
Abstract
Recent studies suggest that signal transduction may have an important role in the development and regulation of the metastatic phenotype. Here, we investigated the role of the epidermal growth factor receptor (EGFR), and protein kinase C (PKC), in the process of reassembly of cadherin-dependent cell-cell adhesion of Caco-2 cells. We used chemical activation of PKC and EGFR with 12-O-tetradecanoylphorbol-13-acetate (TPA), a tumor-promoting agent, pretreatment with protein kinase inhibitors and subcellular fractionation to analyze the effect of the phorbol ester on the redistribution of junctional proteins. Transepithelial resistance (TER), electron microscopy and immunofluorescence analyses were also carried out. Activation with TPA resulted in disassembly of adherens junctions (AJs), but the tight junction (TJ) structure and function remained unaltered. TPA affected E-cadherin levels. In Caco-2 cells at day 2 of culture, when most E-cadherin is not associated with the cytoskeleton, a decrease in the level of this protein was observed as soon as 6 h after TPA addition. However, at day 5 of culture, the major effect observed after 6 h of treatment was a translocation of the protein from the Triton-insoluble to the -soluble fraction. On the other hand, TPA did not significantly affect the E-cadherin-associated proteins α and β-catenins. Potent specific EGFR inhibitors, such as PD153035 and Tyrphostin 25, as well as Calphostin C, an inhibitor of PKC, significantly blocked the effect of TPA on AJs. Furthermore, inhibition of the TPA effect by the PD98059 MAPK inhibitor suggests that activation of this kinase was the final event in the modulation of cadherin-dependent cell-cell adhesion. Pretreatment of cell monolayers with Calphostin C before EGF treatment, one of the ligands of EGFR, blocked the redistribution of E-cadherin caused by EGF. Based on these results, we conclude that both EGFR and PKC activation are involved in TPA-induced cell signaling for modulation of cadherin-dependent cell-cell adhesion and cell shape in Caco-2 cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cadherins comprise a family of widely expressed transmembrane glycoproteins that mediate the initiation, specificity, and maintenance of cell-cell adhesion (Nagafuchi et al. 1987; Takeichi 1993). The adhesive function of E-cadherin requires its attachment to the actin cytoskeleton. This association is mediated by a set of proteins collectively named catenins. In AJs, the intracellular domain of E-cadherin binds directly to β-catenin that, in turn, associates with α-catenin, which is thought to link cadherin complex to the actin cytoskeleton (Cowin and Burke 1996). Although several studies have reported the important role of cadherin as a determinant of cell behavior, cell fate, and probable tumor suppressor gene, the regulation of cell-cell adhesion mediated by this protein, and the recruitment of signaling molecules to complexes at these adhesions, are not yet fully understood. The presence of EGFR, phosphotyrosine phosphatases or tyrosine kinase substrates such as p120 suggests that tyrosine phosphorylation plays a role in the modulation of this complex. This hypothesis has been supported by observations showing that maintenance of the adhesion complex is modulated by the Src family tyrosine kinases, such as pp60C-SRC itself. Receptors with tyrosine kinase activity, such as EGFR, hepatocyte growth factor receptor (HGFR) c-Met, platelet-derived growth factor, and colony-stimulating factor (Hoschchuetzky et al. 1994; Fuji et al. 1996; Shibata et al. 1996), are modulators of the adhesion complex. The insulin-like growth factor-I receptor (IGF-IR), a multifunctional tyrosine kinase, also modulates E-cadherin-mediated cell-cell adhesion (Playford et al. 2000; Mauro et al. 2001). Recently, it was demonstrated that tyrosine kinase induced tyrosine phosphorylation and ubiquitination of components of the E-cadherin complex, causing endocytosis of E-cadherin and disassembly of cell-cell adhesion in epithelial cells (Fujita et al. 2002).
The phorbol ester TPA is a highly potent tumor promoter that activates some isoforms of the important signaling enzyme PKC (Ono et al. 1989), which interferes with both cell proliferation and cell adhesion in a variety of cell types (Lewis et al. 1995; Vuori and Ruoslahti 1993). It has been reported that PKC activation by TPA can also lead to disruption of TJs, causing an increase in paracellular permeability (Soler et al. 1993; Stenson et al. 1993; Farshori and Kachar 1999). However, this effect was not observed in established cell monolayers by other groups (Clarke et al. 2000; Gómez et al. 1999). On the other hand, various studies have demonstrated the involvement of PKC in disassembly of E-cadherin-dependent cell-cell adhesion (Fabre and Garcia de Herreros 1993; Skoudy and Garcia de Herreros 1995; Ratcliffe et al. 1997; Lewis et al. 1995). In addition, it has also been reported that PKC regulates endocytosis and recycling of E-cadherin in cells treated with phorbol ester (Le et al. 2002). It is important to point out that as PKC is an intracellular receptor of TPA, promotion of TPA-induced tumor is thought to be related to the PKC/MAPK pathway (Dong et al. 1994; Young et al. 1999; Li et al. 1996; Huang et al. 1997). However, various studies have reported that TPA-induced PKC activation is a transient process, but that sustained treatment with TPA downregulates PKC activity (Young et al. 1987; Nishizuka 1988; Lu et al. 1997), suggesting that other mechanisms, in addition to the PKC pathway, could mediate TPA-induced signal transduction. Recently, Chen et al. (2001) demonstrated that activation of the EGFR by TPA is a necessary step in TPA-induced signal transduction and cell transformation and, therefore, is involved in TPA-induced tumor promotion. Despite these studies indicating an important role of TPA-induced signal transduction, either in cell transformation or in cell-cell adhesion, the mechanisms underlying this effect are still to be established.
In the present study, we investigated the role of EGFR and PKC activated by TPA as well as the signaling pathways involved in the regulation of the disassembly of intercellular junctions. Using specific kinase inhibitors we show that EGFR and PKC activation by TPA is a necessary step in TPA-induced signal transduction and cadherin-dependent cell-cell adhesion in Caco-2 cells.
Materials and methods
Cell culture and TPA and EGF treatment
Caco-2 cells were cultured in RPMI medium (Sigma) supplemented with 10% fetal bovine serum (FBS) (Hyclone), 60 μg/ml streptomycin and 100 μg/ml penicillin in a humidified atmosphere of 5% CO2. Cells were seeded at 2×104 cells/cm2 and grown for 2–7 days. Culture medium was changed every 24 h to avoid nutrient depletion.
Cell monolayers from different days of culture were treated with 200 nM TPA (Sigma) for 1, 3 and 6 h. Stock solution of TPA was prepared in DMSO and stored at –20°C. When indicated, monolayers were also treated with 20 ng/ml EGF (Sigma) for 12 h.
Pretreatment with protein kinase inhibitors
Before TPA addition, some monolayers were pretreated with protein kinase inhibitors for 1 h at 37°C. The inhibitors were obtained from Sigma (Calphostin C and Tyrphostin 25) or Calbiochem (PD153035 and PD98059) and diluted from stock solutions in DMSO (stored at −20°C) to give final concentrations, in DMEM medium without serum, as indicated: Calphostin, 0.5 μM; Tyrphostin 25, 3 μM; PD153035, 100 nM; PD98059, 50 μM. These concentrations were selected based on previous experiments showing no apparent toxicity to control cells. After 1 h of pretreatment, a fresh medium containing TPA, as well as the protein kinase inhibitors, replaced the bathing solution. To ensure a valid comparison of the relative effect of the four inhibitors tested, pretreatments with each of the inhibitors were performed in parallel.
Cell extraction and immunoblotting
Cell monolayers at 2, 3 and 5 days of culture were treated with 200 nM of TPA for 1, 3 and 6 h. Cell monolayers at 5 days of culture were also pretreated with protein kinase inhibitors before the TPA treatment. After extensive washing, subcellular fractionation to obtain the soluble and the insoluble fractions was carried out as previously described (Skoudy and Garcia de Herreros 1995). Briefly, cells were rinsed in PBS plus 100 mM CaCl2, 100 mM MgCl2, pH 7.6, and homogenized in CSK buffer (50 mM NaCl, 10 mM PIPES, pH 6.8, 3 mM MgCl2, 0.5% Triton X-100, 300 mM sucrose, 1 mM PMSF, 10 μg/ml leupeptin, 1 mM orthovanadate, 1 mg/ml pepstatin) for 10 min at 4°C with gentle stirring. After centrifugation for 10 min, at 4°C, the supernatant constituted the Triton-soluble fraction (TX-100 soluble). The pellet was harvested and solubilized in SDS buffer (20 mM TRIS, pH 7.5, 5 mM EDTA, 2.5 mM EGTA, 1% SDS) and boiled at 100°C for 10 min. After a new centrifugation for 10 min the supernatant (TX-100 insoluble) constituted the cytoskeleton fraction. This fraction usually contained fivefold less protein than the Triton-soluble fraction.
Proteins of cell fractions were electrophoretically separated by SDS-PAGE in 7.5% or 10% gels (Laemmli 1970) and transferred to nitrocellulose sheets using a semidry transfer cell at 10 V for 60 min (Towbin et al. 1979). Then, the nitrocellulose membranes were blocked for 1 h with TBS-T buffer (20 mM TRIS-HCl, pH 7.6, 137 mM NaCl and 0.1% v/v Tween 20) containing 5% low-fat dried milk and incubated for 2 h or overnight with primary antibodies: anti-E-cadherin (dilution 1:5000), anti-α- and β-catenin (dilution 1:4000) (all from Sigma Chemical Co.), and anti-occludin (1 μg/ml) (Zymed Laboratories, Inc.). After washing, membranes were incubated for 1 h with peroxidase-conjugated goat anti-rat or anti-mouse IgG (dilution 1:80,000) or peroxidase-conjugated goat anti-rabbit IgG (Zymed Laboratories, Inc.) diluted 1:1000. Proteins were visualized using an enhanced chemiluminiscence kit (Amersham Pharmacia Biotech., Bucks., UK) and autoradiography.
Immunofluorescence microscopy
Cells were cultured on glass coverslips and following pretreatment with the protein kinase inhibitors, and treatment with TPA or EGF, were washed 3 times with PBS/CM (PBS plus 100 mM CaCl2 and 100 mM MgCl2) and fixed in freshly prepared 2% paraformaldehyde in PBS/CM for 30 min. Subsequently, they were permeabilized for 30 min in PBS/CM containing 0.075% saponin, and 0.2% albumin, and incubated with 50 mM NH4Cl for 10 min. After 1 h incubation in blocking solution (0.2% bovine serum albumin in PBS/CM), the cells were incubated with monoclonal primary anti-E-cadherin antibody diluted 1:1500, for 2 h at room temperature. After washing, the cells were further incubated for 1 h with FITC-conjugated rabbit anti-rat IgG (Sigma) diluted 1:60, in the dark. The coverslips were washed in PBS and mounted using n-propylgallate, and observed in an Axiovert S 100 immunofluorescence microscope equipped with a KS300 image analyzer (Zeiss).
Antibody permeability assay
Macromolecule permeability assay to test TJ functionality was carried out as previously described (Rajasekaran et al. 2001). Confluent cell monolayers grown on glass coverslips were fixed as described above and labeled with the monoclonal antibody against E-cadherin under nonpermeabilized conditions. The cells were washed and stained with FITC-conjugated rabbit anti-rat secondary antibody and visualized using Axiovert S 100 microscopy as described above.
Electron-microscopy analysis
For electron-microscopy analysis, Caco-2 cells were cultured on polycarbonate filters (Transwell, 0.4 μm pore size, Costar, Cambridge, MA, USA) and after the respective treatments they were washed in PBS, and fixed for 1 h in a solution containing 2.5% glutaraldehyde, 1% paraformaldehyde, 0.8% sucrose and 2 mM CaCl2 in 0.1 M cacodylate buffer, pH 7.4. Postfixation was carried out in 1% osmium tetroxide in cacodylate buffer, containing 0.8% potassium ferrocyanide and 5 mM CaCl2. Subsequently, the cells were dehydrated with acetone and embedded in Epon. Ultrathin sections were obtained, stained with uranyl acetate and lead citrate and observed in a Zeiss CEM-900 transmission electron microscope.
For scanning electron microscopy, Caco-2 cells were fixed and postfixed as described above. Subsequently, the samples were dehydrated in graded ethanol, critical-point dried in CO2, coated with chromium in a Penning sputter system in a high vacuum chamber (Gatan-model 681), and viewed in a JEOL-JSM-6340F field emission scanning electron microscope.
Transepithelial electrical resistance
For transepithelial electrical resistance (TER) measurements, cells were plated on polycarbonate filters with a pore size of 0.4 μm. A Millicell ERS system (Millipore Co.) was used to determine the TER value. All TER values were normalized for the area of the filter and were obtained after background subtraction (i.e., filter and bath solution).
Results
TPA affects the expression and distribution of E-cadherin
It is known that functionality of E-cadherin depends on its association with the cytoskeleton. In order to analyze the level of expression and the cell distribution of this protein, soluble and cytoskeleton fractions of Caco-2 cells at different days after seeding, with and without the addition of TPA, were prepared and analyzed by immunoblotting. As shown in Fig. 1A, 2 days after seeding most of the E-cadherin was detected in TX-soluble fraction, thus suggesting a light association with the cytoskeleton. The levels of E-cadherin in the cytoskeleton fraction (TX-100-insoluble fraction) gradually increased with cultivation time, reaching the highest level after 5 days.

Effect of TPA treatment on the cellular levels and the association of E-cadherin with cytoskeleton in Caco-2 cells. A Cells were grown for 2 (D2), 3 (D3) and 5 (D5) days and cell fractions were prepared as described in "Materials and methods." E-cadherin levels were determined by Western blotting with anti-E-cadherin antibody. Observe that the association of E-cadherin with cytoskeleton progressively increases after seeding. B Cells were grown as above, then TPA (200 nM) was added for 1, 3 and 6 h. Cell fractions were prepared and levels and redistribution of E-cadherin determined; 35 μl of the Triton-soluble (S) and 7 μl of the Triton-insoluble (I) fractions were analyzed. Only a band of 120 kDa, corresponding to E-cadherin, was detected
At day 2, when most of the E-cadherin was not associated with the cytoskeleton, an apparent decrease in the level of this protein after 6 h of TPA treatment was observed. The same result was seen at day 3. The disappearance of the insoluble E-cadherin observed before any decrease of the soluble protein suggests its possible translocation from one fraction to the other. The translocation of the protein was more evident at day 5 after 6 h of incubation with TPA (Fig. 1B). However, in this latter case no apparent decrease in the level of the protein was observed.
It has been reported that TPA treatment induces phenotypic changes in epithelial cells (Ben-Ze'v 1986; Skoudy and Garcia de Herreros 1995). In the present work, we observed that cells treated with TPA 2 or 3 days after seeding displayed a fibroblast-like morphology and spreading. Figure 2A–C shows the distribution of E-cadherin in untreated cells after 2, 3 and 5 days of culture, as analyzed by immunofluorescence microscopy. It can be observed that after 5 days of seeding, E-cadherin was mainly localized in the intercellular junction region, indicating its association with the cytoskeleton. Incubation of Caco-2 cells with TPA for 1, 3 and 6 h at 2, 3 and 5 days of culture caused changes in the pattern of distribution of E-cadherin (Fig. 2D–L), as compared to untreated cells. After 2 days of culture after TPA treatment for 3 or 6 h, it was possible to observe that the cells acquired a fibroblast-like morphology with diffuse E-cadherin immunostaining on the cell surface (Fig. 2G, J). At 3 days of culture, when it was possible to see some labeling at the cell-cell junctions, the TPA effect on the redistribution of E-cadherin was more evident also with 3 and 6 h of treatment (Fig. 2H, K). By 3 or 6 h of treatment at 5 days, E-cadherin immunoreactivity appeared diffuse on the cell surface with an eventual punctual labeling, but labeling of the cell-cell contact area was also observed (Fig. 2I, L).

Redistribution of E-cadherin in Caco-2 cells at 2 (D2), 3 (D3) and 5 (5) days of culture after treatment with TPA for 1, 3 and 6 h. Cells were grown on glass coverslips and immunostained with anti E-cadherin antibody. TPA clearly induces alterations in the morphology and distribution of E-cadherin at different days of culture (D–L), as compared to control cells (A–C). Scale bar 20 μm
The effect of TPA is independent of the level and distribution of α- and β-catenin
It is known that catenins bind to E-cadherin and are necessary to its functionality. In order to determine if changes induced by TPA on the levels of E-cadherin are followed by alterations in catenin levels, we used specific monoclonal antibodies to analyze the protein expression by immunoblotting. Figure 3A shows the levels and distribution of α and β-catenin at different days of culture both in TX-soluble and TX-insoluble fractions of cells that were not treated with TPA. An increase in the levels of these proteins with the passage of days of culture was observed. Figure 3B shows that in contrast to what happened with E-cadherin, the distribution of α and β-catenin did not change significantly, either in TX-soluble or TX-insoluble fractions, after TPA treatments in cultures 5 days after seeding. In addition, it was clear that the distribution of α and β-catenin in the cytoskeleton fraction remained unchanged after 6 h of treatment with TPA.

TPA does not modify the cellular levels or the distribution of α- and β-catenins. Untreated (A) fractions of cells grown at 2 (D2), 3 (D3) and 5 (D5) days of culture or (B) fractions treated with 200 nM TPA, obtained from cells at 5 days after seeding, were analyzed by immunoblotting using anti-α-catenin and β-catenin antibodies. In both situations only one band of molecular mass of 105 kDa (α-catenin) and another of 95 kDa (β-catenin) were detected with these antibodies
Inhibitors of protein kinases depress morphological changes and TPA-induced redistribution of E-cadherin
Untreated Caco-2 cells analyzed by scanning electron microscopy were closely apposed and presented numerous apical microvilli (Fig. 4A, C). After 3 h of TPA treatment, long cell processes were observed and many of the cells had a reduced number of microvilli. Also, some cells were separated from each other (Fig. 4B, D). However, microvillar loss was not observed, suggesting that this effect appears to be related to extensive cell elongation.

TPA treatment induces morphological changes as observed by scanning electron microscopy. Caco-2 cells were incubated with or without TPA for 3 h, fixed, critical-point dried and processed for scanning electron microscopy. A, C Control cells incubated in DMEM medium show numerous apical microvilli. After TPA incubation, long cell processes are present, with few microvilli and adjacent cells separated in some regions (B, D). Scale bars 5 μm
We examined the morphological alterations induced by TPA treatment using transmission electron microscopy. Caco-2 cells form a coherent and well-organized monolayer, with a typical junctional complex at the subapical region (Fig. 5A). After addition of TPA to cell monolayers, the AJs region was significantly altered (Fig. 5B). Despite extensive alterations at the AJs region, the desmosomes remained intact. On the other hand, TPA apparently did not affect the TJs. In order to determine if these alterations in AJs took place via tyrosine kinase receptor, the effect of tyrphostin 25, a well-known specific inhibitor of tyrosine kinase, was tested at a concentration (3 μM) that inhibits EGFR. Figure 5C shows the inhibitory effect of tyrphostin 25 on cells treated for 3 h with TPA after 5 days of culture. It could be observed that the junctional complex was significantly reassembled.

Ultrastructure of Caco-2 cell monolayers. Electron micrographs show sections of control cells (A), cells treated with TPA for 3 h (B) or pretreated with tyrphostin 25 (C), and PD153035 (D) before incubation with TPA. Note the enlargement of the area corresponding to the adherent junctions due to the effect of TPA (B) and the stabilization of this structure after treatment with tyrphostin 25 (C) and PD153035 (D) (arrows TJ region, d desmosomes). Scale bar 0.35 μm
This result was further confirmed when cells were pretreated with the specific EGFR inhibitor PD153035 (Fig. 5D). In addition, PD98059 and Calphostin C, inhibitors of MAPK and PKC respectively, also blocked the effect of TPA (not shown).
The TPA effect was also monitored by immunofluorescence microscopy localization of E-cadherin and by immunoblotting. Tyrphostin 25 and PD153035 also blocked the TPA-induced redistribution of E-cadherin, so that the pattern of immunostaining for this protein was similar to that observed in control cells (Fig. 6A, a–d). The specificity of the inhibitory effect by other kinase inhibitors was tested. Figure 6A, e, f shows that PD98059 and Calphostin C also significantly inhibited the effect of TPA treatment. To analyze a possible link between EGFR and PKC signaling, cells were treated with EGF or pretreated with Calphostin C before the EGF treatment. Figure 6B shows that EGF treatment caused alterations in morphology and distribution of E-cadherin in a similar fashion to TPA (Fig. 6B, a). In addition, when cells were pretreated with Calphostin C it was possible to see a significant inhibitory effect of the EGF treatment (Fig. 6B, b).

Immunofluorescence microscopy shows the distribution of E-cadherin in Caco-2 cells, after treatment with TPA and its modulation by inhibitors of protein kinases. A Cell monolayers were incubated in the presence (b) or absence (a) of TPA or after pretreatment with tyrphostin 25 (c), PD153035 (d), PD98059 (e) and Calphostin C (f). B Cell monolayers were treated with EGF (a) or pretreated with Calphostin C (b) and stained for E-cadherin protein. Observe that the effect of TPA on the redistribution of E-cadherin was significantly blocked by pretreatment with all inhibitors tested. Scale bars 10 µm (A), 10 μm (B)
Immunoblotting analysis of TX-100-soluble and -insoluble fractions of cell monolayers pretreated with Tyrphostin 25, PD153035 and PD98059 showed the inhibition of the translocation of E-cadherin to the soluble fraction induced by TPA, thus confirming the above-mentioned result (Fig. 7).

Redistribution of E-cadherin by TPA treatment from the Triton-insoluble fractions to the soluble fraction is blocked by protein kinase inhibitors. Cell fractions of untreated cells (C) or cells incubated with TPA for 1, 3 and 6 h after pretreatment with Tyrphostin 25, PD153035 and PD98059 were fractionated into Triton-insoluble (I) and Triton-soluble (S) fractions. Triton-soluble (35 μl) and Triton-insoluble (7 μl) fractions were loaded and probed for E-cadherin
TPA treatment does not disrupt the paracellular permeability barrier
To determine the functionality of the TJ after TPA treatment, three approaches were used: First, permeability of cell monolayers to ions at 5 days of culture was determined by measuring the transepithelial resistance (TER). Figure 8A shows that TPA treatment of Caco-2 monolayer did not interfere with TER, independently of the presence of the kinase inhibitors Tyrphostin 25 and PD153035. The same results were obtained using Calphostin C and PD98059 (not shown). Second, the permeability of cell monolayers to macromolecules was tested using a macromolecular permeability assay. Anti E-cadherin antibody was added to the apical surface of monolayer grown on glass coverslips and the TJ permeability to the antibody was detected by staining the cells with a secondary antibody under nonpermeabilized conditions (Fig. 8B). Caco-2 control cells (Fig. 8B, A, B), treated with TPA (Fig. 8B, C, D), and pretreated with kinase inhibitors (Fig. 8B, E, H), were impermeable to anti-E-cadherin antibody, indicating the presence of functional TJs. In contrast, cells grown in conditions of low calcium, which are known to functionally impair the TJs, showed clear E-cadherin staining on the plasma membrane, indicating the absence of TJs (Fig. 8B, I, J). Third, the functionality of TJ was also assessed by the presence of highly phosphorylated occludin in the TX-100 fractions of Caco-2 cells after 5 days of culture. As seen in Fig. 8C the presence of a band of higher molecular weight (~80 kDa), corresponding to the hyperphosphorylated occludin form, was observed in the TX-100-insoluble fractions after TPA treatment. The same immunoreactivity profile to this protein was observed in the control group, indicating that occludin remained linked to the cytoskeleton after TPA treatment.

TPA does not perturb the paracellular permeability barrier. A No significant changes in TER are observed following TPA treatment or after pretreatment with Tyrphostin 25 (Tyr) or PD153035, as compared to the control group. A strong decrease in the TER in an experiment in which cells were switched for 2.5 h to low-calcium medium (LC), indicating TJ disruption, is observed. B Confluent monolayers were fixed under nonpermeabilized conditions and incubated from the apical side with anti-E-cadherin antibody and FITC-conjugated secondary antibody. The absence of TJ in cells switched to low-calcium medium resulted in staining of E-cadherin on the plasma membrane (J). In control cells (B), TPA treated (D) or cells pretreated with Tyrphostin 25 (F), and PD153035 (H), functional TJ prevented passage of the antibody and no staining at the basolaterally localized E-cadherin is observed. A, C, E, G, I are phase-contrast micrographs corresponding to B, D, F, H and J, respectively. C Shows that TPA does not modify occludin hyperphosphorylation. TX-100 fractions were prepared as described in "Materials and methods" and probed with antibody specific to occludin. The apparent molecular mass of the two main bands, determined using molecular mass standard, is indicated. Scale bar 10 μM (B)
Discussion
In this study we investigated the distribution and levels of E-cadherin as well as the effect of specific protein kinase inhibitors during the activation of PKC by the phorbol ester TPA. We showed that treatment with TPA resulted in modification of the distribution and levels of E-cadherin in Caco-2 cells. However, the treatment: (a) does not induce changes in paracellular permeability, as revealed by measuring the transepithelial resistance, and the macromolecular permeability assay, (b) does affect the TJ structure, as analyzed by electron microscopy, and (c) caused significant alterations in the phosphorylation state of occludin. Taken together these findings as well as those we previously reported (Morgado-Díaz and De Souza 2001) indicate that both AJs and TJ are independently modulated. Furthermore, the present results indicate that modulation of TJ is not a direct result of PKC activation. It is likely that PKC acts further upstream in the signaling pathway, which causes alterations at the AJs, but not at the TJ.
Immunoblotting analysis showed that the effect of TPA was dependent on the day of culture. Thus, in early cultures (2 days), when E-cadherin is in a soluble state, the phorbol ester caused a significant depletion, but in late cultures (5 days), when the protein is associated with the cytoskeleton, the most evident effect was its translocation to the soluble fraction. Cell fractionation experiments showed also that, under our assay conditions, TPA did not induce any obvious changes on the expression levels of α and β-catenins, independently of the culture time. This later result is in agreement with those reported by van Hengel et al. (1997) and Skoudy and Garcia de Herreros (1995), who did not find changes in the levels of these molecules upon treatment with TPA, despite alterations in the distribution and levels of the E-cadherin in other cell lines. These results suggest that TPA induces disturbance of the E-cadherin-catenin complex by loss of the connection of E-cadherin to the cytoskeleton, but not by loss of expression of the catenin molecules.
Earlier studies have shown that dysfunction in E-cadherin-mediated adhesion can be generated by several mechanisms, including PKC activation (Balda et al. 1993; Denisenko et al. 1994; Stuart and Nigan 1995). Downregulation of PKC has also been considered a fair explanation of transient effects on cell junction functionality, and reversibility upon prolonged TPA treatment (Denisenko et al. 1994; Lewis et al. 1995; Stuart and Nigan 1995). The decrease in the levels of E-cadherin in 2-day cultures (Fig. 1B) could be the result of either a transient loss of cell-cell adhesion or a transient downregulation of the expression of E-cadherin (van Der Wurff et al. 1997) or both. The observed translocation of E-cadherin in later cultures (5 days), induced by TPA treatment, can impair the function of the molecule as a result of post-translational modifications (Frixen et al. 1991; Shiokaki et al. 1995).
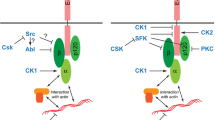
On the other hand, there is considerable evidence implying tyrosine phosphorylation in the disassembly of cadherin-mediated cell-cell contacts. For example, tyrosine phosphorylation of adhesion molecules like E-cadherin and β-catenin due to activation of tyrosine kinases, such as the EGFR, HGFR c-Met, the fibroblast growth factor receptor (FGFR) or Src in epithelial cells, induces cell scattering and a fibroblast-like morphology in epithelial cells (Weidner et al. 1990; Stoker and Gherardi 1991; Behrens et al. 1993; Hamaguchi et al. 1993; Bauer et al. 1998; Owens et al. 2000). Recent studies have reported that loss of cell-cell adhesion is mediated by EGFR signaling (Munshi et al. 2002; Hornia et al. 1999; Jawhari et al. 1999). Although TPA is believed to contribute to PKC activation, it also increases EGFR phosphorylation (Xian et al. 1995; Prenzel et al. 1999). The mechanism by which EGFR is phosphorylated by TPA is still unknown. Here we demonstrated that pretreatment of Caco-2 cell monolayers with Tyrphostin 25 and PD153035 blocked the TPA effect on cell-cell adhesion and on the morphological alterations (e.g., colony dispersion fibroblastic cell shape). This result suggests a direct blockade of the EGFR by the inhibitors or an inhibitory effect through the involvement of an endogenous EGFR-regulated cellular tyrosine kinase. Recently, Chen et al. (2001) showed that blockade of the EGFR by specific inhibitors reduced TPA-induced EGFR phosphorylation and ERK activity, demonstrating that activation of the EGFR is a necessary step in signal transduction induced by TPA. In addition, our results showing that redistribution of E-cadherin was blocked by pretreatment with PD98059 suggest that activation of MAPK is the final event in modulation of E-cadherin-dependent cell-cell adhesion and cell shape. It is known that TPA activates PKC and EGFR and that both may stimulate the Ras/Raf-1 pathway (Marais et al. 1995, 1998; Price et al. 1989; Moran et al. 1991; Denhardt 1996), leading to activation of MAPK. Thus, one may suggest that Ras/Raf-1 proteins are the possible link between MAPK activation and modulation of cell-cell adhesion. However, we do not rule out the possibility that after activation of tyrosine kinases by TPA other mechanisms can also be involved in the redistribution of E-cadherin. For instance, it was recently reported that tyrosine phosphorylation of E-cadherin results in its ubiquitination by the ubiquitin ligase Hakai, inducing its subsequent degradation (Fujita et al. 2002). Furthermore, when cells were pretreated with Calphostin C before EGF treatment, the redistribution of E-cadherin was significantly blocked, indicating a link between EGFR and PKC signaling pathways. We suggest that this link is through PLC-γ, since this molecule is activated by EGF with subsequent PKC activation (Reynolds et al. 1993; Rhee 1991; Goldschmidt-Clermont et al. 1991; Panayotou and Waterfield 1993). Taken together our data suggest that both EGFR and PKC activation are necessary to regulate the disassembly of AJs (Fig. 9).

Proposed model of the mechanism of EGFR and PKC activation in TPA-induced signal transduction and effects on the modulation of E-cadherin-dependent cell-cell adhesion in Caco-2 cells
In conclusion, our observations showing that treatment of Caco-2 cell with TPA causes alterations in the AJs assembly and does not change the TJ structure and function, provide more evidence that these structures are independently regulated. Furthermore, the inhibitory effect of Tyrphostin 25, PD153035, PD98059 and Calphostin C on the AJs disassembly and cell scattering after TPA or EGF treatments, suggests that both EFGR and PKC activation are involved in TPA-induced cell signaling to modulate cadherin-dependent cell-cell adhesion and cell shape. Our results open up the possibility for more detailed studies on the molecular mechanisms involved in the process of regulation of cell-cell adhesion that may lead to identification of novel target for therapeutic suppression of invasion and metastasis.
References
Balda MS, Gonzalez-Mariscal L, Matter K, Cereijido M, Anderson JM (1993) Assembly of the tight junction: the role of diacylglycerol. J Cell Biol 123:293–302
Bauer A, Lickert H, Kemler R, Stappert J (1998) Modification of the E-cadherin-catenin complex in mitotic Madin-Darby canine kidney epithelial cells. J Biol Chem 273:28314–28321
Behrens J, Vaket F, Winterhager E, Van Roy F, Mareel M, Birchmeier W (1993) Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/β-catenin complex in cells transformed with a temperature-sensitive V-SRC gene. J Cell Biol 120:757–766
Ben-Ze'ev A (1986) Tumor promoter-induced disruption of junctional complexes in cultured epithelial cells is followed by the inhibition of cytokeratin and desmoplakin synthesis. Exp Cell Res 164:335–352
Chen N, Ma WY, She QB, Wus E, Liu G, Bode AM, Dong Z (2001) Transactivation of the epidermal growth factor receptor is involved in 12-O-tetradecanoylphorbol-13-acetate-induced signal transduction. J Biol Chem 276:46722–46728
Clarke H, Peralta Soler A, Mullin JM (2000) Protein kinase C activation leads to dephosphorylation of occludin and tight junction permeability increase in LLC-PK1 epithelial sheets. J Cell Sci 113:3187–3196
Cowin PM, Burke B (1996) Cytoskeletal-membrane interactions. Curr Opin Cell Biol 8:56–65
Denhardt DT (1996) Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cell: the potential for multiplex signalling. Biochem J 318:729–747
Denisenko NP, Burighel P, Citi S (1994) Different effects of protein kinase inhibitors on the localization of junctional proteins at cell-cell contact sites. J Cell Sci 107:969–981
Dong Z, Birrer MJ, Watts RG, Matrisian LM, Colburn NH (1994) Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc Natl Acad Sci U S A 91:609–613
Fabre M, Garcia de Herreros A (1993) Phorbol ester-induced scattering of HT-29 human intestinal cancer cells are associated with down-modulation of E-cadherin. J Cell Sci 106:513–522
Farshori P, Kachar B (1999) Redistribution and phosphorylation of occludin during opening and resealing of tight junction junctions in cultured epithelial cells. J Membr Biol 170:147–176
Frame MC (2002) Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta 1602:114–1130
Frixen UH, Beherens J, Sachs M, Eberle C, Voss B, Warda A, Lochner D, Birchmeier W (1991) E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol 113:173–185
Fuji K, Furukawa F, Matsuyoshi N (1996) Ligand activation of overexpressed epidermal growth factor receptor results in colony dissociation and disturbed E-cadherin function in HSC-1 human cutaneous squamous carcinoma cells. Exp Cell Res 223:50–62
Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W (2002) Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol 4:222–231
Goldschmidt-Clermont PJ, Kim JW, Machesky LM, Rhee SG, Pollard TD (1991) Regulation of phospholipase c-gamma-1 by profilin and tyrosine phosphorylation. Science 251:1231–1233
Gómez S, Llosas MM, Verdú J, Roura S, Lloreta J, Fabre M, Herreros AG (1999) Independent regulation of adherens and tight junctions by tyrosine phosphorylation in Caco-2 cells. Biochim Biophys Acta 1452:121–132
Hamaguchi M, Matsuyoshi N, Ohnishi Y, Gotoh B, Takeichi M, Nagai Y (1993) p60v-src causes tyrosine phosphorylation and inactivation of the N-cadherin-catenin cell adhesion system. EMBO J 12:307–314
Hornia A, Lu Z, Sukezane T, Zhong M, Joseph T, Frankel P, Foster DA (1999) Antagonistic effects of protein kinase C alpha and delta on both transformation and phospholipase D activity mediated by the epidermal growth factor receptor. Mol Cell Biol 19:7672–7680
Hoschchuetzky H, Aberle H, KemLer R (1994) β-Catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J Cell Biol 127:16712–16719
Huang C, Schmid PC, Ma WY, Schmid HHO, Dong Z (1997) Phosphatidylinositol-3 kinase is necessary for 12-O-tetradecanoylphorbol-13-acetate-induced cell transformation and activated protein 1 activation. J Biol Chem 272:4187–4194
Jawhari AU, Farthing MJ, Pignatelli M (1999) The E-cadherin/epidermal growth factor receptor interaction: a hypothesis of reciprocal and reversible control of intercellular adhesion and cell proliferation. J Pathol 187:155–157
Laemmli UK (1970) Cleavage of structural proteins during the assembly of head of bacteriophage T4. Nature 227:680–685
Le TL, Joseph SR, Yap AS, Stow JL (2002) Protein kinase C regulates endocytosis and recycling of E-cadherin. Am J Physiol 283:C489–499
Lewis JF, Jensen PI, Johnson KR, Wheelock MJ (1995) E-cadherin mediates adherens junction organization through protein kinase C. J Cell Sci 108:3615–3621
Li JJ, Dong Z, Dawson MI, Colburn NH (1996) Inhibition of tumor promoter-induced transformation by retinoids that transrepress AP-1 without transactivating retinoic acid response element. Cancer Res 56:483–489
Lu Z, Hornia A, Jiang YW, Zang Q, Ohno S, Foster DA (1997) Tumor promotion by depleting cells of protein kinase C delta. Mol Cell Biol 17:3418–3428
Marais R, Light Y, Paterson HF, Marshall CJ (1995) Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J 14:3136–3145
Marais R, Light Y, Mason C, Paterson H, Olson MF, Marshall CJ (1998) Requirement of Ras-GTP-Raf complexes for activation of Raf-1 by protein kinase C. Science 280:109–112
Mauro L, Bartucci M, Morelli C, Ando S, Surmacz E (2001) IGF-I receptor cell-cell adhesion of MCF-7 breast cancer cells requires the expression of junction protein ZO-1. J Biol Chem 276:39892–39897
Moran MF, Polakis P, McCormick F, Pawson T, Ellis C (1991) Protein-tyrosine kinases regulate the phosphorylation, protein interactions, subcellular distribution, and activity of p21ras GTPase-activating protein. Mol Cell Biol 11:1804–1812
Morgado-Díaz JA, De Souza W (2001) Evidence that increased tyrosine phosphorylation causes disassembly of adherents junctions but does not perturb paracellular permeability in Caco-2 cells. Tissue Cell 33:500–513
Munshi HG, Ghosh S, Mukhopadhyay S, Wu YI, Sen R, Green KJ, Stack MS (2002) Proteinase suppression by E-cadherin-mediated cell-cell attachment in premalignant oral keratinocytes. J Biol Chem 277:38159–38167
Nagafuchi A, Shirayoshi Y, Okazaki K, Yasuda K, Takeichi M (1987) Transformation of cell-adhesion properties by exogenously introduced E-cadherin cDNA. Nature 329:341–343
Nishizuka Y (1988) The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature 334:661–665
Ono Y, Fujii T, Igarashi K, Kuno T, Tanaka C, Kikkawa U, Nishizuka Y (1989) Phorbol ester binding to protein kinase C requires a cysteine-rich zinc-finger-like sequence. Proc Natl Acad Sci U S A 86:4868–4871
Owens DW, McLean GW, Wyke AW, Paraskeva C, Parkinson EK, Frame MC, Brunton UG (2000) The catalytic activity of the Src family kinases is required to disrupt cadherin-dependent cell-cell contacts. Mol Biol Cell 11:51–64
Panayotou G, Waterfield M (1993) The assembly of signalling complexes by receptor tyrosine kinases. Bioessays 15:171–177
Playford MP, Bicknell D, Bodmer WF, Macaulay VM (2000) Insulin-like growth factor 1 regulates the location, stability, and a transcriptional activity of beta-catenin. Proc Natl Acad Sci U S A 97:12103–12108
Potempa S, Ridley AJ (1998) Activation of both MAP kinase and phosphatidylinositide 3-kinase by Ras is required for hepatocyte growth factor/scatter factor-induced adherens junction disassembly. Mol Cell Biol 9:2185–2200
Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A (1999) EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402:884–888
Price BD, Morris JDH, Marshall CJ, Hall A (1989) Stimulation of phosphatidylcholine hydrolysis, diacylglycerol release, and arachidonic acid production by oncogenic Ras is a consequence of protein kinase C activation. J Biol Chem 264:16638–16643
Rajasekaran AS, Palmer LG, Quan K, Harper JF, Ball WJ, Bander NH, Peralta Soler A, Rajasekaran AK (2001) Na, K-ATPase β-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol Cell Biol 12:279 –295
Ratcliffe MJ, Rubin LL, Staddon M (1997) Dephosphorylation of the Cadherin-associated p100/p120 proteins in response to activation of protein kinase C in epithelial cells. J Biol Chem 272:31894–31901
Reynolds NJ, Talwar HS, Baldassare JJ, Henderson PA, Elder JT, Voorhees JJ, Fisher GJ (1993) Differential induction of phosphatidylcholine hydrolysis, diacylglycerol formation and protein kinase C activation by epidermal growth factor and transforming growth factor-alpha in normal human skin fibroblasts and keratinocytes. Biochem J 294:535–544
Rhee SG (1991) Inositol phospholipid-specific phospholipase C: interactions of the gamma-1 isoform with tyrosine kinase. Trends Biochem Sci 16:297–301
Shibata T, Ochiai A, Kanai Y, Akimoto S, Gotoh M, Yasui N, Machinami R, Hirohashi S (1996) Dominant negative inhibition of the association between beta-catenin and c-erbβ-2 by N-terminally deleted beta-catenin suppresses the invasion and metastasis of cancer cells. Oncogene 13:883–889
Shiokaki H, Kadowaki T, Doki Y, Inoue M, Tamura S, Oka H, Iwazawa I, Matsui S, Shimaya K, Takeichi M, Mori T (1995) Effect of epidermal growth factor on cadherin-mediated adhesion in a human oesophageal cancer cell line. Br J Cancer 71:750–758
Skoudy A, Garcia de Herreros A (1995) The protein kinase C activator TPA modulates cellular levels and distribution of E-cadherin in HT-29 human intestinal epithelial cells. FEBS Lett 374:415–418
Soler AP, Laughlin KV, Mullin JM (1993) Effects of epidermal growth factor versus phorbol ester on kidney epithelial (LLC-PK) tight junction permeability and cell division. Exp Cell Res 207:398–406
Stenson WF, Easom RA, Riehl TE, Turk J (1993) Regulation of paracellular permeability in Caco-2 cell monolayers by protein kinase C. Am J Physiol 265:G955–G962
Stoker M, Gherardi E (1991) Regulation of cell movement: the mitogenic cytokines. Biochim Biophys Acta 1072:81–102
Stuart RO, Nigan SK (1995) Regulated assembly of tight junctions by protein kinase C. Proc Natl Acad Sci U S A 92:6072–6076
Takeichi M (1993) Cadherins in cancer: implications for invasion and metastasis. Curr Opin Cell Biol 5:806–811
Towbin UT (1979) Electrophoretic transfer of protein from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Nat Acad Sci U S A 76:4350–4354
van Der Wurff AAM, Vermeulen SJT, van Der Linden EPM, Mareel MM, Bosman FT, Frends JM (1997) Patterns of α and β-catenin and E-cadherin expression in colorectal adenomas and carcinomas. J Pathol 182:325–330
van Hengel J, Gohon L, Bruyneel E, Vermeulen S, Cornelissen M, Mareel M, van Roy F (1997) Protein kinase C activation upregulates intercellular adhesion of α-catenin-negative human colon cancer cell variants via induction of desmosomes. J Cell Biol 137:1103–1116
Vuori K, Ruoslahti E (1993) Activation of protein kinase C precedes alpha 5 beta 1 integrin-mediated cell spreading on fibronectin. J Biol Chem 268:21459–21462
Weidner KM, Behrens J, Vanderkerckhove J, Birchmeier W (1990) Scatter factor: molecular characteristics and effect on the invasiveness of epithelial cells. J Cell Biol 111:2097–2108
Xian W, Kiguchi K, Imamoto A, Rupp T, Zilberstein A, DiGiovanni J (1995) Activation of the epidermal growth factor receptor by skin tumor promoters and in skin tumors from SENCAR mice. Cell Growth Differ 6:1447–1455
Young S, Parker PJ, Ullrich A, Stabel S (1987) Down-regulation of protein kinase C is due to an increased rate of degradation. Biochem J 244:775–779
Young MR, Li JJ, Rincón M, Flavell RA, Sathyanarayana BK, Hunziker R, Colburn N (1999) Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc Natl Acad Sci U S A 96:9827–9832
Acknowledgements
We thank Dr. Marcello Barcinski for critically reading the manuscript, Dr. Marcia Attias for assistance with the high-resolution scanning electron microscope, and Dr. Carlos Frederico Leite Fontes and Vanessa Honorato de Oliveira for help with the biochemical assays.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by Fundação Ary Frauzino para Pesquisa e Controle do Câncer (FAF), Instituto Nacional de Câncer de Rio de Janeiro (INCa), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)
Rights and permissions
About this article
Cite this article
Barbosa, L.A., Goto-Silva, L., Redondo, P.A. et al. TPA-induced signal transduction: a link between PKC and EGFR signaling modulates the assembly of intercellular junctions in Caco-2 cells. Cell Tissue Res 312, 319–331 (2003). https://doi.org/10.1007/s00441-003-0727-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-003-0727-z