
Abstract
Isolated or syndromic congenital cataracts are heterogeneous developmental defects, making the identification of the associated genes challenging. In the past, mouse lens expression microarrays have been successfully applied in bioinformatics tools (e.g., iSyTE) to facilitate human cataract-associated gene discovery. To develop a new resource for geneticists, we report high-throughput RNA sequencing (RNA-seq) profiles of mouse lens at key embryonic stages (E)10.5 (lens pit), E12.5 (primary fiber cell differentiation), E14.5 and E16.5 (secondary fiber cell differentiation). These stages capture important events as the lens develops from an invaginating placode into a transparent tissue. Previously, in silico whole-embryo body (WB)-subtraction-based “lens-enriched” expression has been effective in prioritizing cataract-linked genes. To apply an analogous approach, we generated new mouse WB RNA-seq datasets and show that in silico WB subtraction of lens RNA-seq datasets successfully identifies key genes based on lens-enriched expression. At ≥2 counts-per-million expression, ≥1.5 log2 fold-enrichment (p < 0.05) cutoff, E10.5 lens exhibits 1401 enriched genes (17% lens-expressed genes), E12.5 lens exhibits 1937 enriched genes (22% lens-expressed genes), E14.5 lens exhibits 2514 enriched genes (31% lens-expressed genes), and E16.5 lens exhibits 2745 enriched genes (34% lens-expressed genes). Biological pathway analysis identified genes associated with lens development, transcription regulation and signaling pathways, among other functional groups. Furthermore, these new RNA-seq data confirmed high expression of established cataract-linked genes and identified new potential regulators in the lens. Finally, we developed new lens stage-specific UCSC Genome Brower annotation tracks and made these publicly accessible through iSyTE (https://research.bioinformatics.udel.edu/iSyTE/) for user-friendly visualization of lens gene expression/enrichment to prioritize genes from high-throughput data from cataract cases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tissue or cell-specific gene expression information has been important for prioritization of candidate genes associated with developmental defects of the eye (Blackshaw et al. 2001; Lachke et al. 2012b; Kakrana et al. 2018). Thus far, high-throughput transcript profiling analysis of lens development has largely been performed using microarray analysis (Chauhan et al. 2002a, b; Hawse et al. 2003, 2004, 2005; Ivanov et al. 2005; Xiao et al. 2006; Greiling et al. 2009; Lachke et al. 2011, 2012b; Sousounis and Tsonis 2012; Wolf et al. 2013a, b; Agrawal et al. 2015; De Maria and Bassnett 2015; Anand et al. 2015; Kakrana et al. 2018; Siddam et al. 2018). However, microarray-based expression profiling, while effective, is limited by the number/type of probes on an array, which in turn is limited by the existing knowledge of the transcriptome. Furthermore, probe-hybridization kinetics plays a major role in gene expression quantification unlike the direct detection of transcripts using RNA sequencing. Thus, microarrays do not necessarily inform on the full repertoire of the transcriptome in a given cell or tissue type (Anand and Lachke 2017).
To address these shortcomings, recent studies have begun to apply high-throughput RNA sequencing (RNA-seq) to investigate the mouse lens transcriptome. However, these efforts have largely focused on mid to late embryonic and early postnatal stages (Hoang et al. 2014; Manthey et al. 2014a, b; Khan et al. 2015, 2016, 2018; Audette et al. 2016; Wang et al. 2017; Cavalheiro et al. 2017; Budak et al. 2018; Zhao et al. 2018). Further, the principle challenge in leveraging high-throughput expression profiles for disease-associated gene discovery lies in the application of effective downstream analysis for the identification of high-priority candidate genes that potentially function in the morphogenesis, homeostasis or pathology of the specific tissue of interest. Recently, we demonstrated that for mouse lens microarray datasets, an approach termed “in silico WB-subtraction”, involving comparative analysis to a reference mouse whole-embryo body (WB) tissue dataset, allows the identification of genes with lens-enriched expression (Lachke et al. 2012b; Kakrana et al. 2018). Lens-enriched expression is an indicator of the significance of genes to lens biology and is the basis of the webtool iSyTE (integrated Systems Tool for Eye gene discovery), which has led to the identification of several new genes as well as the characterization of multiple regulatory pathways associated with cataract and other eye defects (Lachke et al. 2011, 2012a, b; Kasaikina et al. 2011; Aldahmesh et al. 2012, 2013; Wolf et al. 2013b; Manthey et al. 2014a; Patel et al. 2014, 2017; Agrawal et al. 2015; Dash et al. 2015; Chograni et al. 2015; Greenlees et al. 2015; Evers et al. 2015; Audette et al. 2016; Zhang et al. 2016; Rothe et al. 2017; Cavalheiro et al. 2017; Kakrana et al. 2018; David et al. 2018; Siddam et al. 2018; Krall et al. 2018).
To develop a matching resource for ocular geneticists, we report generation and integrated analyses of new RNA-seq datasets for mouse early embryonic lens at lens pit stage (embryonic day (E) 10.5), at primary fiber cell differentiation stage (E12.5) and at secondary fiber differentiation stages (E14.5 and E16.5). Further, we generated new mouse WB RNA-seq datasets and tested the hypothesis that “in silico WB-subtraction” can be extended to process lens RNA-seq data and prioritize candidates important to lens biology and cataract. Indeed, WB-subtracted datasets for all four lens stages effectively identified all known genes linked to lens development and cataract. When tested for gene ontology (GO) clustering using DAVID analysis, in contrast to unsubtracted lens expression profiles, in silico WB-subtracted lens expression profiles were highly enriched in GO categories for “lens development in camera-type eye” and “structural constituent of eye lens”, among others, indicating the utility of this approach. Furthermore, these analyses led to the identification of several new high-priority candidate genes for the lens. Finally, to enable effective downstream analyses, we developed new lens stage-specific custom UCSC Genome Brower annotation tracks that are publicly accessible through iSyTE (https://research.bioinformatics.udel.edu/iSyTE/), which allow ready visualization and deeper investigation of genes with respect to their expression and enrichment in the lens.
Materials and methods
Tissue collection and RNA isolation
Mice were bred and maintained at the University of Delaware Center for Animal research and the animal protocol was reviewed by the Institutional Animal Care and Use Committee (IACUC). Experiments were performed according to the guidelines established by the Association for Research in Vision and Ophthalmology (ARVO) for the use of animals in ophthalmic and vision research. Mouse embryos were staged by designating the day of the detection of the vaginal plug as embryonic day (E) 0.5. Mouse C57BL/6NTac strain (Taconic Biosciences) lenses were micro-dissected at stages E10.5, E12.5, E14.5 and E16.5. Total RNA was isolated using Qiagen RNAeasy kit from three biological replicates for each stage. Each biological replicate consisted of multiple lenses (E10.5, n = 50 lenses/replicate; E12.5, n = 29 lenses/replicate; E14.5, n = 18 lenses/replicate; E16.5, n = 17 lenses/replicate). Mouse whole embryos at E10.5, E11.5 and E12.5 were dissected and total RNA was isolated from each stage separately in three biological replicates. An equimolar mixture of total RNA from each stage was considered as individual “whole-embryo body (WB)” sample and three independent WB replicates were generated. RNA quality was analyzed using fragment analyzer (Advanced Analytical Technologies, AATI FEMTO Pulse) and samples with RNA quality number (RQN) greater than 8.5 were used for library preparation and RNA sequencing.
Mouse lens transcriptome assembly and analysis
RNA isolated from three biological replicates for each of the E10.5, E12.5, E14.5 and E16.5 mouse lens stages was used to generate strand-specific, paired-end 100 bp-libraries. All libraries were sequenced on an Illumina HiSeq 2500 instrument at the University of Kansas Medical Center Genome Sequencing Facility following standard protocols. Sequencing quality specifically the per-base sequence quality, sequence length distribution, overrepresented adapter/ primer sequences were evaluated using FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adapters were trimmed using the Trimmomatic tool following recommended cutoffs (Bolger et al. 2014). Reads were aligned against Mus musculus reference genome (mm10) using Tophat v2.0.9 (Trapnell et al. 2009) and gene-, cds-, and transcript-level expression were computed through Cufflinks (Trapnell et al. 2012) using the mouse genome annotations (GRCm38.p6 assembly) as reference. The transcript expression raw counts generated using “classic-FPKM” method implemented in Cufflinks were imported in edgeR package (Robinson et al. 2010) for R statistical environment. Lowly expressed reads (< 1 count per million in less than 2 samples) were filtered out, library size was reset and normalization was performed using default Trimmed Mean of M-values (TMM) method implemented in edgeR package, and dispersion estimates were computed using the recommended Cox–Reid profile-adjusted likelihood (CR) method in edgeR. All new RNA-seq data is submitted to NCBI Gene Expression Omnibus (GEO) Database under series GSE119596.
In silico WB subtraction for RNA-seq datasets
The in silico WB-subtraction approach, previously described for estimating lens enrichment of genes from microarray datasets (Lachke et al. 2012b; Kakrana et al. 2018), was applied to the lens RNA-seq datasets. The newly generated RNA-seq WB data was used as reference and the preferential accumulation of transcripts was computed as log2 fold-change (FC) using the “quasi-likelihood” (QF) test included in edgeR package for WB vs. lens stages E10.5, E12.5, E14.5 and E16.5 contrasts. Genes displaying statistically significant preferential accumulation between lens vs. WB (≥1.5-fold, expression cutoff ≥2.0 counts, p < 0.05) were identified using in-house Python scripts and considered as “lens-enriched”. Top 500 lens-enriched genes from each stage were used as an input for gene ontology (GO)-based clustering and comparative analysis, performed using Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resource tool (https://david.ncifcrf.gov/) (Huang et al. 2009).
Lens-enriched gene cluster analysis
Lens-enriched genes (n = 638, normalized expression ≥2 counts-per-million (CPM), lens enrichment ≥1.5 log2 fold-change, i.e., lens stage vs. WB) identified from all four stages (E10.5, E12.5, E14.5, E16.5). These were used for enrichment level-based clustering as previously described (Kakrana et al. 2018); Self-Organizing Tree Algorithm (SOTA) method was used from the “clValid” package available in the Bioconductor (https://www.bioconductor.org). This analyses identified gene clusters exhibiting distinct lens enrichment patterns across E10.5 to E16.4 developmental window.
Web access for lens development RNA-seq data through iSyTE 2.0
Publicly available web-based custom annotation tracks were developed to access gene expression and enrichment scores for E10.5, E12.5, E14.5, and E16.5 lens stages using the University of California at Santa Cruz (UCSC) Genome Browser. Lens expression and enrichment scores were converted into BED (Browser Extensible Data) format and displayed as annotation track in the UCSC genome browser (Mouse GRCm38/mm10 assembly). The custom tracks for Human GRCh38/hg38 assembly were also developed to the corresponding genes in the mouse. The links to access these tracks are made available through iSyTE 2.0 under the tab “Mouse lens RNA-seq” at https://research.bioinformatics.udel.edu/iSyTE/.
Results and discussion
Generation of mouse embryonic lens RNA-seq datasets and transcriptome assembly
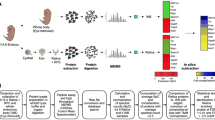
We performed RNA-seq on micro-dissected wild-type mouse lenses at embryonic stages E10.5, E12.5, E14.5 and E16.5 as they inform on key stages of lens development, namely, lens pit (E10.5), primary fiber cell differentiation (E12.5), through secondary fiber cell differentiation (E14.5, E16.5). To perform in silico WB subtraction (see section below), we also generated RNA-seq data from mouse WB as described in detail in the “Methods” section. We designed an appropriate experimental and integrative computational pipeline for these analyses (Fig. 1). Strand-specific, paired-end 100 bp RNA-seq libraries were prepared and sequenced from mouse lenses at the above stages and analyzed using the bioinformatics pipeline described in Fig. 1. A total of 276.6 million reads, on average 18.4 million reads per replicate of lens and WB, were generated and aligned using splice junction mapper, Tophat (Trapnell et al. 2009) (Table 1). On average, 97.7% of the reads were aligned to the Mus musculus reference genome (mm10) (Table 1). Using criteria of p value ≤0.05 (edgeR) (Robinson et al. 2010) and normalized expression counts ≥2 CPM in at least one stage (two replicates) of the lens, we identify a total of 9733 transcripts (mapped at the gene level) from 8480 loci (Table 2, Table S1). Interestingly, the number of distinct transcripts (mapped at the gene level) expressed by E10.5 and E12.5 lenses was higher than the number of distinct transcripts (mapped at the gene level) expressed by E14.5 and E16.5 lens (Table 2). As an important quality control step, principal component analysis (PCA) was performed on expression levels of all the datasets (lens and WB), which showed that individual mouse lens RNA-seq datasets clustered according to their specific stage (Fig. 2a). These data also show that the mouse lens replicates of a specific stage are distinct from those at other stages (Fig. 2a). Further, these data indicate that all the lens datasets, regardless of their stage, are distinct from the WB datasets.

RNA-seq experimental design for mouse embryonic lens transcriptome analysis. A flowchart of experimental design and integrative computational pipeline to determine the mouse lens developmental transcriptome at embryonic stages E10.5, E12.5, E14.5 and E16.5

Quality analysis of RNA-seq data and efficacy of in silico WB subtraction. a Principal component analysis (PCA) of normalized expression counts from RNA-seq datasets for the lens and WB. The plots demonstrate that data generated from individual replicates for specific lens stages [E10.5 (black), E12.5 (red), E14.5 (green) and E16.5 (blue)] clustered in a stage-specific manner and were distinct from the WB datasets that clustered together (turquoise). b The in silico WB-subtraction approach effectively identifies genes with lens-enriched expression that are linked to lens biology. The top 500 genes identified by in silico WB- subtraction and the top 500 genes identified based on just high expression (but which were not subjected to in silico WB subtraction) from E10.5, E12.5, E14.5 and E16.5 mouse lens were examined for significant enrichment of gene ontology (GO) terms. Heat map intensity reflects significant p values for GO term enrichment with high significance represented by red and low significance represented by white. This shows that while non in silico-subtracted genes are enriched for GO terms for housekeeping function and are not specific to the lens, in silico-subtracted genes are significantly enriched for lens-related GO terms
In silico WB subtraction can be effectively applied for RNA-seq data
The principal challenge in high-throughput expression profiling studies is the careful implementation of downstream analytical strategies to effectively prioritize or identify genes that likely function in the tissue of interest. Successful strategies can identify genes linked to cell or tissue morphogenesis/homeostasis and prioritize promising candidates relevant to their associated developmental defects/disease. Recently, we demonstrated that for tissue-specific microarray datasets, an approach termed “in silico WB-subtraction”, involving comparative analysis of the tissue in question to a “reference” WB dataset, provides estimates of tissue-specific enrichment that can be applied for prioritization of candidate genes. For the lens, we showed that tissue enrichment is an excellent predictor of significance to lens biology, which has led to the identification and/or characterization of several new cataract-associated genes (Lachke et al. 2012b; Anand and Lachke 2017; Kakrana et al. 2018). Here, we tested the hypothesis that in silico WB subtraction can be extended to process lens RNA-seq data and prioritize candidates important to lens biology and cataract. RNA-seq datasets on mouse E10.5, E12.5, E14.5 and E16.5 lens tissue was compared to the newly generated WB reference RNA-seq dataset as described in detail in the methods section. Previously, we showed that WB datasets from different developmental stages are similarly effective in identifying lens-enriched genes. Therefore, to make the data interpretation consistent with the previous analysis, as well as, across the four lens developmental stages in this study, we used a single WB reference RNA-seq dataset comprising of the embryonic stages E10.5, E11.5 and E12.5. To test the effectiveness of this strategy to identify lens-relevant genes, we performed gene ontology (GO)-based clustering via DAVID using the top 500 lens-enriched gene profiles from E10.5, E12.5, E14.5 and E16.5 stages generated in this study. In silico WB-subtracted lens expression profiles were enriched in GO categories for “Eye lens protein”, “Structural constituent of eye lens” “Lens development in camera-type eye”, “lens fiber differentiation”, “Eye-development”, and “Embryonic eye-morphogenesis”, indicating the utility of the present approach in identifying the genes relevant to lens biology and development (Fig. 2b). In sharp contrast to lens-enriched genes, the non-subtracted lens expression profiles from all four stages were rich in GO categories for the general functioning of the cell, namely, “Acetylation”, “Structural constituent of ribosome”, “Translation” and “Oxidative phosphorylation”. These data demonstrate that in silico WB subtraction analysis can be successfully applied to lens RNA-seq data to prioritize new candidate genes important for lens biology and cataract.
Lens enrichment identifies known genes linked to lens development and cataract
We evaluated the effectiveness of the in silico WB-subtraction approach by analyzing lens enrichment of genes with known function in lens development, homeostasis and cataract (Cvekl and Zhang 2017). A vast majority of genes (58/61 genes; 95%) associated with cataract or lens defects were found to be highly enriched in the lens at one or more stages (Fig. 3). Further, all of these genes (n = 61) were significantly expressed in the lens datasets (Fig. 3). These include established lens-associated genes that encode crystallins (Cryaa, Cryab, Cryba1, Cryba2, Cryba4, Crybb1, Crybb2, Crybb3, Cryga, Crygb, Crygc, Crygd, Crygs), transcription factors (Foxe3, Hsf4, Mab21l1, Maf, Mafg, Meis1, Meis2, Pax6, Pitx3, Prox1, Six3, Sox1, Sox2, Sox11, Tfap2a, Zeb2), structural proteins (Bfsp1, Bfsp2), gap-junction proteins (Gja3, Gja8), RNA-binding proteins (Caprin2, Celf1, Tdrd7), enzymes (Adam9, Adamts18, Dnase2b, Pxdn), and proteins with other functions (Fig. 3). We next examined the lens enrichment of genes as the lens develops from E10.5 through E16.5 stages. Using stringent criteria of lens enrichment cutoff ≥1.5-fold-change (lens vs. WB), lens expression cutoff ≥2 CPM expression (p < 0.05), we identified 1401 lens-enriched genes at E10.5 (Table S2), 1937 lens-enriched genes at E12.5 (Table S3), 2514 lens-enriched genes at E14.5 (Table S4), and 2745 lens-enriched genes at E16.5 (Table S5) (Fig. 4). This analysis showed that while the total number of lens-expressed genes showed a reduction between E10.5 and E16.5, the number and percent of lens-enriched genes progressively increased with development, with E10.5 having 17% lens-enriched genes and E16.5 having 34% lens-enriched genes (Fig. 4). Finally, the present RNA-seq analysis is more effective in identifying the human cataract-linked genes FYCO and PXDN (Fig. 3), compared to our previous microarray analysis on embryonic lens tissue (Lachke et al. 2012b).

In silico WB subtraction and expression analysis effectively identified genes linked to cataract and lens development. Genes known for lens development and cataract formation were examined for lens expression and lens enrichment in the RNA-seq data. Genes are grouped based on gene ontology (GO) terms and presented as a heatmap. Lens expression scores (expression counts) are presented as ‘red’ for high expression to ‘white’ for low expression. Lens-enriched expression scores (fold-change) are presented as ‘red’ for high enrichment to ‘green’ for low enrichment

Lens-enriched gene expression dynamics in mouse embryonic development. Lens-enriched genes at different embryonic stages were identified by in silico WB-subtraction approach using criteria of: expression counts ≥2.0, ≥1.5-fold-change in the lens compared to WB, p < 0.05. a At E10.5, 17% (n = 1401) genes showed lens enrichment. b At E12.5, 22% (n = 1937) genes showed lens enrichment. c At E14.5, 31% (n = 2514) genes showed lens enrichment. d At E16.5, 34% (n = 2745) genes showed lens enrichment
Dynamics of “lens-enriched” gene expression in embryonic development
To gain biological insights from the cohort of lens-enriched genes identified by in silico WB-subtraction, we examined these candidates from all the embryonic stages using the Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resource. Expectedly, this analysis revealed enrichment of gene groups with known function in the lens, such as “Lens development in camera-type eye” and “Structural constituent of eye lens” in all the stages (Fig. 5). Further, this analysis also revealed gene groups with functional significance in the lens as it transits from early to late developmental stages. For example, “Homeodomain” and “Notch signaling pathway” were enriched at E10.5 (Fig. 5a, Table S6), as would be expected based on earlier studies (Ashery-Padan et al. 2000; Liu et al. 2006; Rowan et al. 2008; Saravanamuthu et al. 2012). Further, gene groups for “Transcription regulation”, “Cell–cell adherens junction”, “Phosphatidylinositol signaling”, “Basic-leucine zipper domain” and “Pleckstrin homology-like domain” were identified in all four stages, indicating the general importance of these factors in lens development. For example, “Cell–cell adherens junctions” at E10.5 included genes such as Cdh1, Epha2, and Ezr that are known to be important in the lens. Interestingly, “Transcription regulation” at E10.5 includes 175 genes involved in the process of transcription, which in addition to the known key lens regulators (e.g., Foxe3, Pax6, Pitx3, Prox1, Six3, Tfap2a), also identifies new transcription factor genes to be expressed in the lens. At E10.5, these are Crebzf (CREB/ATF bZIP transcription factor), Gabpa (GA repeat binding protein, alpha), Sox7 [SRY (sex determining region Y)-box 7], Atf1, Atf2 (activating transcription factors), Alx3 (aristaless-like homeobox 3), Sall4 (sal-like 4) and Tcf3 (transcription factor 3) (Table S6). Interestingly, “Pleckstrin homology (PH)-like domain” was identified in 50 genes expressed at E10.5. PH domains are present in a variety of signaling proteins and also function as “lipid-binding” domains. These genes encoded Rho GTPase activating proteins (Arhgap21, Arhgap24, Arhgap27), Rho guanine nucleotide exchange factors (Arhgef5, Arhgef6, Arhgef16, Arhgef19, Arhgef28) and Pleckstrin homology domain-containing proteins (Plekha1, Plekha5, Plekhg1, Phlda1, Phlda2) (Table S6). In addition to the above pathways, KEGG analysis of the E10.5 lens-enriched genes identified several signaling pathways such as “cGMP-PKG signaling pathway”, “G-protein coupled receptor signaling”, “Jak-STAT signaling”, “MAPK signaling” and “TNF signaling pathway”, among others, which suggests their importance to early lens developmental events (Fig. 5).

Identification of gene ontology (GO), KEGG pathway and Interpro terms for lens-enriched genes. Enrichment of gene groups related to lens function from GO analysis of lens-enriched genes identified at mouse embryonic stages a E10.5, b E12.5, c E14.5 and d E16.5. The genes were subjected to GO annotation clustering method implemented in DAVID bioinformatics tool to identify various GO terms, KEGG pathway and InterPro entries
Starting from stage E12.5, gene groups for “Cell differentiation”, “Actin binding”, “Gap junction” and “Spectrin/alpha-actinin” were identified among lens-enriched genes, which correlate with fiber cell differentiation (Fig. 5) (Bassnett et al. 2011). The “Actin binding” category included myosin-encoding genes Myo6, Myo7a, Myo7b as well as other understudied genes in the lens such as Ablim1, Ablim2, Mylk and Sptbn1. At stages E12.5, E14.5, and E16.5, gene groups for “Fibronectin, type III”, “Glutathione metabolism”, “PDZ domain”, “Spectrin/alpha-actinin” and “ErbB signaling pathway” are enriched (Fig. 5, Tables S7, S8, S9). This highlights interesting new candidates in these stages, for example, the “ErbB signaling pathway” comprises of receptor tyrosine kinases (RTKs) that are involved in the regulation of diverse cellular aspects including survival, proliferation and differentiation. Interestingly, only at stages E14.5 and E16.5, genes for the categories “Intracellular ribonucleoprotein complex”, “Catenin complex”, “PPAR signaling pathway”, “EF-hand like domain” and “Src homology-3 domain” were identified, indicating the importance of these genes in these later stages.
Additionally, genes displaying highly dynamic expression patterns that correlate with the specific lens developmental stage(s) were identified as well. For example, at early lens stages E10.5 and E12.5, the gene group belonging to “Epidermal growth factor-like domain” was enriched in the lens, which was not represented in later stages. Interestingly, only at stage E12.5, the gene categories “Semaphorin-plexin signaling pathway” and “Calcium signaling pathway” were enriched (Table S7). The latter pathway identified 18 candidates that include genes for the calcium voltage-gated channel subunit alpha proteins (Cacna1d, Cacna1a), calcium/calmodulin-dependent protein kinase II alpha protein (Camk2a) and phospholipases (Plcd3, Plcb1, Plcb4) that require calcium as a cofactor. Further, at stages E12.5 and E14.5, “biosynthesis of unsaturated fatty acids” was enriched (Tables S7, S8), while only stage E16.5 showed enrichment of the gene groups for “basement membrane” and “phospholipid biosynthetic process”, which correlates with lens differentiation (Table S9). Moreover, gene groups for “autophagosome assembly” and “mitophagy” were identified only at E16.5; genes identified in these autophagy/mitophagy-related GO clusters, for example, Atg4a, Atg7, Pink1, have been previously reported in the lens (Brennan et al. 2012) and were also identified by a recent RNA-seq-based study of isolated lens fiber cells (Zhao et al. 2018). In addition to validating these known autophagy-related genes, our lens RNA-seq data identifies high expression in the lens of new candidate genes such as Atg4c, Atg9a, Ctsd, Pacs2 and Wipi1 (Table S9). Finally, in agreement with previous reports (Zhao et al. 2018), we find that the autophagy/mitophagy-related genes Bnip3, Gabarapl1, Mtor, Pink1, Plekhm1, and Rblcc1 show an overall increase in expression with progression of lens development (Table S1, Table S5).
In addition to the dynamics of these lens-enriched genes, the present lens RNA-seq data also validates the “lens signature genes” previously identified by microarray analysis (Kakrana et al. 2018). Indeed, the lens signature genes Aldoc, Dhx32, Fabp5, Gprc5b, Grifin, Gstm1, Hmgn3, Mboat1, Mocs2, Npl, Ogn, Pgam2, Tmem40 and Zbtb8b are found to be highly expressed in the lens by the present analysis as well (Table S1).
Lens-enriched genes exhibit distinct clusters based on developmental expression pattern
We next sought to identify specific gene clusters that share dynamics of expression patterns as the lens develops from E10.5 through E16.5. To achieve this goal, we first identified the top lens-enriched genes (n = 638) in all four stages of lens development based on the filtering criteria of expression score > 2 CPM and lens enrichment score > 1.5-fold-change (p < 0.05). Next, we applied Self-Organizing Tree Algorithm (SOTA) method to identify clusters (n = 7) with distinct expression patterns across the four stages. These clusters revealed remarkably distinct expression patterns (Fig. 6, Table S10) and included: 66 genes in Cluster 1, 26 genes in Cluster 2, 11 genes in Cluster 3, 207 genes in Cluster 4, 223 genes in Cluster 5, 56 genes in Cluster 6 and 49 genes in Cluster 7. Cluster 1 (for lens pit formation) showed high expression of genes at E10.5 and comparatively low expression at other stages (E12.5, E14.5 and E16.5). Cluster 2 (for primary fiber cell differentiation) genes showed high expression at E12.5 and low at other stages. Cluster 3 (from lens pit formation to fiber cell differentiation) genes had high expression at E10.5, E12.5 and E14.5 and low expression at E16.5. Similarly, Cluster 4 (for primary and secondary fiber cell differentiation) genes had high expression at E12.5, E14.5 and E16.5 and low expression at E10.5. Cluster 5 (for secondary fiber cell differentiation and elongation) genes had high expression at E14.5 and E16.5 and low expression at E10.5 and E12.5. Cluster 6 and Cluster 7 had varied expression at all stages, and therefore did not classify as distinct compared to Clusters 1–5 (Fig. S1).

Developmental expression clusters reveal dynamics of lens-enriched genes. Self-organizing tree algorithm (SOTA)-based clustering of 638 common lens-enriched genes at stages E10.5, E12.5, E14.5 and E16.5 were used to identify stage-specific clusters. a Heatmap of cluster (1–5) genes showing dynamic expression (normalized expression generated by SOTA) in the lens at stages E10.5, E12.5, E14.5 and E16.5. Transcription factors identified in each cluster are listed. b Clusters of genes with distinct expression patterns in the developing lens are shown as Cluster 1 (green), Cluster 2 (blue), Cluster 3 (purple), Cluster 4 (brown) and Cluster 5 (red). y-axis represents normalized expression generated by SOTA and x-axis represents mouse lens embryonic stage. The red line represents the data trend average for the genes in each cluster
Analysis of genes in these Clusters using the AnimalTFDB 2.0 database (Zhang et al. 2015) identified several new genes related to transcription regulation in distinct clusters, which may potentially function in embryonic lens development. For example, in Cluster 1, Pax6 was identified among the other new TF genes L3mbtl3, Myef2 and Zfp507. In Cluster 2, five new TF genes Arnt2, Mef2a, Platr25, Tgif1 and Zfp369 were identified, and in Cluster 3, Zbtb33 was identified as a new TF candidate gene for further investigation. Cluster 4 contained known lens TFs genes such as Foxe3, Hmx1, Maf, Mitf, Pitx3, Prox1 and Zeb2 and predicted new TF genes such as Aff4, Dnajc1, Etv1, Kmt2c, Mbd2, Mta3, Pbrm1, Satb1, Sox13, Srebf1, Zbtb8b, Zfp385a, Zfp445, Zfp672, Zfp84, Zfp874a, Zfp932 and Zfp933. Cluster 5 identified 12 new TFs, namely, Bhlhe41, Dmtf1, Gabpa, Plag1, Rcor3, Ttf1, Zbtb20, Zbtb26, Zbtb26, Zbtb6, Zfp280d, Zfp420 and Zfp62. In addition to these TFs, SOTA clustering approach revealed co-factors potentially important in transcription regulation in the lens. For example, in Cluster 1, Ccnk, Mtf2, Mrip1, Ogt, Rbm15, Taf9 and Tbl1x were identified. Cluster 3 identified Ino80b and Rnf2, Cluster 4 identified Anp32a, Ezr, Kdm5a, Lancl2, Malt1, Med10, Med12l, Sirt7 and Trim, and Cluster 5 identified Ash2l, Brms1l, Clu, Ddx5, Hspa8, Jade1 and Mlk2. Several other interesting candidate genes were highlighted in stage-specific clusters. For example, Sfpq (splicing factor proline and glutamine rich) and a known syndromic microphthalmia gene Stra6 (stimulated by retinoic acid 6) were identified in cluster 1). Thus, SOTA clustering analysis of genes based on lens-enrichment profiles led to the identification of several new potential regulators in lens development.
Web-based public resource to prioritize lens-enriched candidate genes from RNA-seq data
Next, we sought to make this rich knowledge of RNA-seq-based lens gene expression widely applicable and easily accessible to the research community for prioritization of gene candidates that are relevant to lens biology and cataract through our iSyTE 2.0 tool. To achieve this goal, we developed a new interactive web page that allows direct public access to both, the RNA-seq-based lens expression as well as lens enrichment data, in a user-friendly manner. The interactive web page named as “RNA-seq Lens-enrichment” is accessible under the under “Lens Gene Expression” section of iSyTE 2.0 web resource at https://research.bioinformatics.udel.edu/iSyTE/ppi/expression.php. The “RNA-seq Lens enrichment” page provides direct access to our eight newly developed custom iSyTE tracks for lens gene expression and enrichment at E10.5, E12.5, E14.5 and E16.5 loaded to the University of California at Santa Cruz (UCSC) Genome Browser (Fig. 7). Four new custom iSyTE tracks provide color codes for lens enrichment based on in silico WB-subtracted gene expression profiles at E10.5, E12.5, E14.5 and E16.5. Further, four separate custom tracks provide color codes for lens gene expression (without in silico WB subtraction). Thus, lens enrichment as well as lens expression for individual genes can now be viewed in the mouse genome GRCm38/mm10 assembly in the context of the other rich genome-level information available. Functionally, the user can browse a specific genomic interval, such as a mapped interval, and use the color codes for lens expression or enrichment to effectively visualize and prioritize the best candidate genes in the region of interest for further investigation (Fig. 7). Alternately, a user can search for specific genes, such as those from exome-seq data, to estimate their lens expression and enrichment. Concurrent visualization of these tracks for four embryonic stages allows analysis of the dynamic expression of genes as lens development progresses. As an example to illustrate the utility of these RNA-seq-based custom tracks, the human cataract-linked gene BFSP2 shows high lens enrichment at early embryonic stages (e.g., E10.5) prior to its progressive upregulated expression in the lens at later stages of development (Fig. 7).

RNA-seq data-based custom tracks for UCSC browser to visualize lens expression and enrichment. To access the RNA-seq-based lens expression and lens enrichment data, we developed UCSC custom tracks on the human genome GRCh38/hg38 and the mouse genome GRCm38/mm10 assembly and have made these publicly accessible through iSyTE 2.0 at https://research.bioinformatics.udel.edu/iSyTE/. Under the “RNA-seq lens-enriched” tab, a link is given that allows access to eight UCSC custom tracks. Four tracks provide visual representation of lens gene expression and four tracks provide visual representation of lens gene enrichment data for mouse embryonic stages E10.5, E12.5, E14.5 and E16.5. A color key is provided on iSyTE 2.0 to estimate lens enrichment (numbers indicate fold-change gene expression in the lens compared to WB) and expression (numbers indicate counts for expression). As an example, the human cataract-linked gene BFSP2 can be analyzed for expression and enrichment in the lens by following steps 1 through 3
Conclusion
The present study reports the development and utility of high-throughput RNA-seq-based developmental transcriptomic profiles for mouse lens at the embryonic stages E10.5, E12.5, E14.5 and E16.5. These data demonstrate that in silico WB-subtraction analysis can be successfully applied to lens RNA-seq data to prioritize genes important for lens biology and cataract. The rich early lens developmental expression information generated in this study is made publicly accessible as a new web resource through iSyTE 2.0 in form of user-friendly custom tracks on the UCSC Genome Browser. The inclusion of several key embryonic stages facilitates effective visualization of the developmental dynamics of gene expression and enrichment in the lens. Further, visualization of this data in the UCSC Genome Browser allows comparative analysis with other publicly available genome-level resources such as evolutionary conservation, SNP positions, and epigenetic information. Thus, this new lens RNA-seq data and its accessibility through iSyTE 2.0 serves as a new integrative resource for prioritization of lens defects and/or cataract-linked candidate genes identified by other high-throughput analyses such as exome seq and GWAS. Importantly, by generating a new RNA-seq based WB dataset and showing that RNA-seq datasets can be effectively WB subtracted, we have developed a new resource that can be used by the research community to apply in silico subtraction to a wide range of cells and tissues for prioritization of genes with high tissue-enriched expression.
References
Agrawal SA, Anand D, Siddam AD et al (2015) Compound mouse mutants of bZIP transcription factors Mafg and Mafk reveal a regulatory network of non-crystallin genes associated with cataract. Hum Genet 134:717–735. https://doi.org/10.1007/s00439-015-1554-5
Aldahmesh MA, Khan AO, Mohamed JY et al (2012) Genomic analysis of pediatric cataract in Saudi Arabia reveals novel candidate disease genes. Genet Med 14:955–962. https://doi.org/10.1038/gim.2012.86
Aldahmesh MA, Alshammari MJ, Khan AO et al (2013) The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) is caused by mutations in ADAMTS18. Hum Mutat 34:1195–1199. https://doi.org/10.1002/humu.22374
Anand D, Lachke SA (2017) Systems biology of lens development: a paradigm for disease gene discovery in the eye. Exp Eye Res 156:22–33. https://doi.org/10.1016/j.exer.2016.03.010
Anand D, Agrawal S, Siddam A et al (2015) An integrative approach to analyze microarray datasets for prioritization of genes relevant to lens biology and disease. Genomics Data 5:223–227. https://doi.org/10.1016/j.gdata.2015.06.017
Ashery-Padan R, Marquardt T, Zhou X, Gruss P (2000) Pax6 activity in the lens primordium is required for lens formation and for correct placement of a single retina in the eye. Genes Dev 14:2701–2711
Audette DS, Anand D, So T et al (2016) Prox1 and fibroblast growth factor receptors form a novel regulatory loop controlling lens fiber differentiation and gene expression. Development 143:318–328. https://doi.org/10.1242/dev.127860
Bassnett S, Shi Y, Vrensen GFJM (2011) Biological glass: structural determinants of eye lens transparency. Philos Trans R Soc Lond B Biol Sci 366:1250–1264. https://doi.org/10.1098/rstb.2010.0302
Blackshaw S, Fraioli RE, Furukawa T, Cepko CL (2001) Comprehensive analysis of photoreceptor gene expression and the identification of candidate retinal disease genes. Cell 107:579–589
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Brennan LA, Kantorow WL, Chauss D et al (2012) Spatial expression patterns of autophagy genes in the eye lens and induction of autophagy in lens cells. Mol Vis 18:1773–1786
Budak G, Dash S, Srivastava R et al (2018) Express: a database of transcriptome profiles encompassing known and novel transcripts across multiple development stages in eye tissues. Exp Eye Res 168:57–68. https://doi.org/10.1016/j.exer.2018.01.009
Cavalheiro GR, Matos-Rodrigues GE, Zhao Y et al (2017) N-myc regulates growth and fiber cell differentiation in lens development. Dev Biol 429:105–117. https://doi.org/10.1016/j.ydbio.2017.07.002
Chauhan BK, Reed NA, Yang Y et al (2002a) A comparative cDNA microarray analysis reveals a spectrum of genes regulated by Pax6 in mouse lens. Genes Cells 7:1267–1283
Chauhan BK, Reed NA, Zhang W et al (2002b) Identification of genes downstream of Pax6 in the mouse lens using cDNA microarrays. J Biol Chem 277:11539–11548. https://doi.org/10.1074/jbc.M110531200
Chograni M, Alkuraya FS, Ourteni I et al (2015) Autosomal recessive congenital cataract, intellectual disability phenotype linked to STX3 in a consanguineous Tunisian family. Clin Genet 88:283–287. https://doi.org/10.1111/cge.12489
Cvekl A, Zhang X (2017) Signaling and gene regulatory networks in mammalian lens development. Trends Genet 33:677–702. https://doi.org/10.1016/j.tig.2017.08.001
Dash S, Dang CA, Beebe DC, Lachke SA (2015) Deficiency of the RNA binding protein Caprin2 causes lens defects and features of Peters anomaly. Dev Dyn. https://doi.org/10.1002/dvdy.24303
David D, Anand D, Araújo C et al (2018) Identification of OAF and PVRL1 as candidate genes for an ocular anomaly characterized by Peters anomaly type 2 and ectopia lentis. Exp Eye Res 168:161–170. https://doi.org/10.1016/j.exer.2017.12.012
De Maria A, Bassnett S (2015) Birc7: a late fiber gene of the crystalline lens. Investig Ophthalmol Vis Sci 56:4823–4834. https://doi.org/10.1167/iovs.15-16968
Evers C, Paramasivam N, Hinderhofer K et al (2015) SIPA1L3 identified by linkage analysis and whole-exome sequencing as a novel gene for autosomal recessive congenital cataract. Eur J Hum Genet 23:1627–1633. https://doi.org/10.1038/ejhg.2015.46
Greenlees R, Mihelec M, Yousoof S et al (2015) Mutations in SIPA1L3 cause eye defects through disruption of cell polarity and cytoskeleton organization. Hum Mol Genet 24:5789–5804. https://doi.org/10.1093/hmg/ddv298
Greiling TMS, Stone B, Clark JI (2009) Absence of SPARC leads to impaired lens circulation. Exp Eye Res 89:416–425. https://doi.org/10.1016/j.exer.2009.04.008
Hawse JR, Hejtmancik JF, Huang Q et al (2003) Identification and functional clustering of global gene expression differences between human age-related cataract and clear lenses. Mol Vis 9:515–537
Hawse JR, Hejtmancik JF, Horwitz J, Kantorow M (2004) Identification and functional clustering of global gene expression differences between age-related cataract and clear human lenses and aged human lenses. Exp Eye Res 79:935–940. https://doi.org/10.1016/j.exer.2004.04.007
Hawse JR, DeAmicis-Tress C, Cowell TL, Kantorow M (2005) Identification of global gene expression differences between human lens epithelial and cortical fiber cells reveals specific genes and their associated pathways important for specialized lens cell functions. Mol Vis 11:274–283
Hoang TV, Kumar PKR, Sutharzan S et al (2014) Comparative transcriptome analysis of epithelial and fiber cells in newborn mouse lenses with RNA sequencing. Mol Vis 20:1491–1517
Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. https://doi.org/10.1038/nprot.2008.211
Ivanov D, Dvoriantchikova G, Pestova A et al (2005) Microarray analysis of fiber cell maturation in the lens. FEBS Lett 579:1213–1219. https://doi.org/10.1016/j.febslet.2005.01.016
Kakrana A, Yang A, Anand D et al (2018) iSyTE 2.0: a database for expression-based gene discovery in the eye. Nucleic Acids Res 46:D875–D885. https://doi.org/10.1093/nar/gkx837
Kasaikina MV, Fomenko DE, Labunskyy VM et al (2011) Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. J Biol Chem 286:33203–33212. https://doi.org/10.1074/jbc.M111.259218
Khan SY, Hackett SF, Lee M-CW et al (2015) Transcriptome profiling of developing murine lens through RNA sequencing. Investig Ophthalmol Vis Sci 56:4919–4926. https://doi.org/10.1167/iovs.14-16253
Khan SY, Hackett SF, Riazuddin SA (2016) Non-coding RNA profiling of the developing murine lens. Exp Eye Res 145:347–351. https://doi.org/10.1016/j.exer.2016.01.010
Khan SY, Ali M, Kabir F et al (2018) Identification of novel transcripts and peptides in developing murine lens. Sci Rep. https://doi.org/10.1038/s41598-018-28727-w
Krall M, Htun S, Anand D et al (2018) A zebrafish model of foxe3 deficiency demonstrates lens and eye defects with dysregulation of key genes involved in cataract formation in humans. Hum Genet 137:315–328. https://doi.org/10.1007/s00439-018-1884-1
Lachke SA, Alkuraya FS, Kneeland SC et al (2011) Mutations in the RNA granule component TDRD7 cause cataract and glaucoma. Science 331:1571–1576. https://doi.org/10.1126/science.1195970
Lachke SA, Higgins AW, Inagaki M et al (2012a) The cell adhesion gene PVRL3 is associated with congenital ocular defects. Hum Genet 131:235–250. https://doi.org/10.1007/s00439-011-1064-z
Lachke SA, Ho JWK, Kryukov GV et al (2012b) iSyTE: integrated systems tool for eye gene discovery. Investig Ophthalmol Vis Sci 53:1617–1627. https://doi.org/10.1167/iovs.11-8839
Liu W, Lagutin OV, Mende M et al (2006) Six3 activation of Pax6 expression is essential for mammalian lens induction and specification. EMBO J 25:5383–5395. https://doi.org/10.1038/sj.emboj.7601398
Manthey AL, Lachke SA, FitzGerald PG et al (2014a) Loss of Sip1 leads to migration defects and retention of ectodermal markers during lens development. Mech Dev 131:86–110. https://doi.org/10.1016/j.mod.2013.09.005
Manthey AL, Terrell AM, Lachke SA et al (2014b) Development of novel filtering criteria to analyze RNA-sequencing data obtained from the murine ocular lens during embryogenesis. Genomics Data 2:369–374. https://doi.org/10.1016/j.gdata.2014.10.015
Patel N, Khan AO, Mansour A et al (2014) Mutations in ASPH cause facial dysmorphism, lens dislocation, anterior-segment abnormalities, and spontaneous filtering blebs, or Traboulsi syndrome. Am J Hum Genet 94:755–759. https://doi.org/10.1016/j.ajhg.2014.04.002
Patel N, Anand D, Monies D et al (2017) Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract. Hum Genet 136:205–225. https://doi.org/10.1007/s00439-016-1747-6
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Rothe M, Kanwal N, Dietmann P et al (2017) An Epha4/Sipa1l3/Wnt pathway regulates eye development and lens maturation. Development 144:321–333. https://doi.org/10.1242/dev.147462
Rowan S, Conley KW, Le TT et al (2008) Notch signaling regulates growth and differentiation in the mammalian lens. Dev Biol 321:111–122. https://doi.org/10.1016/j.ydbio.2008.06.002
Saravanamuthu SS, Le TT, Gao CY et al (2012) Conditional ablation of the Notch2 receptor in the ocular lens. Dev Biol 362:219–229. https://doi.org/10.1016/j.ydbio.2011.11.011
Siddam AD, Gautier-Courteille C, Perez-Campos L et al (2018) The RNA-binding protein Celf1 post-transcriptionally regulates p27Kip1 and Dnase2b to control fiber cell nuclear degradation in lens development. PLoS Genet 14:e1007278. https://doi.org/10.1371/journal.pgen.1007278
Sousounis K, Tsonis PA (2012) Patterns of gene expression in microarrays and expressed sequence tags from normal and cataractous lenses. Hum Genomics 6:14. https://doi.org/10.1186/1479-7364-6-14
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-SEq. Bioinformatics 25:1105–1111. https://doi.org/10.1093/bioinformatics/btp120
Trapnell C, Roberts A, Goff L et al (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. https://doi.org/10.1038/nprot.2012.016
Wang Y, Terrell AM, Riggio BA et al (2017) β1-integrin deletion from the lens activates cellular stress responses leading to apoptosis and fibrosis. Investig Ophthalmol Vis Sci 58:3896–3922. https://doi.org/10.1167/iovs.17-21721
Wolf L, Gao CS, Gueta K et al (2013a) Identification and characterization of FGF2-dependent mRNA: microRNA networks during lens fiber cell differentiation. G3 (Bethesda) 3:2239–2255. https://doi.org/10.1534/g3.113.008698
Wolf L, Harrison W, Huang J et al (2013b) Histone posttranslational modifications and cell fate determination: lens induction requires the lysine acetyltransferases CBP and p300. Nucleic Acids Res 41:10199–10214. https://doi.org/10.1093/nar/gkt824
Xiao W, Liu W, Li Z et al (2006) Gene expression profiling in embryonic mouse lenses. Mol Vis 12:1692–1698
Zhang H-M, Liu T, Liu C-J et al (2015) AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res 43:D76–D81. https://doi.org/10.1093/nar/gku887
Zhang Y, Fan J, Ho JWK et al (2016) Crim1 regulates integrin signaling in murine lens development. Development 143:356–366. https://doi.org/10.1242/dev.125591
Zhao Y, Zheng D, Cvekl A (2018) A comprehensive spatial-temporal transcriptomic analysis of differentiating nascent mouse lens epithelial and fiber cells. Exp Eye Res 175:56–72. https://doi.org/10.1016/j.exer.2018.06.004
Acknowledgements
This work was supported by the National Institutes of Health awards R01EY021505 (S.L.) from National Eye Institute, and R03DE024776 (S.L., I.S.) from National Institute of Dental and Craniofacial Research. We thank the University of Kansas Medical Center Genomics Core for generating the sequence data sets. The Genomics Core is supported by the University of Kansas School of Medicine, the Kansas Intellectual and Developmental Disability Research Center (NIH U54 HD090216) and the Molecular Regulation of Cell Development and Differentiation COBRE (5P20GM104936).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Anand, D., Kakrana, A., Siddam, A.D. et al. RNA sequencing-based transcriptomic profiles of embryonic lens development for cataract gene discovery. Hum Genet 137, 941–954 (2018). https://doi.org/10.1007/s00439-018-1958-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-018-1958-0