William’s Medium E/10% FCS. The culture medium for the IHI1 cell line was additionally supplemented with 20 ng/ml basic fibroblast growth factor and 20 ng/ml epidermal growth factor.
Human venous blood samples were taken from healthy volunteers.
Isolation and amplification of nucleic acid
Cellular RNA was isolated from human cell lines and from blood samples and lung tissue with the peqGold RNAPure Isolation protocol (Peqlab, Erlangen, Germany) according to the manufacturer’s instructions. From the total RNA sample, 5 μg was taken to synthesize cDNA with the RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas, St. Leon-Rot, Germany).
Genomic DNA was isolated by using the QIAamp DNA Blood Kit (Qiagen, Hilden, Germany) according to the manufacture’s instructions.
The sequences of polymerase chain reaction (PCR) primer pairs for long-fragment and quantitative PCR are given in Table 1. All primers were custom-synthesized by MWG Biotech (Ebersberg, Germany). The primers were designed without mismatches to common SNPs to exclude false-negative results. PCR was, in general, performed at 94°C for 5 min, then 38 cycles at 94°C for 30 s, variable T annealing (see Table 1) for 30 s, at 72°C for 1 min and extension at 72°C for 10 min. The PCR mixture contained 1.25 mM MgCl2, 200 μM each dNTP, 0.24 μM each primer and 1 U BIOTAQ DNA polymerase (Bioline, Luckenwalde, Germany). The PCR mixture without the cDNA template served as a negative control to exclude false-positive data. PCR for the detection of multiple GSTT1-PCR products was performed with 1 U FIREPol DNA polymerase (Solis BioDyne, Tartu, Estonia).
Quantitative real-time PCR was performed on Rotorgene (LTF-Labortechnik, Wasserburg/B., Germany). PCR was carried out at 94°C for 8 min, then 40 cycles at 94°C for 30 s, variable T annealing (see Table 1) for 30 s and at 72°C for 30 s. The PCR mixture contained 1.25 mM MgCl2, 200 μM each dNTP, 0,24 μM each primer, 1 U BIOTAQ DNA polymerase and a 5 fold concentrate of fluorogenous substrate SYBRgreen (MoBiTec, Göttingen, Germany). Specific PCR products were examined according to their expected melting curve peak in comparison with the non-template control.
Cloning strategies
Multiple PCR products were simultaneously cloned into pGEM-T vector (Promega, Mannheim, Germany) and transformed into Escherichia coli K12 JM109 strain. The presence of different gene transcripts was checked by PCR on white colonies. Colonies with different gene transcripts were further amplified by overnight incubation. The plasmids were thereafter isolated by the Nucleospin Plasmid Kit (BD Biosciences, Heidelberg, Germany) according to the manufacture’s instructions and sequenced by using the Big-Dye terminator kit on an ABI377 automated sequencer (PerkinElmer Instruments GmbH, Rodgau-Jügesheim, Germany).
Genotyping for GSTM1, GSTT1 and GSTP1 alleles
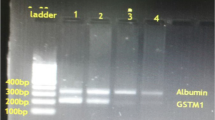
The analysis of genetic polymorphism for the GSTM1 and GSTT1 genes was simultaneously performed by using a multiplex PCR approach. Briefly, 10 ng genomic DNA was amplified in a 20 μl reaction mixture containing 10 pmol of each of the following primers: GSTM1_for 5′-GTGGGAGACAGAAGAAGAGAAGA-3′, GSTM1_rev 5′AGAGGCCAGAGCTGATGAAGG-3′, GSTT1_for 5′-ACCCTGGCAGAGTTGGATGTGACC-3′, GSTT1_rev 5′-GTGGAAGACAGGGTGGGGATGGT-3′. As an internal positive control, the RON gene was co-amplified with the primers RON_for 5′CTAGTGGGGGAGGTGGAGCAGATA-3′ and RON_rev 5′-AAGCAGGTCCAGCCCAAGAACTAA-3′. PCR was performed at 94°C for 5 min, then 38 cycles at 94°C for 30 s, at 60°C for 30 s, at 72°C for 1 min and extension at 72°C for 10 min. The PCR mixture contained 1.25 mM MgCl2, 200 μM each dNTP, 10 pmol each primer and 1 U BIOTAQ DNA polymerase. We used a PCR mixture without cDNA template as a negative control to exclude false-positive data. The multiplex PCR products were then electrophoretically analysed on a 2% ethidium-bromide-stained agarose gel (QA-Agarose, Qbiogene, Heidelberg, Germany). The presence or absence of the GSTM1 and GSTT1 genes was detected by the presence or absence of a 688-bp (corresponding to GSTT1) and a 378-bp (corresponding to GSTM1) PCR product. The internal positive control generated a 220-bp (corresponding to RON) PCR band.
The GSTP1 I 105V polymorphism was detected by means of the restriction fragment length polymorphism approach. Genomic DNA was amplified by PCR with the GSTP1_I4_for 5′-CTGCCCCCGGAGCCCTTTTGTTTA-3′ and GSTP1_I5_rev 5′-CTCGCCCCCATGACCCGTTACTTG-3′ primer pair giving a 655-bp PCR product. The PCR product was consecutively restricted by BsmAI endonuclease for 2 h at 37°C. The reaction mixture was analysed by agarose gel electrophoresis. A 3-band and 4-band restriction pattern was identified as 105 Ile and Val, respectively. The frequency of the GST polymorphisms (GSTM1, GSTT1, GSTP1) of the cell lines was compared with data from human populations (Gawronska-Szklarz et al. 1999; Stanulla et al. 2000; Spurdle et al. 2001).
GST activity
Activity of GST was measured according to Sharma et al. (1997). Briefly, exponentially growing cells plated for 24 h were homogenized by sonication. The homogenate was assayed for GST in a modified microplate enzyme assay by using CDNB (1-chloro-2,4-dinitrobenzene) as a substrate. A concurrent Bredford protein assay (KMF Laborchemie, Lohmar, Germany) was performed to determine the specific enzyme activity. GST activity was calculated based on the formula proposed by Sharma et al. (1997).
Results
The human lung and hepatocyte cell lines were initially genotyped for the most common genetic polymorphism of the GST enzymes affecting their activity (Table 2).
The most common polymorphic genotypes for the GST genes M1 (GSTM1*0), P1 (GSTP1*A/*B) and T1 (GSTT1*0) were found within the tested human lung and hepatocyte cell lines. The homozygous GSTT1*0 allele was present in 25% of the lung cell lines. This was comparable to an expected frequency of 22% for a Caucasian population. In contrast, a frequency of 75% and 25% for the homozygous GSTM1*0 allele and the combination of GSTM1*0/GSTT1*0 alleles among the lung cell lines was approximately 1.5-fold and two-fold higher than the expected frequency of 55% and 10%-14%, respectively. Therefore, the homozygous GSTM1 null and/or the GSTM1/T1 double-null genotype may represent a risk for cancerous transformation of pulmonary epithelial cells. The GSTP1*A and GSTP1*B alleles were present at an expected frequency of about 53% for homozygous GSTP1*A, 11% for homozygous GSTP1*B and 35% for heterozygous GSTP1*A/GSTP1*B.
The cell lines were subsequently analysed for GST transcript expression. A long-fragment RT-PCR technique, amplifying the whole coding sequence region of the appropriate gene, was performed. The presence or absence of a PCR product of the five GST classes is summarized in Table 2. The data indicate that the GST transcripts in general are frequently expressed under unstimulated in vitro conditions. Class alpha, theta and zeta are commonly expressed in both lung and hepatocyte cell lines. Class pi is preferentially expressed in lung cell lines. The hepatocyte cell lines differ in expression of class pi. The hepatoma-derived cell lines HepG2, IHH1 and HH1 are distinguished from the IHFL1 and IHI1 cell lines by the absence of class pi. Class mu represented by M1 and M3 is much less frequently present among the investigated cell lines. In summary, the expression pattern of GST transcripts demonstrates that the five involved GST classes were not uniformly distributed among the tissue-specific cell lines.
The quantity of GST transcripts was subsequently investigated by means of the real-time PCR approach (Fig. 1). Lung cell lines were subdivided into three populations. An approximately ten-fold lower (NCI-H82, NCI-H322) and five-fold higher (A549, KNS62, Colo699) total GST mRNA expression could be distinguished compared with the mean of the remaining seven lung cell lines. The total GST mRNA expression in hepatocyte cell lines was lower than in lung cells. An approximately six-fold and two-fold lower total GST mRNA expression was shown for the three hepatoma-derived hepatocyte cell lines and IHFL1/IHI1 cell lines, respectively.

Summarized total glutathione-S-transferase (GST) mRNA expression of various human lung and hepatocyte cell lines. Quantitative real-time PCR data for GSTA, GSTM1, GSTM3, GSTP1, GSTT1 and GSTZ1 are normalized to the housekeeping gene hypoxanthine phosphoribosyl transferase (HPRT). Lung cell lines express an enhanced mRNA expression compared with hepatocyte cell lines. Values are given as means±SEM
Normal human lung tissue expresses GST transcripts as follows: approximately 95% GSTP1, 3% GSTT1, 2% GSTZ1 and less than 0.1% GSTM1, GSTM3 and GSTA. A specification of the five classes revealed a 70%, 24%, 6% and less than 1% content of GSTP1, GSTZ1, GSTT1 and GSTM1/GSTM3/GSTA, respectively, in lung cell lines (Fig. 2). Data from the NCI-H322 cell line were excluded from this calculation because of their almost absent expression of GST transcripts. A different expression pattern was obtained for the hepatoma-derived hepatocyte cell lines. For these cell lines, a 60%, 35%, 4% and less than 1% content of GSTT1, GSTZ1, GSTM3 and GSTP1/GSTM1/GSTA was calculated, respectively. Thus, GSTP1 and GSTZ1 represented the two dominant GST transcripts for the lung cell lines and the hepatocyte cell lines IHFL1 and IHI. In contrast, the GSTT1 and GSTZ1 transcripts were dominant for the hepatoma-derived hepatocyte cell lines. The proportion of GST transcripts most similar to that of the lung tissue was represented by the two lung cell lines Colo699 and MR65. By comparing lung with hepatoma-derived hepatocyte cell lines, the relative mRNA expression for GSTP1, GSTZ1, GSTT1, GSTM3 and GSTA was approximately 2000-fold and five-fold increased, equally expressed and three-fold and 12-fold decreased, respectively.

Relative quantitative GST mRNA amounts in various human lung and hepatocyte cell lines. Lung cell lines mainly express GSTP1 (70%), followed by GSTZ1 (24%) and GSTT1 (6%). In contrast, the three hepatoma-derived cell lines HepG2, IHH1 and HH1 mainly express GSTT1 (60%), followed by GSTZ1 (35%)
The human cell lines were subsequently evaluated for GST activity (Table 2). CDNB was used as a particularly suitable substrate for the dominantly expressed pi class. Additionally, it served as a substrate for GST class alpha and mu. The NCI-H322 lung cell line was the sole cell line that did not show CDNB-promoted GST activity. The highest GST activity was expressed by the lung cell line Colo699, which exceeded the mean of activity 2.5-fold. In comparison with the lung cell lines, the GST activity of the hepatoma-derived hepatocyte cell lines was marginally lower. In contrast, hepatocyte cell lines IHFL1 and IHI1 possessed a GST activity level similar to that of lung cell lines.
Between CDNB-promoted GST activity and normalized GSTP1 mRNA expression, a significant correlation was calculated with a coefficient of determination of R2=0.77 (P<0.001; Fig. 3). All data were logarithmically transformed. No correlation was seen between GST activity and mRNA expression of the remaining CDNB-associated GSTA and GSTM. Surprisingly, the GSTP1 genotype was not reflected by GST activity, as an expected three-fold to four-fold diminished catalytic activity for the homozygous GSTP1*B/*B genotype of the two cell lines A549 and IHI1 (443±60 mU/mg) was not detected in comparison with the GSTP1*A/*A genotype (678±98 mU/mg, n=6).

Correlation of CDNB-promoted GST activity and mRNA expression of GSTP1. Data are logarithmically transformed. The two parameters significantly correlate with a coefficient of determination of R2=0.77 (P<0.001)
Surprisingly, the long-fragment PCR approach generated additional PCR products for GSTT1. At least four additional PCR products for GSTT1 were obtained in the cDNA templates. To exclude any possibility of amplifying pseudogenes attributable to genomic DNA contamination, a control PCR with the GSTT1 primer pair on genomic DNA was performed. The control PCR was free of unexpected PCR products. Cloning strategies offered three new alternative splicing variants, plus a recently described variant (accession no. CR456499), as depicted in Fig. 4. In addition to the expected transcript of 634 bp, additional transcripts with 546, 485, 397 and 218 bp, respectively, were detected. The new 485-bp transcript was characterized by the absence of exon 2 and a 5′-part of exon 3, the 397-bp transcript by the simultaneous absence of exon 2 and 3, and the 218-bp transcript by the simultaneous absence of exon 2 to 4. Sequences were submitted to the GeneBank (accession nos. AJ 874719, AJ 867757 and AJ 867758). All the new splice variants would encode truncated GSTT1 proteins. The splicing variants were not considered to be a sign of cell culture or cancerous cell lines as they were also detected in healthy lung and blood tissues (Fig. 5).

Multiple alternatively spliced transcripts of GSTT1. The splicing variants (lane 3) include the three new variants (accession nos. AJ874719, AJ867757 and AJ867758). Exon numbering and accession number as in the EMBL database. The resulting polymerase chain reaction (PCR) and putative translation products are indicated (grey boxes similarity to wildtype protein, black boxes non-similarity to wildtype protein). To exclude false-positive GSTT1 PCR products by contamination with genomic DNA, a PCR for GSTT1 (lane 1) and RON (lane 2, positive control) was performed on genomic DNA. GSTT1-specific PCR products are not present within the range of the splicing variants (arrows unspecific PCR product attributable to primer amplification, M 100-bp DNA ladder marker)

Expression of GSTT1 splicing variants in human tissue (numbers the various subjects, M 100-bp DNA ladder marker). Splicing variants are present in lung tissue (a) and blood (b).
Discussion
The aim of the study has been to characterize different human cell lines for their use in subsequent in vitro approaches to exposure to environmental xenobiotics. We have favoured cell lines over primary cell cultures because of their standardized availability. However, the dedifferentiation of the cell lines evoked by permanent proliferation may be a disadvantage for the stimulation and transcription of the genes necessary for metabolism and/or detoxification of xenobiotics. Therefore, we have examined the gene transcripts and enzyme activity of five major GST classes as important phase II enzymes of biotransformation of different human cell lines.
We first genotyped the cell lines for the most prominent genetic polymorphisms GSTM1*0, GSTP1*A/*B and GSTT1*0, which are correlated to specific clinical endpoints. A GSTM1*0 gene deletion polymorphism is described as being associated with increased susceptibility to rheumatoid arthritis (Yun et al. 2005). The GSTT1*0 genotype is more frequent among patients with advanced alcoholic liver disease (Ladero et al. 2005) and serves as a risk factor for cardiovascular injuries among smokers (Doney et al. 2005) and the GSTP1*A/*B substitution polymorphism is responsible for a significantly increased risk of physician-diagnosed asthma in children (Lee et al. 2005).
However, a single polymorphism of a GSTM gene should not be sufficient to explain an affected enzymatic activity because of the overlap in substrates of the five mu-class genes (GSTM1-M5; Comstock et al. 1994). Linkages between polymorphisms have to be considered as a reason for an affected GSTM activity. In this respect, two distinct interactions have been described between GSTM1 and GSTM3. First, individuals with GSTM1 null express lower levels of GSTM3 (Nakajima et al. 1995). Second, individuals that express GSTM1, GSTM1*A and GSTM1*B may confer different susceptibilities to several cancers (Fryer et al. 2000) because of linkage with alleles in GSTM3 (Inskip et al. 1995). These findings indicate that the effect of GSTM1 on susceptibility may be influenced by the expression of GSTM3.
The 12 studied lung cell lines enabled us to obtain all possible genotypes for GSTM1, GSTP1 and GSTT1. However, regarding GSTM1/T1 genotypes, not all combinations were present. The GSTM1 wildtype/GSTT1 null genotype combination was not present. The GSTM1 null genotype was much more frequently present in lung cancer cell lines. Therefore, this null genotype has to be considered as a sign of cancerous feature of the lung cell lines. However, this assumption has not been approved by the International Collaborative Study on Genetic Susceptibility to Environmental Carcinogens (GSEC; Raimondi et al. 2005). In this study, a GSTT1 deletion is mentioned as being a risk factor for lung cancer in Caucasian non-smokers, but not a GSTM1 deletion and only when the analysis was restricted to studies including healthy controls. Further studies have noted the absence of correlation between the GSTM1 null allele and cancer risk. The GSTM1 null genotype in combination with the CYP1A1 wildtype and GSTT1 non-null genotypes even acts protectively against lung cancer (Raimondi et al. 2005). Other types of cancer, such as bladder cancer do not significantly correlate with null alleles of the GSTM1 and GSTT1 (Srivastava et al. 2005).
In contrast, recently published studies indicate a significant correlation of GSTM1 null with cancer diseases. The null allele is associated with Kazakh’s esophageal squamous cell cancer (Lu et al. 2005). In combination with a GSTT1 deletion, it increases the risk of developing a transverse or rectal tumour (Ates et al. 2005).
The gene expression of the GST classes has been characterized by various PCR techniques. All genotyped deletions have been confirmed at the level of the transcript.
The five GST classes are non-homogeneously expressed among the cell lines. The GST transcript expression levels are broad and range from overall loss (NCI-H322) to gain (Colo699). Here, we should note that the variability in any gene expression is not a disadvantageous physiological feature of a cell line but corresponds to a significant variability of enzyme activity within a human population. The GST transcript expression level of the lung cell lines in vitro is similar to that of human tissue. The relationship between GSTP1:GSTZ1:GSTT1 transcript expression is about 20:1:1 for lung tissue (n=5), 26:4:1 for lung cell lines (n=12) and 19:1.7:1 for blood leukocytes (n=9). This means that the basal expression of GST transcripts is not influenced by in vitro culture conditions. Therefore, the human cell lines are suitable for in vitro stimulation experiments focusing on GST enzymes.
The cell lines have been further characterized at the GST protein level by a GST-specific CDNB-substrate reaction. The CDNB-promoted GST activity is the summarized activity of the GSTA, GSTM and GSTP classes. Because of an approximately 600-fold over-expression of GSTP1, the GSTA and GSTM classes do not influence this activity under in vitro conditions. This is confirmed by hepatoma-derived hepatocyte cell lines, which show a 2000-fold lower GSTP1 expression than lung cell lines and consequently express only background activity. GSTP1 activity significantly correlates with GSTP1 mRNA expression and, therefore, the transcript level is valuable for an approximate estimation of GSTP1 enzyme activity. GST activity should additionally be influenced by the polymorphism of the GST gene. Surprisingly, the expected three-fold to four-fold reduction of CDNB or other substrate-specific GSTP1 activity was not apparent for a GSTP1*B/*B homozygous genotype (Ali-Osman et al. 1997; Sundberg et al. 1998).
The hepatoma-derived hepatocyte cell lines only express a CDNB-promoted background activity, undoubtedly because of the negligible GSTP1 expression. On the other hand, the two immortalized hepatocytes IHFL1 and IHI1 demonstrate a similar GST activity to the lung cell lines. This remaining GSTP1 activity of the two hepatocyte cell lines seems to be a sign of their differentiation.
Long-fragment PCR was performed to evaluate the expression of a complete transcript. In consequence, an appropriate full protein should have been translated. This PCR approach generated a specific transcript, with one exception. PCR for GSTT1 exhibited a multiple transcript pattern. Subsequent cloning and sequencing strategies suggested that the PCR products were multiple, alternatively spliced transcripts. All of them were characterized by aberrant skipping exons, which would have caused truncation of the protein. A comparison with the genome sequence shows that the introns are well supported and follow the consensual [gt-ag] rule. This rule is also maintained for AJ874719, wherein an [ag] motif within the 5′ sequence of exon 3 was used as the splicing boundary. This alternative 3′-splice site results in the shortening of exon 3. The GSTT1 splicing variants do not seem to be cell-culture-specific or tissue-specific because of their occurrence in the cancerous cell lines and in healthy lung tissue and blood. Therefore, the splicing variants cannot be considered as a sign of abnormality. The presence of multiple spliced transcripts indicates a mistake within the splicing enhancers or inhibitors necessary for this gene. The physiological role of the GSTT1 splicing variants in affecting GSTT1 activity has not been clarified by the present study. However, the data on different transcripts are important for future quantitative PCR design strategies and reflect the biological complexity of post-transcriptional modifications.
In conclusion, we described various human cell lines with respect to their GST expression. GST characteristics are valuable for discriminating the variations that are frequently represented within a human population. These GST variations shall be taken into account for human in vitro stimulation approaches as they might influence individual susceptibilities to toxic exposure.
References
Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB (1979) Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 282:615–616
Ali-Osman F, Akande O, Antoun G, Mao JX, Buolamwini J (1997) Molecular cloning, characterization, and expression in Escherichia coli of full-length cDNAs of three human glutathione S-transferase pi gene variants. Evidence for differential catalytic activity of the encoded proteins. J Biol Chem 272:10004–10012
Asahi Y, Kubonishi I, Imamura J, Kamioka M, Matsushita H, Furihata M, Ohtsuki Y, Miyoshi I (1996) Establishment of a clonal cell line producing granulocyte colony-stimulating factor and parathyroid hormone-related protein from a lung cancer patient with leukocytosis and hypercalcemia. Jpn J Cancer Res 87:451–458
Ates NA, Tamer L, Ates C, Ercan B, Elipek T, Ocal K, Camdeviren H (2005) Glutathione S-transferase M1, T1, P1 genotypes and risk for development of colorectal cancer. Biochem Genet 43:149–163
Autrup H (2000) Genetic polymorphisms in human xenobiotica metabolising enzymes as susceptibility factors in toxic response. Mut Res 464:65–76
Bepler G, Koehler A, Kiefer P, Havemann K, Beisenherz K, Jaques G, Gropp C, Haeder M (1988) Characterization of the state of differentiation of six newly established human non-small-cell lung cancer cell lines. Differentiation 37:158–171
Brower M, Carney DN, Oie HK, Gazdar AF, Minna JD (1986) Growth of cell lines and clinical specimens of human non-small cell lung cancer in a serum-free defined medium. Cancer Res 46:798–806
Comstock KE, Widersten M, Hao XY, Henner WD, Mannervik B (1994) A comparison of the enzymatic and physicochemical properties of human glutathione transferase M4-4 and three other human mu class enzymes. Arch Biochem Biophys 311:487–495
Crawford EL, Khuder SA, Durham SJ, Frampton M, Utell M, Thilly WG (2000) Normal bronchial epithelial cell expression of glutathione transferase P1, glutathione transferase M3, and glutathione peroxidase is low in subjects with bronchogenic carcinoma. Cancer Res 60:1609–1618
Doney AS, Lee S, Leese GP, Morris AD, Palmer CN (2005) Increased cardiovascular morbidity and mortality in type 2 diabetes is associated with the glutathione S transferase theta-null genotype: a Go-DARTS study. Circulation 111:2927–2934
Ellison M, Woodhouse D, Hillyard C, Dowsett M, Coombes RC, Gilby ED, Greenberg PB, Neville AM (1975) Immunoreactive calcitonin production by human lung carcinoma cells in culture. Br J Cancer 32:373–379
Falany CN (1997) Enzymology of human cytosolic sulfotransferases. FASEB J 11:206–216
Flamant C, Henrion-Caude A, Boelle PY, Bremont F, Brouard J, Delaisi B, Duhamel JF, Marguet C, Roussey M, Miesch MC, Boule M, Strange RC, Clement A (2004) Glutathione-S-transferase M1, M3, P1 and T1 polymorphisms and severity of lung disease in children with cystic fibrosis. Pharmacogenetics 14:295–301
Fryer AA, Bianco A, Hepple M, Jones PW, Strange RC, Spiteri MA (2000) Polymorphism at the glutathione S-transferase GSTP1 locus. A new marker for bronchial hyperresponsiveness and asthma. Am J Resp Crit Care Med 161:1437–1442
Gaspar P, Moreira J, Kvitko K, Torres M, Moreira A, Weimer T (2004) Cyp1A1, Cyp2E1, GSTM1, GSTT1, GSTP1, and TP53 polymorphisms: do they indicate susceptibility to chronic obstructive pulmonary disease and non-small-cell lung cancer? Genet Mol Biol 27:133–138
Gawronska-Szklarz B, Wojcicki M, Kuprianowicz A, Kedzierska K, Kedzierski M, Gornik W, Pawlik A (1999) CYP2D6 and GSTM1 genotypes in a Polish population. Eur J Clin Pharmacol 55:389–392
Gazdar AF, Linnoila RI, Kurita Y, Oie HK, Mulshine JL, Clark JC, Whitsett JA (1990) Peripheral airway cell differentiation in human lung cancer cell lines. Cancer Res 50:5481–5487
Glatt H (2000) Sulfotransferases in the bioactivation of xenobiotics. Chem Biol Interact 129:141–170
Guy CA, Hoogendoorn B, Smith SK, Coleman S, Donovan MC, Buckland PR (2004) Promoter polymorphisms in glutathione-S-transferase genes affect transcription. Pharmacogenetics 14:45–51
Harries LW, Stubbins MJ, Forman D, Howard GC, Wolf CR (1997) Identification of genetic polymorphisms at the glutathione S-transferase pi locus and association with susceptibility to bladder, testicular and protate cancer. Carcinogenesis 18:641–644
Hayes JD, Strange RC (2000) Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology 61:154–166
Heidtmann HH, Salge U, Havemann K, Kirschke H, Wiederanders B (1993) Secretion of a latent, acid activatable cathepsin L precursor by human non-small cell lung cancer cell lines. Oncol Res 5:441–451
Henrion-Caude A, Flamant C, Roussey M, Housset C, Flahault A, Fryer AA, Chadelat K, Strange RC, Clement A (2002) Liver disease in pediatric patients with cystic fibrosis is associated with glutathione S-transferase P1 polymorphism. Hepatology 36:913–917
Indulski JA, Lutz W (2000) Metabolic genotype in relation to individual susceptibility to environmental carcinogens. Int Arch Occup Environ Health 73:71–85
Inskip A, Elexperu-Camiruaga J, Buxton N, Dias PS, MacIntosh J, Campbell D, Jones PW, Yengi L, Talbot JA, Strange RC, Fryer AA (1995) Identification of polymorphism at the glutathione S-transferase, GSTM3 locus: evidence for linkage with GSTM1*A. Biochem J 312:713–716
Ji X, Blaszczyk J, Xiao B, O’Donnell R, Hu X, Herzog C, Singh SV, Zimniak P (1999) Structure and function of residue 104 and water molecules in the xenobiotic substrate-binding site in human glutathione S-transferase P1-1. Biochemistry 38:10231–10238
Ladero JM, Martinez C, Garcia-Martin E, Fernandez-Arquero M, Lopez-Alonso G, Concha EG de la, Diaz-Rubio M, Agundez JA (2005) Polymorphisms of the glutathione S-transferases mu-1 (GSTM1) and theta-1 (GSTT1) and the risk of advanced alcoholic liver disease. Scand J Gastroenterol 40:348–353
Lee YL, Hsiue TR, Lee YC, Lin YC, Guo YL (2005) The association between glutathione s-transferase p1, m1 polymorphisms and asthma in Taiwanese schoolchildren. Chest 128:1156–1162
Lieber M, Smith B, Szakal A, Nelson-Rees W, Todaro G (1976) A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int J Cancer 17:62–70
Little CD, Nau MM, Carney DN, Gazdar AF, Minna JD (1983) Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 306:194–196
Lu XM, Zhang YM, Lin RY, Arzi G, Wang X, Zhang YL, Zhang Y, Wang Y, Wen H (2005) Relationship between genetic polymorphisms of metabolizing enzymes CYP2E1, GSTM1 and Kazakh’s esophageal squamous cell cancer in Xinjiang, China. World J Gastroenterol 11:3651–3654
Mann CL, Davies MB, Boggild MD, Alldersea J, Fryer AA, Jones PW (2000) Glutathione S-transferase polymorphisms in MS: their relationship to disability. Neurology 54:552–557
Mannervik B, Danielson UH (1988) Glutathione transferases—structure and catalytic activity. CRC Crit Rev Biochem 23:283–337
Mattey DL, Hassell AB, Plant M, Dawes PT, Ollier WR, Jones PW (1999) Association of polymorphism in glutathione S-transferase loci with susceptibility and outcome in rheumatoid arthritis: comparison with the shared epitope. Ann Rheum Dis 58:164–168
Menegon A, Board PG, Blackburn AC, Mellick GD, Le Couteur DG (1998) Parkinson’s disease, pesticides, and glutathione transferase polymorphisms. Lancet 352:1344–1346
Nakajima T, Elovaara E, Anttila S, Hirvonen A, Camus AM, Hayes JD, Ketterer B, Vainio H (1995) Expression and polymorphism of glutathione S-transferase in human lungs: risk factors in smoking-related lung cancer. Carcinogenesis 16:707–711
Olshan AF, Weissler MC, Watson MA, Bell DA (2000) GSTM1, GSTT1, GSTP1, CYP1A1, and NAT1 polymorphisms, tobacco use, and the risk of head and neck cancer. Cancer Epidemiol Biomarkers Prev 9:185–191
Piipari R, Nurminen T, Savela K, Hirvonen A, Mantyla T, Anttila S (2003) Glutathion S-transferases and aromatic DNA adducts in smoker’s bronchoalveolar macrophages. Lung Cancer 39:265–272
Raimondi S, Boffetta P, Anttila S, Brockmoller J, Butkiewicz D, Cascorbi I, Clapper ML, Dragani TA, Garte S, Gsur A, Haidinger G, Hirvonen A, Ingelman-Sundberg M, Kalina I, Lan Q, Leoni VP, Marchand LL, London SJ, Neri M, Povey AC, Rannug A, Reszka E, Ryberg D, Risch A, Romkes M, Ruano-Ravina A, Schoket B, Spinola M, Sugimura H, Wu X, Taioli E (2005) Metabolic gene polymorphisms and lung cancer risk in non-smokers. An update of the GSEC study. Mutat Res [Epub ahead of print]
Rebbeck TR (1997) Molecular epidemiology of the human glutathione S-transferase genotypes GSTM1 and GSTT1 in cancer susceptibility. Cancer Epidemiol Biomarkers Prev 6:733–743
Sharma S, Nemecz SK, Zhu S, Stelle VE (1997) Identification of chemopreventive agents by screening for induction of glutathione-S-transferase as a biomarker. Methods Cell Sci 19:49–52
Spurdle AB, Webb PM, Purdie DM, Chen X, Green A, Chenevix-Trench G (2001) Polymorphisms at the glutathione S-transferase GSTM1, GSTT1 and GSTP1 loci: risk of ovarian cancer by histological subtype. Carcinogenesis 22:67–72
Srivastava DS, Mishra DK, Mandhani A, Mittal B, Kumar A, Mittal RD (2005) Association of genetic polymorphism of glutathione S-transferase M1, T1, P1 and susceptibility to bladder cancer. Eur Urol 48:339–344
Stanulla M, Schrappe M, Brechlin AM, Zimmermann M, Welte K (2000) Polymorphisms within glutathione S-transferase genes (GSTM1, GSTT1, GSTP1) and risk of relapse in childhood B-cell precursor acute lymphoblastic leukemia: a case-control study. Blood 95:1222–1228
Sundberg K, Johansson AS, Stenberg G, Widersten M, Seidel A, Mannervik B, Jernström B (1998) Differences in the catalytic efficiencies of allelic variants of glutathione transferase P1-1 towards carcinogenic diol epoxides of polycyclic aromatic hydrocarbons. Carcinogenesis 19:433–436
Takaki T (1980) An epithelial cell line (KNS-62) derived from a brain metastasis of bronchial squamous cell carcinoma. J Cancer Res Clin Oncol 96:27–33
Thier R, Taylor JB, Pemble SE, Humphreys WG, Persmark M, Ketterer B, Guengerich FP (1993) Expression of mammalian glutathione S-transferase 5-5 in Salmonella typhimurium TA1535 leads to base-pair mutations upon exposure to dihalomethanes. Proc Natl Acad Sci USA 90:8576–8580
Turner AM, Zsebo KM, Martin F, Jacobsen FW, Bennett LG, Broudy VC (1992) Nonhematopoietic tumor cell lines express stem cell factor and display c-kit receptors. Blood 80:374–381
Yun BR, El-Sohemy A, Cornelis MC, Bae SC(2005) Glutathione S-transferase M1, T1, and P1 genotypes and rheumatoid arthritis. J Rheumatol 32:992–997
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bauer, M., Herbarth, O., Aust, G. et al. Expression patterns and novel splicing variants of glutathione-S-transferase isoenzymes of human lung and hepatocyte cell lines. Cell Tissue Res 324, 423–432 (2006). https://doi.org/10.1007/s00441-005-0150-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-005-0150-8