
Abstract
Low levels of the survival of motor neuron (SMN) protein cause the neurodegenerative disease spinal muscular atrophy (SMA). SMA is a pediatric disease characterized by spinal motor neuron degeneration. SMA exhibits several levels of severity ranging from early antenatal fatality to only mild muscular weakness, and disease prognosis is related directly to the amount of functional SMN protein that a patient is able to express. Current therapies are being developed to increase the production of functional SMN protein; however, understanding the effect that natural stresses have on the production and function of SMN is of critical importance to ensuring that these therapies will have the greatest possible effect for patients. Research has shown that SMN, both on the mRNA and protein level, is highly affected by cellular stress. In this review we will summarize the research that highlights the roles of SMN in the disease process and the response of SMN to various environmental stresses.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Protein coding genes account for an estimated 20,000 genes in the human genome; however, the sheer number of protein isoforms dramatically overshadows this number. A major source of protein diversity is alternative splicing of mRNA. Splicing is the process by which immature pre-mRNA is processed to remove introns or non-protein-coding regions. Alternative splicing defines the system by which mature mRNA arising from a common gene can vary from transcript to transcript. This occurs by inclusion or exclusion of elements within the pre-mRNA, resulting in alternative protein-coding message. Therefore, alternative splicing generates protein diversity through interpreting a single genomic sequence in many different ways through alternative inclusion or exclusion of mRNA elements, known as exons. Ultimately, the inclusion and exclusion of exons lead to different protein isoforms arising from the same pre-mRNA transcript. Deep-sequencing of RNA has revealed that alternative splicing occurs in mRNAs from 95–100% of human protein coding genes that contain more than one exon, underscoring the importance of this process in development, cellular homeostasis and, when disrupted, in disease (Gerstein et al. 2014; Wang et al. 2008; Pan and Shai 2008; reviewed in Nilsen and Graveley 2010).
mRNA splicing is an essential step in normal gene expression that relies on a highly complex and dynamic mechanism comprising multiple cis- and trans-acting factors; thus, mutations in any one of these key elements may have profound effects on the health of the cell. The occurrence of mRNA splicing aberrations that contribute to a range of human disorders is well documented, with cancer, muscular dystrophy, retinitis pigmentosa, cardiomyopathies, amyotrophic lateral sclerosis, and spinal muscular atrophy among the most prominent examples (reviewed in Scotti and Swanson 2016).
Spinal muscular atrophy (SMA): the disease
Spinal muscular atrophy is an autosomal recessive pediatric neurodegenerative disease in which splicing alterations are associated with disease pathology. SMA is characterized by degeneration of alpha spinal motor neurons, which results in the denervation of target muscles, that consequently atrophy due to lack of stimulation. Interestingly, though all cells in an SMA patient exhibit decreased levels of the survival of motor neuron (SMN) protein, mainly motor neurons and certain populations of cortical neurons appear to be severely affected by this deficit (d’Errico et al. 2013). Due the pronounced effects of motor neuron degeneration in patients, much of the research on SMN deficiency has focused on these cells in SMA. However, with the development of numerous mouse models of SMA, there is growing evidence that other cell types and tissues are affected by the disease (reviewed in Shababi et al. 2014; Nash et al. 2016). While both males and females are affected by SMA, there is evidence supporting the existence of sex-specific manifestations of the disease that have not been extensively explored. Both humans and mouse models of SMA exhibit developmental defects in the male reproductive tract, with male sterility being observed in some mouse models (Howell et al. 2017; Riessland et al. 2010; Richert et al. 1986). While the genetic etiology of SMA is relatively simple, the reason that some cell types exhibit increased susceptibility to SMN deficiency is not completely understood. To approach this question, it will be important to consider the specific microenvironment of these cell types and to identify circumstances, such as cellular stress responses, that could exacerbate the phenotype in this context.
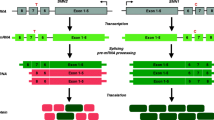
SMN is encoded by two genes in humans: SMN1 and SMN2. The two genes are nearly identical, but a single base substitution in exon 7 accounts in large part for a deviation in the mRNA splicing patterns of the two genes (Lorson 1999; Cartegni and Krainer 2002; Kashima and Manley 2003; Lefebvre et al. 1995; Monani 1999) (Fig. 1). mRNA processing requires the mRNA message to be configured correctly by the spliceosome, which removes introns and determines which exons will be included in the final message. These decisions are determined by splice site strength, supportive or inhibitive sequences within the transcript, known as enhancers or repressors, respectively, and the availability and activity of protein accessory factors that bind these sequences. At the DNA level, the 6th nucleotide of exon 7 in SMN1 is a cytosine that encodes an mRNA splicing enhancer sequence that functions in spliceosome recruitment and facilitates exon recognition. The respective residue in SMN2 is a thymine, which alters the enhancer to become a repressor in the resultant mRNA, which blocks the spliceosome from including the exon (Lorson 1999; Cartegni and Krainer 2002; Kashima and Manley 2003; Lefebvre et al. 1995; Monani 1999). While the C to T substitution between SMN1 and SMN2 expn 7 is translationally silent, the result is an mRNA splicing pattern in which over 90% of SMN1 transcripts include exon 7, while a substantial portion of SMN2 transcripts lack exon 7, depending on cell type and environmental context. This splicing alteration is functionally significant as transcripts lacking exon 7 produce truncated and unstable protein, which ultimately results in lower levels of functional SMN arising from the SMN2 locus (Pellizzoni 1999; Burnett et al. 2009; Lorson 1998). While the role of the exon 7 C>T transition in causing exon exclusion is clear, there are many other sequences within SMN1 and SMN2 that also influence the final spliced products of the SMN transcripts. These regions of influence can extend a considerable distance from the exon itself and exert their effect through pre-mRNA secondary structure that modulates spliceosomal activity (Singh et al. 2015). These modes of splicing modulation are reviewed extensively by Singh et al. (2017b). Indeed, as the field advances, the spectrum of SMA-causing splicing mutations continues to broaden and we likely will continue to discover more sites of splicing modulation within SMN1 and SMN2.

(figure adapted from Burghes and Beattie 2009)
SMN1 and SMN2 splicing. SMN1 and SMN2 genes contain a C to T nucleotide alteration in exon7. This alteration changes the splicing of the pre-mRNA, leading to exon inclusion in SMN1 and predominant exon exclusion in SMN2. This alters the isoforms of protein produced, with SMN1 producing mainly full-length protein, which oligomerizes and is stable, and SMN2 producing mainly truncated proteins (depicted by missing wedge), which do not oligomerize and are rapidly degraded. The small amount of full-length protein that can be produced from SMN2, however, is identical to SMN1 and the two populations can interact
SMA is caused by mutation or deletion of SMN1 and retention of SMN2 (Lefebvre et al. 1995). The complete absence of SMN protein is embryonic lethal, but the presence of SMN2 in the absence of SMN1 in humans results in low levels of SMN protein, giving rise to SMA. A correlation between SMA severity and SMN protein levels has been observed both in SMA patients and in mouse models of SMA (Harada et al. 2002; Lefebvre et al. 1997; Hsieh-Li et al. 2000; Monani et al. 2000; Coovert et al. 1997). SMN levels can vary widely among SMA patients and consequently, SMA is a disease of variable severity ranging from Type I, the most severe, with paralysis and death as early as birth, to Type IV, which is mild, with muscular weakness but no effect on life span. The most significant correlative factor across the spectrum of disease severity is the copy number of SMN2 genes (McAndrew 1997; Burghes 1997). The SMN2 gene locus is amplified in some individuals, with improved disease outcomes in those with higher copy numbers (McAndrew 1997; Burghes 1997). Furthermore, mouse models with higher copy numbers of a human SMN2 transgenes exhibited increased life span compared to littermates with fewer copy numbers (Hsieh-Li et al. 2000; Monani et al. 2000; Le et al. 2005). Therefore, SMN levels directly correlate to disease phenotypes (Lefebvre et al. 1997; Coovert et al. 1997). For individuals with fewer copies of SMN2, it is critical to understand how splicing of this transcript responds to more subtle cues, both for understanding the disease and also for developing therapeutics. Splicing is a dynamic and malleable process that is influenced not only by development and tissue-specific cues, but also by environmental stresses. Environmental stresses are known to activate stress pathways, which in turn activate signaling cascades. These signaling cascades are mediated largely through kinase activation, with phosphorylation playing a key role. Many trans-acting splicing factors can be phosphorylated, which affects their cellular localization and activity. This can in turn alter the activity of the key splicing machinery (Stamm 2008). We review here the effect of environmental stresses on mRNA splicing and protein modification. We will focus on these stresses in the scope of SMA pathology, but these effects may be relevant to any number of other diseases and splicing products.
SMA: SMN function and the etiology of the disease
Years of extensive research into how SMN deficiency causes SMA have produced an expanding list of cellular processes that involve SMN function. In addition to a host of relatively well-defined roles in RNA metabolism, SMN has been implicated in signal transduction, intracellular trafficking, DNA recombination, endocytosis and autophagy (reviewed in Singh et al. 2017a, b). Though the role of SMN is widespread and diverse, its most well characterized function in the cell is that of snRNP assembly.
SMN in snRNP assembly
SMN is required for mRNA processing in all cells. Genetic depletion of SMN protein levels is pre-implantation embryonic-lethal in mouse (Schrank 1997), while in zebrafish and Drosophila, translation-blocking morpholinos or genetic mutation to decrease functional Smn levels results in larval lethality (and a portion of zebrafish embryo lethality occurring between gastrulation and somitogenesis) (McWhorter et al. 2003; Boon et al. 2009; Winkler et al. 2005; Hao et al. 2013; Rajendra 2007; Chan et al. 2003). These studies from distantly related organisms illustrate the highly conserved and developmentally essential nature of SMN.
SMN functions in the biogenesis of small nuclear ribonucleic proteins (snRNPs) that are building blocks of the splicing machinery (Eggert et al. 2006; Pellizzoni 2007; Gubitz et al. 2004; Pellizzoni et al. 2002; Meister et al. 2001; Fischer et al. 1997; Liu et al. 1997; Ogawa et al. 2009). RNA components of snRNPs, snRNAs U1, U2, U4, and U5 are assembled with Sm protein pentameric rings and U6 assembling with an LSm ring (Pellizzoni et al. 2002; Wan et al. 2005). These snRNPs are critical to carry out the catalysis of splicing and are assisted by the Sm ring, which are brought together and assembled by the SMN complex (SMN protein and Gemin proteins 2–8) (Pellizzoni et al. 2002; Meister et al. 2000; Otter et al. 2007; Carissimi et al. 2006a, b). Reduced SMN protein levels decrease snRNP assembly in vitro and in vivo, and the cases in which snRNP assembly is most dramatically decreased corresponds to the most severe disease types (Workman et al. 2009; Shpargel et al. 2005; Sun et al. 2005; Bühler et al. 1999; Monani et al. 2003; Ogawa et al. 2007; Kotani et al. 2007; Clermont et al. 2004; Cuscó et al. 2004; Parsons 1998). Hence, reduced SMN complex levels and activity is a well-established cause of SMA (Shpargel et al. 2005; Sun et al. 2005; Ogawa et al. 2007; Kotani et al. 2007; Clermont et al. 2004; Workman et al. 2009; Alías et al. 2009; Cuscó et al. 2004; Prior 2007; Bühler et al. 1999; Rochette et al. 1997; Lorson 1998; Hahnen et al. 1997; Wang et al. 1998; Talbot et al. 1997; Seng et al. 2015).
It was recently demonstrated that, in addition to SMN deficiency per se as a cause of SMA, missense mutations that alter the ability of Sm proteins to bind or be released from the SMN complex can also cause the disease. Structural analysis of reconstituted SMN complexes in vitro, using either wild type SMN or SMN (E134K), a common SMA-causing mutant isoform that substitutes a lysine for glutamate at residue 134, found that the wild-type and mutant complexes were indistinguishable (Sun et al. 2005; Neuenkirchen et al. 2015). Furthermore, both complexes bound Sm protein similarly. However, upon addition of U1 snRNA, the transfer of Sm ring to snRNA was severely diminished in the SMN (E134K) samples. The lower transfer efficiency was found to be due to a much higher dissociation constant (K d) of SMN (E134K) compared to wild-type SMN, indicating tighter binding or misarrangement (Neuenkirchen et al. 2015). These experiments suggest that, in addition to Sm assembly onto snRNPs, Sm dissociation during the RNA remodeling of the splicing process may also play a role in the SMA disease phenotype. It should be noted that these experiments were performed in vitro in the absence of ATP. While the formation of snRNP assembly may be ATP-independent in vitro (Neuenkirchen et al. 2015; Raker 1996, 1999; Sumpter et al. 1992), the complex is ATP-dependent in vivo (Kleinschmidt 1989; Meister et al. 2001; Temsamani et al. 1991; Meister and Fischer 2002; Pellizzoni et al. 2002; Meister et al. 2000; Otter et al. 2007; Carissimi et al. 2006a, b). Therefore, the results should be verified in vivo to conclusively determine the role of Sm dissociation in addition to the formation of the SMN complex and association with Sm protein.
SMN and the minor spliceosome
The improper formation of the core spliceosomal subunits is clearly a manifestation of decreased SMN protein levels characteristic of the SMA disease state. Although it seems logical that the snRNP deficiency would lead to improper splicing, the role of downstream splicing events in SMA disease progression remains controversial. It has been shown that gemin levels and snRNP assembly are decreased in spinal cord from severe SMA mice (SMN2 +/+; mSmn −/−) at postnatal day 3 (pnd3) and correlate with disease severity. Interestingly, the reduced SMN levels and snRNP assembly preferentially affects the levels of the U11 snRNP, an essential component of the minor spliceosome (Turunen et al. 2013). This finding lends credence to the hypothesis that the minor spliceosome is preferentially affected in SMA and affects the removal of U12-type introns that comprise less than 1% of all introns in human cells (Gabanella et al. 2007).
In one study, the decrease in minor spliceosomal subunits was indeed linked to aberrant splicing of transcripts containing the U12-type subtype of introns in Drosophila third instar larvae expressing the Smn 73Ao loss-of-function allele compared to their wild-type counterparts (Lotti et al. 2012). Eighteen of the twenty-three U12 intron-containing genes in Drosophila were mis-spliced in the Smn 73Ao mutant. Mis-splicing of one of the affected U12 intron-containing genes, stasimon, correlated with motor neuron pathology, where knock down of stasimon resulted in NMJ dysfunction that was, in turn, rescued by restoring stasimon expression. Overexpression of stasimon was also sufficient to rescue axonal pathfinding and outgrowth defects in Smn knockdown zebrafish (Lotti et al. 2012). It should be noted, however, that stasimon expression was not able to rescue the locomotion or viability defects.
In contrast, an earlier study that examined Drosophila third instar larvae from a cross between a mutant line carrying the null allele Smn D, caused by an insertion between the start codon and the Tudor domain (Rajendra 2007), and a mutant line carrying the Smn X7 allele that is deleted for most of the Smn region (Chang et al. 2008), showed decreased minor and major spliceosomal components but did not observe the same splicing defect specifically of minor intron-containing mRNA (Praveen et al. 2012). Of seven transcripts that were also identified by Lotti et al. in the study described above, only 2 exhibited similar levels of aberrant intron retention in response to the reduced Smn level in Smn DxSmn x7 larvae. Moreover, low level expression of a wild type SMN transgene could rescue motility and viability defects without restoring normal snRNA levels or snRNP assembly, suggesting independent roles for SMN in motor function and snRNP assembly (Praveen et al. 2012). It was further shown that the changes in U12-intron containing transcripts could more likely stem from a developmental delay in the Smn mutant larvae since the splicing pattern of the transcripts varied more with developmental time than Smn mutant status (Garcia et al. 2013). Currently, the debate over the role of misspliced transcripts in the etiology of SMA remains unresolved.
More recently, genome-wide studies of splicing defects were performed in two different SMA mouse models: the severe Taiwanese model at PND1 and PND5, and an ASO-inducible SMA mouse model induced at 8 weeks and analyzed at 20 and 30 days post SMN depletion (Jangi et al. 2017; Doktor et al. 2017). Both studies report splicing defects in all tissues analyzed, specifically aberrant intron retention of both major and minor introns. Intron retention increased over time and the transcriptional profile was recapitulated with siRNA knock down of SMN in human HeLa cells, arguing against the influence of mouse development in inducing these changes (Doktor et al. 2017). Although minor introns are present in less than 1% of all genes, the sensitivity of U12 introns to low levels of SMN protein is illustrated by these studies, as evidenced by the large proportion of the minor intron containing genes that undergo mis-splicing in SMA mice. The most common changes observed in spinal cord mRNA after SMA disease induction is intron retention, with U2-type intron retention in 4628 transcripts (3.54% of major introns) and U12-type intron retention in 152 transcripts (31.54% of minor introns) (Jangi et al. 2017). However, the fact that these U12 intron-containing transcripts are prone to mis-splicing in the presence of decreased SMN levels may be an association and not causative of disease, since the number of mis-spliced U2 intron-containing mRNAs far exceeds the number of U12 intron-containing transcripts. Therefore, more detailed study of the most highly affected transcripts will be necessary to determine their function and the relationship of the splice variants to SMA pathology. It will be important to ask whether the changes in splicing cause SMA disease phenotype or are a consequence of disease progression.
Additional roles for SMN
In addition to SMN localization in the cell body where snRNP assembly occurs, in neurons SMN is also found within dendrites and axons. This raises the question of whether SMN protein has additional roles in axons independent of snRNP assembly that could contribute to the SMA disease phenotype. In a maternal-zygotic Smn mutant zebrafish line, transgenic expression of Smn specifically in motor neurons, driven by the mnx1 or hb9 promoter, is sufficient to rescue axonal and dendritic morphological defects not only in motor neurons and dorsal root ganglia, but also Schwann cell myelination defects (Hao et al. 2015). Additionally, in a Drosophila model of SMA, restoration of basal levels of SMN in muscles or motor neurons did not improve muscle morphology or neuronal physiology. However, SMN expression in cholinergic interneurons and proprioceptive neurons rescues motor defects (Imlach et al. 2012). These findings from zebrafish and Drosophila SMA models suggest that SMA may be a disease affecting cells in a non-cell autonomous way, where normal motor neuron SMN levels are required for functional motor-sensory circuit establishment (Hao et al. 2015; Imlach et al. 2012).
SMN has also been implicated in mRNP trafficking (Fallini et al. 2012; Burghes and Beattie 2009; Pellizzoni 2007; Donlin-Asp et al. 2016). SMN and other members of the SMN complex (gemin6, gemin7, gemin2, and gemin3) colocalize in axons and dendrites but do not associate with splicing-specific proteins (Sm proteins) (Zhang et al. 2006; Sharma et al. 2005). However, co-staining of only two binding partners has been visualized at a time and it has not yet been shown that three or more of these endogenous proteins colocalize in axons together. Sm proteins are generally thought to function exclusively in splicing; however, SmB and SmD3 have also been shown to be involved in mRNA localization in Drosophila. Specifically, SmB and SmD3 colocalize with mRNPs that transport oskar mRNA in oocytes (Gonsalvez et al. 2010). SmD3 GFP-insertion mutants cause a delocalization of oskar mRNA deposition at the pole in oocytes. Similarly, SMN regulates the localization of several axonal mRNAs and mRNA-binding proteins (Sanchez et al. 2013; Rossoll et al. 2003; Fallini et al. 2012, 2014; Akten et al. 2011; Fallini et al. 2011). Colocalization of SMN and HuD in primary motor neuron axons was shown using immunohistochemistry, which was validated by SMN immunoprecipitation of rat spinal cord lysate and then probing for HuD (Fallini et al. 2011). Since HuD is a known mRNA-binding protein, immunoprecipitation was performed in the presence of RNAse A, which indicated that mRNA is not required for SMN-HuD binding (Fallini et al. 2011). Furthermore, SMN knock down by shRNA prevents mRNA from being transported to the axons, demonstrating a critical role for SMN in the distribution of mRNA (Fallini et al. 2011). However, it should be kept in mind that these conclusions are based on the overexpression of tagged proteins, and further in vivo work to analyze the role of the endogenous SMN in mRNA transport would be beneficial.
In addition to its a role in transporting mRNAs for local translation, SMN has been implicated in the regulation of translation itself. An in vitro transcription and translation assay utilizing rabbit reticulocyte lysates coupled with a luciferase assay show that SMN protein can act as a translation inhibitor, with this function being dependent on an intact Tudor domain (Sanchez et al. 2013). The effect of SMN on translation was found to affect the protein synthesis of the methyltransferase CARM1 (Sanchez et al. 2013). CARM1 stabilizes SMN mRNA through HuD interaction (Hubers et al. 2011). Therefore, CARM1 translational regulation by SMN represents a possible negative feedback loop. As this is not a global regulatory phenomenon, further experiments will be required to determine whether the translation of other mRNAs is affected by SMN activity and whether these phenomena are related to disease manifestation. In contrast, knockdown of SMN levels decreases local translation in axons, evidenced by nascent protein detection with the methionine analog, AHA (Fallini et al. 2016). Actin levels and growth-associated protein 43 (GAP43) that is associated with growth cones, are also decreased in axons both with SMN knock-down as well as in SMA versus control motor neurons. Hence, SMN may function both in repressing translation as well as transporting mRNA. These roles are not mutually exclusive, with translation silencing being required for transport of mRNA granules. It is important to note that the effect SMN has on translation is likely to differ between mRNAs from different genes and will require further analysis in the future. It is becoming increasingly evident that the multiple functions of SMN throughout a variety of cell types are likely to contribute in varying degrees to the SMA disease phenotype and that increasing SMN levels will likely ameliorate a broad range of defects.
Regulation of SMN splicing
Although a clear correlation exists between SMN2 copy number and SMN protein levels, there are cellular processes that additionally affect SMN protein levels. One example is the splicing efficiency of the SMN2 gene itself. Splicing of SMN2 mRNA does not occur at a fixed rate, but is highly dynamic and context-dependent. One critical determinant of final the splicing outcome is the combinatorial binding of splicing factors to the region in and around exon 7 that determines whether the exon will be included in the transcript. Trans-acting factors bind pre-mRNA directly or as complexes with other proteins and can influence splicing by either promoting or blocking interaction with the spliceosome (Kashima and Manley 2003; Cartegni et al. 2006; Vezain et al. 2010; Hua et al. 2008; Singh et al. 2006a, b; Hofmann 2000; Hofmann and Wirth 2002; Cléry et al. 2011; Chen et al. 2008; Pedrotti et al. 2010; Young et al. 2002; Passini et al. 2011) and function as positive and negative splicing factors, respectively. Sequences in the mRNA to which splicing regulators bind to facilitate inclusion are called splicing enhancers, while regions to which splicing regulators bind to prevent spliceosome recognition of target sequences are splicing silencers. The balance of positively and negatively acting protein–RNA interactions will largely influence the activity the spliceosome (reviewed in Saltzman et al. 2011). Thus, while most SMN2 mRNA lacks exon 7, the splicing efficiency can be skewed to favor exon 7 inclusion by increasing the activity of splicing-promoting factors, or to exacerbate exon exclusion by facilitating elements that promote exon 7 skipping.
Many of the identified splicing factors fall into two main classes of splicing regulators, serine arginine rich (SR) proteins and heterogeneous nuclear ribonuclear proteins (hnRNPs) (reviewed in Saltzman et al. 2011). The activity of these proteins is context dependent and many of them may regulate splicing either positively or negatively depending on the target sequence to which they are bound, post-translational modifications, and binding partners. The most well-characterized positively acting factors affecting SMN2 exon 7 splicing are serine/arginine-rich splicing factor 1 (SRSF1) and transformer 2 protein homolog beta (Tra2B) (Hofmann 2000; Young et al. 2002). The most well-characterized negatively acting splicing factors are heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) and src-associated substrate in mitosis 68 (Sam68) (Fig. 2) (Hua et al. 2008; Singh et al. 2006a, b; Kashima and Manley 2003; Cartegni et al. 2006; Vezain et al. 2010; Pedrotti et al. 2010). Other factors that are also known to bind SMN mRNA and affect splicing of exon 7 are SRSF9, hnRNP G, and hnRNP Q1 (Table 1).

SMN1 and SMN2 exon 7 splicing regulators. SMN1 is characterized by predominantly positive splicing regulation. SRSF1 and TRA2B bind directly to exon 7 splicing enhancer regions SE1 and SE2, respectively. TRA2B allows the cooperative binding of hnRNP G and SRSF9, which add further positive influence for exon recognition. These factors help to recruit U1 and U2 snRNPs to the exon boundaries. This snRNP recognition is critical for the inclusion of this exon. On the other hand, SMN2 transcripts contain a nucleotide alteration from SMN1; C>U in SE1 region (marked in red). This nucleotide alteration changes the preferred protein-binding partner from the positive SRSF1 to the negative hnRNP A1. Similarly, SAM68 is also capable of binding this region and exerting negative influence. Two more hnRNP A1 sites are known to induce exon 7 exclusion, located in SE2 region and the intronic silencing element, ISS-N1. A GC-rich region overlapping the 5′end of ISS-N1 also associates with long-distance interaction (LD-1); the negatively acting proteins decrease the recognition of the exon by U1 and U2 snRNPs, leading to exon 7 exclusion. hnRNP M binds to sequence in SE2 that overlaps the TRA2B binding site and promotes exon 7 inclusion in both SMN1 and SMN2 transcripts
The splicing factors that are critical for SMN exon 7 splicing, SRSF1, Tra2B, hnRNP A1, and Sam68 (Hofmann 2000; Young et al. 2002; Hua et al. 2008; Singh et al. 2006a, b; Kashima and Manley 2003; Cartegni et al. 2006; Vezain et al. 2010; Pedrotti et al. 2010) are all RNA-binding proteins that recognize specific sequences in pre-mRNA to facilitate or repress recognition of exon 7 (Fig. 2). Transcripts from both SMN1 and SMN2 include the same cis-acting regulatory sites in their sequences that are bound by splicing factors to facilitate or block the recognition and inclusion of exon 7. These regulatory regions are known as splicing enhancer 1 (SE1), splicing enhancer 2 (SE2), and intronic splicing silencer 1 (ISSN1) (Lorson 1999; Hofmann 2000; Kashima and Manley 2003; Cartegni et al. 2006; Vezain et al. 2010; Hua et al. 2008; Singh et al. 2006a, b). The difference in splicing patterns of SMN1 and SMN2 transcripts is caused by a single genomic C-to-T transition in exon 7, resulting in a C-to-U transition in the mRNA (Lorson 1999; Kashima and Manley 2003; Cartegni et al. 2006; Vezain et al. 2010). In SMN1, the positive splicing factor, SRSF1 binds to SE1, with further positive exon recognition from TRA2B binding to SE2 (Cartegni and Krainer 2002; Hofmann 2000; Young et al. 2002). TRA2B binding to SE2 is required for the additional recruitment and binding of the positively acting splicing factors, hnRNP G and SRSF9. (Hofmann and Wirth 2002; Young et al. 2002; Passini et al. 2011; Cléry et al. 2011). The presence of these factors on the pre-mRNA promotes the recruitment of the initial splice site recognition snRNPs, U1 and U2, that bind to the 5′ end of intron 6 and 3′ end of the intron 7, respectively. The snRNP binding is essential for proper definition of the exon. In SMN2 transcripts, the C-to-U substitution in SE1 results in decreased binding affinity for SRSF1 and increased affinity for hnRNP A1, that in turn decreases exon definition by U1 and U2 (Lorson 1999; Cartegni and Krainer 2002). This strong influence by hnRNP A1 overpowers the ability of TRA2B to bind SE2. SAM68 is also found to bind in the region around SE1 and decrease exon definition (Pedrotti et al. 2010). Additionally, more negative splicing influence of exon 7 arises from hnRNP A1 binding to the intronic ISS-N1 region (Singh et al. 2006a, b; Hua et al. 2008). In addition to these proximal RNA regulatory regions, variants in downstream sequences have been found to alter the efficiency of SMN exon 7 splicing by disrupting the normal secondary structure of the transcript, underscoring the potential for long-range effects in splicing regulation (Singh et al. 2015, 2017b).
Effects of stress on splicing
While several splicing factors are known to promote exon 7 inclusion in SMN1 and exon 7 exclusion in SMN2 splicing, the relative abundance of final spliced mRNA product is also affected by stresses and cell signaling processes, such as hypoxia, starvation, and temperature changes (Bebee et al. 2012; Sahashi et al. 2012). An example of this effect may be seen under the influence of oxidative stress induced by paraquot treatment, which has been shown to result in multiple aberrant SMN2 splicing products in murine tissues (Seo et al. 2016). Protein modification affects the activity and localization of splicing factors and these changes are highly regulated. For instance, SRSF1 requires phosphorylation to promote spliceosome assembly, but must be dephosphorylated to catalyze splicing (Cao 1997). Not only is activity regulated by phosphorylation, but also physical availability. For instance, SR proteins are found both in the cytoplasm and in the nucleus, but nuclear localization is necessary for their influence on splicing (Saltzman et al. 2011). It is known that phosphorylation is required for the SR proteins to re-enter the nucleus (Lai et al. 2001). Furthermore, SR proteins are stored in dense nuclear granules, called nuclear speckles, and require phosphorylation to be removed from the speckle and relocalized to sites of transcription and splicing (Misteli 1998). Phosphorylation events affecting splicing factor availability and activity have been seen for SRSF1 as mentioned above, but also Tra2B (Stoilov et al. 2004), hnRNP A1 (van der Houven van Oordt et al. 2000) (Guil et al. 2006), and Sam68 (Lukong et al. 2005; Paronetto et al. 2006; Matter et al. 2002), all of which directly impact SMN splicing.
Importantly, different cellular contexts and environmental conditions alter the location, activity, or expression of these factors through activation of signaling cascades that indirectly alter splicing. Signaling cascades that are known to affect SMN or SR protein and hnRNP phosphorylation are AKT (oxidative stress and starvation; phosphorylation of hnRNP A1), p38MAPK (UV stress, heat shock, osmotic stress, anisomycin; stabilization of SMN mRNA and relocalization of hnRNP A1), and PKA pathways (neuronal differentiation, oxidative stress) (van der Houven van Oordt et al. 2000; Guil et al. 2006; Lukong et al. 2005; Paronetto et al. 2006; Matter et al. 2002; Jo et al. 2008; Farooq et al. 2009; Obata et al. 2000; Cao et al. 2011). These pathways are activated by various growth and stress signals. Furthermore, cells that expend large amounts of energy, such as neurons and muscles undergo elevated levels of oxidative and other stresses. Indeed, it has been shown that a feature common of many motor neuron diseases is excitotoxicity, mitochondrial damage, and calcium handling aberrations (Saxena et al. 2011). Environmental stresses that affect several aspects of splicing may likewise exacerbate the SMA disease state (summarized in Fig. 3 and described below). With so many pathways as potential areas for splicing factor alteration, this begs the question, what environmental stresses associated with the SMA disease phenotype affect splicing, and how can these pathways be manipulated to foster improved disease outcomes?

SMN protein and RNA processes are affected by environmental and microenvironmental stresses. The range of possible SMN-mediated effects produced by stress is likely dependent on the type and magnitude of stress. While starvation and hypoxia reduce SMN protein levels by increasing aberrant splicing of SMN mRNA, oxidative stress may effectively reduce SMN function through inactivation resulting from covalent binding of oligomers. The net result of reduced functional SMN would be predicted to impact the full-range processes in which SMN participates. In contrast, metabolic stress resulting from bacterial infection may have a more limited effect due to the specific aggregation of SMN and snRNA
Hypoxic stress
Patients with severe SMA often do not live past 2 years and ultimately succumb to respiratory failure and hypoxic distress due to muscle weakness. Concordantly, a hallmark of late-stage disease in a mouse model of severe SMA is increased levels of the hypoxic markers, Hif1alpha (Bebee et al. 2012) and Hif3alpha (Zhang et al. 2008). Additionally, hypoxic treatment of cultured SMA patient fibroblasts leads to increased levels of exon 7 skipping and subsequent reduced levels of stable, functional SMN protein. Importantly, this splicing alteration was found to be due to upregulation of the negative splicing factor hnRNP A1 (Bebee et al. 2012). While the mechanism of hnRNP A1 upregulation was not addressed in that study, hypoxia has also been shown to activate the AKT pathway, which phosphorylates hnRNP A1 (Jo et al. 2008). Further investigation of the possible involvement of the AKT pathway may identify additional mechanisms that link hypoxic stress and the regulation of SMN2 splicing by hnRNP A1.
Notably, hyperbaric hyperoxia treatment of the SMA mice improved motor function and body weight (Bebee et al. 2012). The authors proposed that a feed-forward cycle contributes to the progression of the SMA disease state, in which motor neuron denervation of intercostal and diaphragm muscles leads to breathing impairment and hypoxia. This in turn decreases the proportion of full-length SMN2 transcript, thereby decreasing the level of functional SMN protein, which in turn leads to further motor neuron damage. Consistent with this notion, vascular malformations in muscle and spinal cord have been described in three different SMA mouse models, Taiwanese, SMN∆7, and Burghes’ severe model (Somers et al. 2016; Nobutoki et al. 2015; Somers et al. 2012; Le et al. 2005; Monani et al. 2000; Hsieh-Li et al. 2000). Somers et al. analyzed muscle capillary beds in the severe Burghes mouse model at PND1, PND3, and PND5. The caudal band of the levator auris longus (LALc) that exhibits extensive NMJ pathology was stained for vascular beds. Samples from SMA mice exhibited much less densely organized capillary beds, with larger diameter capillaries and less intramuscular vascularization than wild-type samples at PND3 and PND5 but not at PND1. This decreased capillary bed density was similarly observed in the transverse abdominal muscle, that exhibits substantial muscular atrophy, and LAL rostral band (LALr), that shows mild muscular atrophy, indicating decreased capillary bed density did not correlate with only severely affected muscle types (Somers et al. 2012). Additionally, the Taiwanese and SMN∆7 SMA mouse models were also analyzed for capillary bed infiltration in muscle and spinal cord (Somers et al. 2016; Le et al. 2005; Monani et al. 2000). Spinal cord vasculature at pre-symptomatic stages matches that of non-disease littermates. However, over time, capillary beds in control animals grow denser while in SMA mice, they remain sparse, at approximately half the density of the controls (Somers et al. 2016). A similar vascular deficiency was observed in human SMA patient muscle biopsies, with a capillary-to-filament ratio approaching tenfold lower in SMA patients than controls (Somers et al. 2016). However, other similarly denervating conditions were not examined in these studies, so whether this defect is due to denervation or results from reduced SMN in blood vessels remains unclear. Regardless of the primary cause, this condition creates a hypoxic microenvironment in the mouse spinal cord, as well as a disrupted blood–spinal cord barrier (Somers et al. 2016; Nobutoki et al. 2015).
Taken together, these findings identify hypoxia as a significant contributor to SMA disease onset and progression. SMA may lead to a hypoxic environment in patients through capillary bed deficiency as well as respiratory distress suffered by many SMA patients. These conditions may create a hypoxic environment that reduces the level of full-length SMN2 transcripts as was observed in mice.
Oxidative stress
Neurons and other cells with high energy demands are subject to oxidative stress that results from a buildup of free radicals, including superoxide, hydrogen peroxide and other reactive oxygen species (ROS) as a byproduct of energy production from the mitochondrial electron transport chain. The well-established role of oxidative stress as a factor in the pathology of several neurodegenerative diseases (reviewed in Niedzielska et al. 2016; Kim et al. 2015) has prompted investigation into whether it contributes to SMA pathology as well.
One of the first studies to indicate that SMN deficiency results in oxidative stress used siRNA-mediated knock-down of SMN in NSC-34 cells, a motor neuron-like hybrid cell line, to demonstrate that 72 h after transfection, with a 66% reduction in SMN protein, cells showed increased levels of ATP, cytochrome C oxidase and hydrogen peroxide, indicating a defect in mitochondrial energy production (Acsadi et al. 2009). The knock-down cells also displayed increased mitochondrial membrane potential and a reduced number of cells with neurite outgrowth. Similar results were seen in long-term cultures of motor neurons derived from in vitro differentiation of WT human embryonic stem cells (hESCs) (Wang et al. 2013). In lines where SMN expression was knocked-down with stably transfected shRNA, mitochondrial superoxide production was significantly increased, as measured by MitoSOX staining. These cultures also showed increased caspase 3/7 activity relative to control cells, and long-term cultures (6 weeks) showed a greater loss of motor neurons, as measured by staining for HB9, a motor neuron marker. In cultures treated with the antioxidant N-acetylcysteine (NAC), caspase 3/7 levels and motor neuron survival returned to control levels. While the SMN knock-down cultures showed a dramatic reduction in axon length, it is unclear whether this effect was due to oxidative stress since the effect of NAC treatment was not analyzed.
To better understand how SMN deficiency could induce oxidative stress, two recent studies examined mitochondrial dysfunction in vitro and in vivo. Using live cell imaging of motor neurons derived from SMA type 1 iPSCs, Xu et al (2016) found a reduction in size, number, and axonal transport of mitochondria in the SMA cells compared to WT cells. NAC treatment returned mitochondrial numbers and transport to WT levels. The mitochondrial defects were not observed in telencephalic glutamatergic (forebrain) neurons derived from the same iPSCs, suggesting that the impaired mitochondrial transport was specific to motor neurons. After long-term (6 weeks) culture, the SMA motor neurons displayed an increased number of axonal swellings and increased caspase 3/7 activity relative to WT cells, reflecting increased degeneration and apoptosis. NAC treatment ameliorated these defects as well, suggesting that oxidative stress that accompanies mitochondrial dysfunction in SMA motor neurons contributes to motor neuron degeneration. Similar results were obtained by Miller et al. (2016) using motor neurons isolated by cell sorting from spinal cords of SMA mice (∆7 SMA or Smn−/−;SMN2 tg/−) at P9. The mutant motor neurons showed reduced mitochondrial transport and oxygen consumption, as well as increased oxidative stress, as measured by live-imaging of signal from transfected mito-roGFP, a redox-sensitive GFP targeted to mitochondria. No differences in mitochondrial transport or oxygen consumption were observed between SMA and WT midbrain neurons, suggesting a higher susceptibility of motor neurons to mitochondrial dysfunction due to SMN deficiency. Ultrastructural analysis by electron microscopy of mitochondria in SMA mouse spinal cords at P3 (presymptomatic) and P9 (symptomatic) found reduced size and increased mitochondrial edema at P3, and additional morphological abnormalities at P9, indicating that mitochondrial dysfunction may be present before disease symptoms are manifest in vivo.
Additional in vivo evidence of compromised mitochondrial function due to SMA deficiency recently came from analysis of a zebrafish model of SMA (Boyd et al. 2017). Morpholino-induced knock-down of SMN expression in embryos resulted in reduced ATP-linked respiration and lower levels of ATP synthase subunit alpha (APT5A), a subunit of mitochondrial ATP synthase. The SMA morphants display a well-characterized defect in axonal outgrowth of motor neurons (McWhorter et al.2003) and this phenotype could be partially rescued by overexpression of the glycolytic enzyme Pgk1, suggesting that ATP deficiency due to mitochondrial dysfunction contributes to the impaired axonal outgrowth.
Although these studies, taken together, implicate mitochondria dysfunction, bioenergetic deficiency and oxidative stress in SMA motor neuron pathology, very little is currently known about the specific mechanisms by which SMN deficiency affects mitochondria. It should be noted that the occurrence of oxidative stress in SMN-deficient cells is still unresolved. For example, Patitucci et al. (2016), analyzing motor neurons and astrocytes derived from SMA patient iPSCs, found no evidence of increased oxidative stress in the SMA cells. Surprisingly, staining for ROS using DHE indicated lower ROS in SMA motor neurons than WT cells. This may be accounted for by the dramatically elevated levels of catalase, that decomposes hydrogen peroxide, detected the SMA cells. siRNA-mediated knockdown of catalase expression resulted in increased ROS levels in SMA motor neurons, but other markers of oxidative stress were not induced. What triggers the dramatic induction of catalase in the SMA motor neurons in vitro is not known.
In addition to SMA deficiency potentially giving rise to oxidative stress, conversely, SMN function may be impaired by oxidative stress. A study using an in vitro snRNP assembly high-throughput screen found that the ROS-generating compounds beta-lapachone, menadione, and peroxide inhibit the assembly of Sm rings onto snRNAs (Wan et al. 2008). Closer examination showed that under oxidative stress, SMN forms disulfide bonds between multiple SMN proteins, which was postulated to render them unable to function in snRNP assembly. These data suggest that SMN function is sensitive to oxidizing environments. However, these experiments were performed either in vitro in the absence of reducing agents, or in lysed cells, where conditions almost certainly do not maintain the in vivo redox state of SMN upon cellular lysis. Thus, more experiments will be required to determine whether in vivo, where glutathione levels are maintained in the millimolar range, a sufficient imbalance of oxidizing and reducing agents exists to cause SMN crosslinking.
Finally, a novel way in which SMN deficiency may render cells vulnerable to oxidative stress was proposed with the recent discovery that the SMN complex binds SECIS-binding protein 2 (SBP2), which is required for specialized translation of mRNA encoding selenoproteins (Gribling-Burrer et al. 2017). Incorporation of the amino acid selenocysteine into nascent protein requires SBP2 binding to a Selenocysteine Insertion Sequence (SECIS) in the 3′ UTR of mRNA. Selenoproteins are known to be involved in protection from oxidative stress through their oxidoreductase activity (reviewed in Labunskyy et al. 2014; Papp et al. 2010). mRNA levels for several selenoproteins were significantly lower in spinal cords from SMN-deficient versus WT mice, while in the brain, there was no difference (Gribling-Burrer et al. 2017). If the lower transcript levels are found to result in lower selenoprotein levels in the spinal cord, it would represent a possible mechanism by which SMA motor neurons are made more susceptible to damage from oxidative stress. Thus, SMN deficiency could promote oxidative damage both through mitochondrial dysfunction, as discussed above, as well as by impairing mechanisms that are protective against oxidative stress. If the elevated oxidative stress were sufficient to cause SMN inactivation through cross-linking, it would represent another feed-forward loop, as discussed above with regard to hypoxia, in which SMN deficiency causes a stress condition that, in turn, further reduces levels of functional SMN.
Adaptation to temperature changes
The heat shock response (HSR) is a highly conserved, global stress response that a cell undergoes when faced with thermal stress. Thermal stress causes aberrant protein folding and leads to cellular dysfunction and is, therefore, counteracted by the induction of precise protein folding quality control mechanisms (reviewed in Balchin et al. 2016). At the other end of the temperature spectrum, hypothermia and cold-shock are additional sources of cellular stress. Cold-shock suppresses transcription and may also be associated with oxidative stress due to associated changes in reactive oxygen species (Al-Fageeh et al. 2006). A large collaborative study analyzed the differences in care for SMA patients in North America, Australasia, and Europe (Bladen et al. 2014). Within the data from this study, Tsai et al. identified a correlation between climate and SMA type III ambulation, in which regions with cooler climates (Germany/Austria, Switzerland, and the UK) have patients with a more extended period of ambulation than those in countries with warmer climates (Argentina, Hungary, Ukraine, and Serbia) (Tsai et al. 2016; Bladen et al. 2014). The interpretation of these data by Tsai et al. did not take into consideration differences in health care systems of these countries or dietary or cultural considerations that may also contribute to the phenotypic severity (Tsai et al. 2016). Additionally, it is not clear that the classification of cooler vs. warmer climates is well delimited, since the “warmer” regions experience the following average temperatures for hottest and coldest months, respectively: Argentina (76–53 °F at most equatorial location and 56–35 °F at its most southern regions), Hungary (70–30 °F), Ukraine (66–24 °F), Serbia (80 °F–39 °C). Likewise, the “cooler” countries experience the following yearly temperatures: Germany/Austria (72 °C–38 °F), Switzerland (65–32 °F), UK (65–42 °F).
These concerns notwithstanding, Tsai et al. (2016) did find that repeated transient hypothermia treatment of a severe SMA mouse model (Taiwanese) ameliorated disease manifestations in early postnatal SMA pups. Specifically, hypothermia treatment consisted of exposing neonatal pups to crushed ice for 50 s either daily or every 3 days during which their core body temperature dropped from 33 to 20 °C. The pups were then warmed to restore normal body temperature within 5 min. Following this treatment, SMA pups displayed improved muscle morphology in the quadriceps, intercostal, and diaphragm muscles, as well as improved motor end plate occupancy in the quadriceps. While clearly beneficial to muscle biology, the hypothermia treatment led to only a modest increase in average lifespan and no SMA animals survived beyond 14 days of age. The authors found a correlation between activated p38MAPK and increased SMN protein levels in the brain and spinal cord of hypothermia-treated SMA mice. Consistent with this result, p38 MAPK was previously shown to increase the stability of SMN transcripts, leading to increased SMN protein (Farooq et al. 2009). However, there appeared to be an uncoupling of the protein and mRNA upregulation. For instance, in the spinal cord, SMN transcript levels were significantly upregulated with daily hypothermia treatment while transcript levels were not upregulated with hypothermia treatments given every three days. On the other hand, spinal cord SMN protein levels were not increased significantly with daily treatments, but were significantly upregulated with the every third day treatment (Tsai et al. 2016). Therefore, while the mechanism needs clarification, there is a significant increase in the SMN protein levels of brain and spinal cord upon daily hypothermia treatment, which may indicate interplay between a potential cold shock mechanism and other stress pathways known to upregulate p38 MAPK. These data suggest that the cold-shock pathway may be harnessed to increase SMN protein levels and as a potential therapeutic intervention for SMA.
Metabolic stress
Recently, it was shown that intracellular bacterial infection by Shigella, Salmonella, and Listeria leads to aggregation of snRNA and the SMN complex (Tsalikis et al. 2015). The aggregation of these factors rendered them temporarily inactive. This response was caused by metabolic stress (amino acid starvation and ER stress) triggered by membrane damage. The formation of these aggregate bodies limits the availability of snRNA and greatly affects their functional association with the Sm protein ring. Hence, bacterial infection may reduce splicing capacity due to decreased snRNP availability. Because decreased snRNP assembly and splicing are known to be hallmarks of SMA, it may also contribute to the increased mortality due to infections in the SMA patient population (Cobben 2008; Finkel and Richard 2013). However, most infections experienced by SMA patients are respiratory, which are most often caused by Streptococcus pneumonia, Haemphilus species, Staphylococcus aureus, and Mycobacterium tuberculosis (Speert 2006). These bacteria include both intracellular and extracellular pathogens and therefore, the effect of infection by both intracellular and extracellular bacteria should be similarly analyzed with regard to snRNA and SMN complex aggregation and relevance to SMA.
Another metabolic stress recently found to be linked to SMN deficiency is oxidative phosphorylation. A transcriptional profiling study revealed that mRNA involved in ribosomal processes and oxidative phosphorylation were preferentially decreased in an intermediate-severity SMA mouse model (Murray et al. 2015). Oxidative phosphorylation is critical for production of energy from the mitochondria, and importantly, this study compared vulnerable and non-vulnerable motor neuron populations (as previously determined by NMJ loss), all at pre-symptomatic time points. Interestingly, the p53 pathway, which is known to trigger cell death, was activated preceding NMJ loss, indicating that metabolic deficiencies may program cell death before NMJ denervation.
Starvation stress
Another environmental stress that affects SMN mRNA processing is starvation, which is also a significant morbidity associated with SMA. Prolonged weight loss begins a starvation-signaling cascade, in which AKT plays a role. It has been shown in a mild SMA mouse model that starvation exacerbates SMN2 mis-splicing (Sahashi et al. 2012). Furthermore, when starvation occurs during early postnatal development, the mis-splicing is greater than in adults undergoing starvation (Sahashi et al. 2012). Similar results were observed in all tissues analyzed (spinal cord, heart, liver). Though no regulatory splicing factors were implicated in this study, it is reasonable to hypothesize that due to signaling similarities between starvation and hypoxia, the key modulator in this context is hnRNP A1. Further research will elucidate common mechanisms by which these stress pathways affect SMN2 splicing.
SMN in cytoplasmic stress granules
Stress granules (SG) are dense aggregates of protein and RNA that appear transiently in the nucleus and cytoplasm in response to a variety of stress conditions. Different types of SGs have distinct compositions depending on their cellular sublocalization, cell type, and the type of stress under which they appear (Aulas et al. 2017; Mahboubi and Stochaj 2017). Nuclear SGs, also called nuclear stress bodies, are known to include the heat-shock response transcription factors HSF1/2, as well as pre-mRNA processing factors including HAP (hnRNP A1 interacting protein), hnRNP M, Sam68, and SR splicing factors (Denegri 2001). Cytoplasmic SGs commonly include mRNA bound to stalled translation initiation complexes with phosphorylated eIF2a initiation factor, along with the RNA-binding proteins TIA-1 and TIAR that are associated with repression of translation, in addition to perhaps dozens of other RNA-binding and non-RNA-binding proteins in various combinations under different conditions (Damgaard and Lykke-Anderson 2011; reviewed in Panas et al. 2016; Mahboubi and Stochaj 2017). Broadly, cytoplasmic stress granules are thought to provide a mechanism to very rapidly alter or reprioritize gene expression by transiently sequestering a subset of mRNAs from translation and allowing specific stress response genes to be expressed (reviewed in Anderson and Kedersha 2007).
SMN immunofluorescence signal co-localizes with TIA-1 and TIAR in cytoplasmic SGs that appear in HELA cells in response to heat shock, arsenite or UV irradiation (Hua and Zhou 2004). Further, this study found that overexpressed SMN co-immunoprecipitates with TIA-1/R proteins in extracts from heat shocked HeLa cells. P19 embryonal carcinoma cells in which SMN expression is knocked down by a stably transfected shRNA directed against SMN display a reduced number of SG-positive cells in response to arsenite, and a reduced level of phosphorylated eIF2a and lower cell viability in response to arsenite or hydrogen peroxide treatment (Zou et al. 2011). These studies demonstrate that SMN functions in SGs that promote cell survival under stress conditions. Although numerous studies have investigated the subcellular localization of SMN in neurons and other cells types, mainly in vitro, questions remain regarding the presence and/or role of SMN specifically in stress granules in relevant cell types in vivo. The many SMA mouse models currently available provide a resource to address the question of how SMN deficiency affects stress granule formation and function in relevant cell types during disease progression.
In vivo evidence that SMN participates in SG formation in motor neurons recently came from a study of a mouse model of amyotropic lateral sclerosis (ALS), a neurodegenerative disease that, like SMA, is marked by loss of motor neurons and progressive muscle weakness. SMN was found to be upregulated and associated with HuR-positive SGs that appear in motor neurons of the lumbar spinal cord and brain in male TDP-43A315T transgenic mice, a model of ALS (Perera et al. 2016). TAR DNA binding protein 43 (TDP-43) is a multifunctional DNA- and RNA-binding protein involved in regulation of transcription, translation, and alternative splicing (reviewed in Ratti and Buratti 2016). Moreover, TDP-43 binds to SMN in co-IP experiments, colocalizes with SMN-containing nuclear gems, and is recruited into SGs in response to arsenite stress in vitro (Liu-Yesucevitz et al. 2010). Aberrant cytoplasmic aggregates of TDP-43, that may result from persistent SGs (Parker et al. 2012), are present in motor neurons in a large majority of ALS and frontotemporal lobar degeneration (FTLD) patients (Neumann et al. 2006). Transgenic overexpression of SMN, specifically in neurons of the TDP-43A315T mice, rescued motor neuron loss and delayed onset of hindlimb paralysis in females, but not males (Perera et al. 2016). The reason for the sex-specific effects on SG formation and response to SMN overexpression is not clear, but TDP-43A315T males showed a significantly earlier onset of weight loss and hindlimb paralysis than females and androgen receptor levels were reduced by nearly 50% in spinal cords of TDP-43A315T versus WT males, while they were not different between TDP-43A315T and WT females.
These results add to a growing body of evidence indicating that despite important differences between SMA and ALS, including genetic etiology, age of onset, and specific neurons affected, the two disorders share common underlying disease mechanisms in motor neurons (reviewed in Achsel et al. 2013). Lowering SMN levels in a mouse model of ALS by crossing Smn +/− animals to the SOD1G93A transgenic model of ALS resulted in poorer rotorod performance and reduced lifespan in the SOD1G93A; Smn +/− animals compared to SOD1G93A alone (Turner et al. 2009). Likewise, genetic analysis of ALS patients previously indicated that reduced levels of SMN increased susceptibility to and severity of ALS (Veldink et al. 2005). By contrast, a more recent study that found a significant association between SMN1 gene duplications (but not SMN1 deletions or SMN2 copy number) and sporadic ALS leaves the question of how SMN levels contribute to ALS pathogenesis in humans unresolved (Blauw et al. 2012).
Fused in sarcoma (FUS), a DNA and RNA-binding protein that shuttles between the nucleus and cytoplasm, and functions in DNA repair, transcription, splicing, and mRNA transport (Qiu et al. 2014; Reber et al. 2016; Yasuda et al. 2017), is yet another link between the neurodegenerative diseases SMA and ALS. Mutations in FUS are associated with cases of familial ALS and aberrant FUS-positive aggregates have been detected in motor and spinal neurons (Kwiatkowski et al. 2009; Vance et al. 2009). Exogenously expressed FUS carrying ALS-associated mutations localizes to SGs in vitro in response to oxidative, thermal, mitochondrial, and ER stress (reviewed in Aulas and Vande Velde 2015). Physical interaction between WT FUS and SMN has been demonstrated by co-IP in lysates from HeLa cells, neural-derived cell lines N2a and NSC-34 cells, as well as mouse brain (Yamazaki et al. 2012; Groen et al. 2013; Sun et al. 2015). Groen et al. found that expression of several different ALS-associated mutant forms of FUS in mouse primary cortical neurons causes mislocalization of SMN into FUS-positive cytoplasmic aggregates, with reduced SMN detected in axons. Neurons transfected with the R521C FUS mutant showed reduced axon branching and growth cone area compared to WT FUS, and this defect could be rescued by overexpression of SMN. The authors speculate that mutant FUS traps SMN in cytoplasmic aggregates, thereby preventing SMN from performing its normal function in mRNA transport along axons. An abnormally enhanced interaction between SMN and mutant FUS was confirmed by Sun et al. (2015) and they further observed a reduced number of SMN-containing nuclear gems in ALS patient fibroblasts with FUS mutations and in spinal motor neurons of transgenic mice expressing ALS-associated R521C FUS.
The physical interaction between SMN, TDP-43 and FUS, their association with SGs or aberrant aggregates in motor neuron disease, along with their role in RNA metabolism, specifically splicing, all point toward dysregulated splicing as a factor in motor neuron loss in SMA and ALS. The protective function of SGs in regulating RNA metabolism under stress conditions may be compromised either by SMN deficiency in SMA or by their abnormal persistence and sequestration of SMN in ALS.
Concluding remarks
SMN transcripts and protein are sensitive to external and internal stimuli and responsive to a variety of stresses. We have reviewed here the responses of SMN mRNA and protein to hypoxic stress, oxidative stress, changes in temperature, metabolic stress, and starvation. The mechanisms of cellular response to these stress states share regulatory steps or key factors. The fact that these pathways either affect SMN directly or factors that are critical for the proper production of SMN protein is important for cell maintenance. This is an especially critical piece of information as the splicing of SMN2 and protein status relates directly to the severity of SMA, and the disease state can in turn lead directly back to splicing and protein mishandling aberrations in SMN. We propose that these stresses that decrease the amount of SMN levels can act in a feed-forward cycle, in which an accumulation of stresses resulting from disease progression further exacerbates the disease pathology.
The understanding of stress manifestations in disease will help us to understand disease progression in SMA as well as related neurodegenerative diseases. This understanding can extend even beyond the realm of neurodegeneration and find applications in diseases such as cancers that also revolve around these stress pathways.
It has been shown in mouse models of SMA that SMN restoration as therapy for the disease loses efficacy as the disease progresses and that earlier time points are more efficacious (Robbins et al. 2014; Lutz et al. 2011; Le et al. 2011). Even complete restoration of SMN in severe SMA mice is unable to rescue them if the addition of SMN is after this therapeutic window, indicating that after a certain point, the secondary symptoms of SMA and not just the original cause of the disease must be treated. Therefore, if we are aware of the stress pathways that predispose SMA individuals to the highest level of morbidity, we can customize treatment options for these secondary ailments in addition to restoring SMN protein levels in these individuals.
As clinical trials advance and treatment options improve for these patients, we are entering a new era of improved prognosis for SMA. Understanding and mitigating the cellular stresses left in the wake of lowered SMN levels will be the next step for patients and families.
References
Achsel T, Barabino S, Cozzolino M, Carrì MT (2013) The intriguing case of motor neuron disease: ALS and SMA come closer. Biochem Soc Trans 41(6):1593–1597. doi:10.1042/BST20130142
Acsadi G, Lee I, Li X, Khaidakov M, Pecinova A, Parker GC, Hüttemann M (2009) Mitochondrial dysfunction in a neural cell model of spinal muscular atrophy. J Neurosci Res 87(12):2748–2756. doi:10.1002/jnr.22106
Akten B, Kye MJ, Hao LT, Wertz MH, Singh S, Nie D, Huang J et al (2011) Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA Cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci USA 108(25):10337–10342. doi:10.1073/pnas.1104928108
Al-Fageeh MB, Smales CM (2006) Control and regulation of the cellular responses to cold shock: the responses in yeast and mammalian systems. Biochem J 397(2):247–259. doi:10.1042/BJ20060166
Alías L, Bernal S, Fuentes-Prior P, Barceló MJ, Also E, Martínez-Hernández R, Rodríguez-Alvarez FJ et al (2009) Mutation update of spinal muscular atrophy in Spain: molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum Genet 125(1):29–39. doi:10.1007/s00439-008-0598-1
Balchin D, Hayer-Hartl M, Hartl FU (2016) In vivo aspects of protein folding and quality control. Science 353(6294):aac4354. doi:10.1126/science.aac4354
Bebee TW, Dominguez CE, Samadzadeh-Tarighat S, Akehurst KL, Chandler DS (2012) Hypoxia is a modifier of SMN2 splicing and disease severity in a severe SMA mouse model. Hum Mol Genet 21(19):4301–4313. doi:10.1093/hmg/dds263
Bladen CL, Thompson R, Jackson JM, Garland C, Wegel C, Ambrosini A, Pisano P et al (2014) Mapping the differences in care for 5,000 spinal muscular atrophy patients, a survey of 24 National Registries in North America, Australasia and Europe. J Neurol 261(1):152–163. doi:10.1007/s00415-013-7154-1
Boon K-L, Xiao S, McWhorter ML, Donn T, Wolf-Saxon E, Bohnsack MT, Moens CB, Beattie CE (2009) Zebrafish survival motor neuron mutants exhibit presynaptic neuromuscular junction defects. Hum Mol Genet 18(19):3615–3625. doi:10.1093/hmg/ddp310
Boyd PJ, Tu WY, Shorrock HK, Groen EJN, Carter RN, Powis RA, Thomson SR, Thomson D, Graham LC, Motyl AAL, Wishart TM, Highley JR, Morton NM, Becker T, Becker CG, Heath PR, Gillingwater TH (2017) Bioenergetic status modulates motor neuron vulnerability and pathogenesis in a zebrafish model of spinal muscular atrophy. PLoS Genet 13(4):e1006744. doi:10.1371/journal.pgen.1006744
Bühler D, Raker V, Lührmann R, Fischer U (1999) Essential role for the tudor domain of SMN in spliceosomal U snRNP assembly: implications for spinal muscular atrophy. Hum Mol Genet 8(13):2351–2357
Burghes AH (1997) When is a deletion not a deletion? When it is converted. Am J Hum Genet 61(1):9–15. doi:10.1086/513913
Burghes AHM, Beattie CE (2009) Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 10(8):597–609. doi:10.1038/nrn2670
Burnett BG, Muñoz E, Tandon A, Kwon DY, Sumner CJ, Fischbeck KH (2009) Regulation of SMN protein stability. Mol Cell Biol 29(5):1107–1115. doi:10.1128/MCB.01262-08
Cao W, Jamison SF, Garcia-Blanco MA (1997) Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. RNA 3(12):1456–1467
Cao W, Sohail M, Liu G, Koumbadinga GA, Lobo VG, Xie J (2011) Differential effects of PKA-controlled CaMKK2 variants on neuronal differentiation. RNA Biol 8(6):1061–1072. doi:10.4161/rna.8.6.16691
Carissimi C, Saieva L, Gabanella F, Pellizzoni L (2006a) Gemin8 is required for the architecture and function of the survival motor neuron complex. J Biol Chem 281(48):37009–37016. doi:10.1074/jbc.M607505200
Carissimi C, Saieva L, Baccon J, Chiarella P, Maiolica A, Sawyer A, Rappsilber J, Pellizzoni L (2006b) Gemin8 is a novel component of the survival motor neuron complex and functions in small nuclear ribonucleoprotein assembly. J Biol Chem 281(12):8126–8134. doi:10.1074/jbc.M512243200
Cartegni L, Krainer AR (2002) Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 30(4):377–384. doi:10.1038/ng854
Cartegni L, Hastings ML, Calarco JA, de Stanchina E, Krainer AR (2006) Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet 78(1):63–77. doi:10.1086/498853
Chan YB, Miguel-Aliaga I, Franks C, Thomas N, Trülzsch B, Sattelle DB, Davies KE, van den Heuvel M (2003) Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet 12(12):1367–1376
Chang HC-H, Dimlich DN, Yokokura T, Mukherjee A, Kankel MW, Sen A, Sridhar V et al (2008) Modeling spinal muscular atrophy in Drosophila. PLoS One 3(9):e3209. doi:10.1371/journal.pone.0003209
Chen H-H, Chang J-G, Lu R-M, Peng T-Y, Tarn W-Y (2008) The RNA binding protein hnRNP Q modulates the utilization of exon 7 in the survival motor neuron 2 (SMN2) gene. Mol Cell Biol 28(22):6929–6938. doi:10.1128/MCB.01332-08
Cho S, Moon H, Loh TJ, Oh HK, Cho S, Choy HE, Song WK, Chun JS, Zheng X, Shen H (2014) hnRNP M facilitates exon 7 inclusion of SMN2 pre-mRNA in spinal muscular atrophy by targeting an enhancer on exon 7. Biochim Biophys Acta 1839(4):306–315. doi:10.1016/j.bbagrm.2014.02.006
Clermont O, Burlet P, Benit P, Chanterau D, Saugier-Veber P, Munnich A, Cusin V (2004) Molecular analysis of SMA patients without homozygous SMN1 deletions using a new strategy for identification of SMN1 subtle mutations. Hum Mutat 24(5):417–427. doi:10.1002/humu.20092
Cléry A, Jayne S, Benderska N, Dominguez C, Stamm S, Allain FH-T (2011) Molecular basis of purine-rich RNA recognition by the human SR-like protein Tra2-Β1. Nat Struct Mol Biol 18(4):443–450. doi:10.1038/nsmb.2001
Cobben JM, Lemmink HH, Snoeck I, Barth PA, van der Lee JH, de Visser M (2008) Survival in SMA type I: a prospective analysis of 34 consecutive cases. Neuromuscul Disord NMD 18(7):541–544. doi:10.1016/j.nmd.2008.05.008
Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, Burghes AH (1997) The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 6(8):1205–1214
Cuscó I, Jesus Barceló M, del Río E, Baiget M, Tizzano EF (2004) Detection of novel mutations in the SMN Tudor domain in type I SMA patients. Neurology 63(1):146–149
d’Errico P, Boido M, Piras A, Valsecchi V, De Amicis E, Locatelli D, Capra S, Vagni F, Vercelli A, Battaglia G (2013) Selective vulnerability of spinal and cortical motor neuron subpopulations in delta7 SMA mice. PLoS One 8(12):e82654. doi:10.1371/journal.pone.0082654
Denegri M, Chiodi I, Corioni M, Cobianchi F, Riva S, Biamonti G (2001) Stress-induced nuclear bodies are sites of accumulation of pre-mRNA processing factors. Mol Biol Cell 12(11):3502–3514
Doktor TK, Hua Y, Andersen HS, Brøner S, Liu YH, Wieckowska A, Dembic M, Bruun GH, Krainer AR, Andresen BS (2017) RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns. Nucleic Acids Res 45(1):395–416. doi:10.1093/nar/gkw731
Donlin-Asp PG, Bassell GJ, Rossoll W (2016) A role for the survival of motor neuron protein in mRNP assembly and transport. Curr Opin Neurobiol 39:53–61. doi:10.1016/j.conb.2016.04.004
Eggert C, Chari A, Laggerbauer B, Fischer U (2006) Spinal muscular atrophy: the RNP connection. Trends Mol Med 12(3):113–121. doi:10.1016/j.molmed.2006.01.005
Fallini C, Zhang H, Su Y, Silani V, Singer RH, Rossoll W, Bassell GJ (2011) The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(a) mRNA in primary motor neuron axons. J Neurosci 31(10):3914–3925. doi:10.1523/JNEUROSCI.3631-10.2011
Fallini C, Bassell GJ, Rossoll W (2012) Spinal muscular atrophy: the role of SMN in axonal mRNA regulation. Brain Res 1462:81–92. doi:10.1016/j.brainres.2012.01.044
Fallini C, Rouanet JP, Donlin-Asp PG, Guo P, Zhang H, Singer RH, Rossoll W, Bassell GJ (2014) Dynamics of survival of motor neuron (SMN) protein interaction with the mRNA-binding protein IMP1 facilitates its trafficking into motor neuron axons. Dev Neurobiol 74(3):319–332. doi:10.1002/dneu.22111
Fallini C, Donlin-Asp PG, Rouanet JP, Bassell GJ, Rossoll W (2016) Deficiency of the survival of motor neuron protein impairs mRNA localization and local translation in the growth cone of motor neurons. J Neurosci 36(13):3811–3820. doi:10.1523/JNEUROSCI.2396-15.2016
Farooq F, Balabanian S, Liu X, Holcik M, MacKenzie A (2009) P38 mitogen-activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR. Hum Mol Genet 18(21):4035–4045. doi:10.1093/hmg/ddp352
Finkel RS (2013) Electrophysiological and motor function scale association in a pre-symptomatic infant with spinal muscular atrophy type I. Neuromuscul Disord NMD 23(2):112–115. doi:10.1016/j.nmd.2012.09.006
Fischer U, Liu Q, Dreyfuss G (1997) The SMN–SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 90(6):1023–1029
Gabanella F, Butchbach MER, Saieva L, Carissimi C, Burghes AHM, Pellizzoni L (2007) Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One 2(9):e921. doi:10.1371/journal.pone.0000921
Garcia EL, Lu Z, Meers MP, Praveen K, Matera AG (2013) Developmental arrest of drosophila survival motor neuron (Smn) mutants accounts for differences in expression of minor intron-containing genes. RNA 19(11):1510–1516. doi:10.1261/rna.038919.113
Gerstein MB, Rozowsky J, Yan K-K, Wang D, Cheng C, Brown JB, Davis CA et al (2014) Comparative analysis of the transcriptome across distant species. Nature 512(7515):445–448. doi:10.1038/nature13424
Gonsalvez GB, Rajendra TK, Wen Y, Praveen K, Matera AG (2010) Sm proteins specify germ cell fate by facilitating oskar mRNA localization. Development 137(14):2341–2351. doi:10.1242/dev.042721
Gubitz AK, Feng W, Dreyfuss G (2004) The SMN complex. Exp Cell Res 296(1):51–56. doi:10.1016/j.yexcr.2004.03.022
Guil S, Long JC, Cáceres JF (2006) hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol Cell Biol 26(15):5744–5758. doi:10.1128/MCB.00224-06
Hahnen E, Schönling J, Rudnik-Schöneborn S, Raschke H, Zerres K, Wirth B (1997) Missense mutations in exon 6 of the survival motor neuron gene in patients with spinal muscular atrophy (SMA). Hum Mol Genet 6(5):821–825
Hao LT, Duy PQ, Jontes JD, Wolman M, Granato M, Beattie CE (2013) Temporal requirement for SMN in motoneuron development. Hum Mol Genet 22(13):2612–2625. doi:10.1093/hmg/ddt110
Hao LT, Duy PQ, Jontes JD, Beattie CE (2015) Motoneuron development influences dorsal root ganglia survival and Schwann cell development in a vertebrate model of spinal muscular atrophy. Hum Mol Genet 24(2):346–360. doi:10.1093/hmg/ddu447
Harada Y, Sutomo R, Sadewa AH, Akutsu T, Takeshima Y, Wada H, Matsuo M, Nishio H (2002) Correlation between SMN2 copy number and clinical phenotype of spinal muscular atrophy: three SMN2 copies fail to rescue some patients from the disease severity. J Neurol 249(9):1211–1219. doi:10.1007/s00415-002-0811-4
Hofmann Y, Wirth B (2002) hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-Beta1. Hum Mol Genet 11(17):2037–2049
Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B (2000) Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc Natl Acad Sci USA 97(17):9618–9623. doi:10.1073/pnas.160181697
Howell MD, Ottesen EW, Singh NN, Anderson RL, Singh RN (2017) Gender-specific amelioration of SMA phenotype upon disruption of a deep intronic structure by an oligonucleotide. Mol Ther 25(6):1328–1341. doi:10.1016/j.ymthe.2017.03.036
Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H (2000) A mouse model for spinal muscular atrophy. Nat Genet 24(1):66–70
Hua Y, Zhou J (2004) Survival motor neuron protein facilitates assembly of stress granules. FEBS Lett 572(1–3):69–74
Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR (2008) Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet 82(4):834–848. doi:10.1016/j.ajhg.2008.01.014
Hubers L, Valderrama-Carvajal H, Laframboise J, Timbers J, Sanchez G, Côté J (2011) HuD interacts with survival motor neuron protein and can rescue spinal muscular atrophy-like neuronal defects. Hum Mol Genet 20(3):553–579. doi:10.1093/hmg/ddq500
Imlach WL, Beck ES, Choi BJ, Lotti F, Pellizzoni L, McCabe BD (2012) SMN is required for sensory-motor circuit function in Drosophila. Cell 151(2):427–439. doi:10.1016/j.cell.2012.09.011
Jangi M, Fleet C, Cullen P, Gupta SV, Mekhoubad S, Chiao E, Allaire N et al (2017) SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc Natl Acad Sci USA 114(12):E2347–E2356. doi:10.1073/pnas.1613181114
Jo OD, Martin J, Bernath A, Masri J, Lichtenstein A, Gera J (2008) Heterogeneous nuclear ribonucleoprotein A1 regulates cyclin D1 and C-Myc internal ribosome entry site function through Akt signaling. J Biol Chem 283(34):23274–23287. doi:10.1074/jbc.M801185200
Kashima T, Manley JL (2003) A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet 34(4):460–463. doi:10.1038/ng1207
Kim GH, Kim JE, Rhie SJ, Yoon S (2015) The role of oxidative stress in neurodegenerative diseases. Exp Neurobiol 24(4):325–340. doi:10.5607/en.2015.24.4.325
Kleinschmidt AM, Patton JR, Pederson T (1989) U2 small nuclear RNP assembly in vitro. Nucleic Acids Res 17(12):4817–4828
Kotani T, Sutomo R, Sasongko TH, Sadewa AH, Gunadi, Minato T, Fujii E et al (2007) A novel mutation at the N-terminal of SMN tudor domain inhibits its interaction with target proteins. J Neurol 254(5):624–630. doi:10.1007/s00415-006-0410-x
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323(5918):1205–1208. doi:10.1126/science.1166066
Labunskyy VM, Hatfield DL, Gladyshev VN (2014) Selenoproteins: molecular pathways and physiological roles. Physiol Rev 94(3):739–777. doi:10.1152/physrev.00039.2013
Lai MC, Lin RI, Tarn WY (2001) Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc Natl Acad Sci USA 98(18):10154–10159. doi:10.1073/pnas.181354098
Le TT, Pham LT, Butchbach MER, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AHM (2005) SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet 14(6):845–857. doi:10.1093/hmg/ddi078
Le TT, McGovern VL, Alwine IE, Wang X, Massoni-Laporte A, Rich MM, Burghes AHM (2011) Temporal requirement for high SMN expression in SMA mice. Hum Mol Genet 20(18):3578–3591. doi:10.1093/hmg/ddr275
Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80(1):155–165
Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J (1997) Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16(3):265–269. doi:10.1038/ng0797-265
Liu Q, Fischer U, Wang F, Dreyfuss G (1997) The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 90(6):1013–1021
Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, Petrucelli L, Wolozin B (2010) Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5(10):e13250. doi:10.1371/journal.pone.0013250
Lorson CL, Strasswimmer J, Yao JM, Baleja JD, Hahnen E, Wirth B, Le T, Burghes AH, Androphy EJ (1998) SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet 19(1):63–66. doi:10.1038/ng0598-63
Lorson CL, Hahnen E, Androphy EJ, Wirth B (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA 96(11):6307–6311
Lotti F, Imlach WL, Saieva L, Beck ES, Hao LT, Li DK, Jiao W et al (2012) An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151(2):440–454. doi:10.1016/j.cell.2012.09.012
Lukong KE, Larocque D, Tyner AL, Richard S (2005) Tyrosine phosphorylation of Sam68 by breast tumor kinase regulates intranuclear localization and cell cycle progression. J Biol Chem 280(46):38639–38647. doi:10.1074/jbc.M505802200
Lutz CM, Kariya S, Patruni S, Osborne MA, Liu D, Henderson CE, Li DK et al (2011) Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J Clin Investig 121(8):3029–3041. doi:10.1172/JCI57291
Mahboubi H, Stochaj U (2017) Cytoplasmic stress granules: dynamic modulators of cell signaling and disease. Biochim Biophys Acta 1863(4):884–895. doi:10.1016/j.bbadis.2016.12.022
Matter N, Herrlich P, König H (2002) Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 420(6916):691–695. doi:10.1038/nature01153
McAndrew PE, Parsons DW, Simard LR, Rochette C, Ray PN, Mendell JR, Prior TW, Burghes AH (1997) Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet 60(6):1411–1422. doi:10.1086/515465
McWhorter ML, Monani UR, Burghes AHM, Beattie CE (2003) Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol 162(5):919–931. doi:10.1083/jcb.200303168
Meister G, Fischer U (2002) Assisted RNP assembly: SMN and PRMT5 complexes cooperate in the formation of spliceosomal UsnRNPs. EMBO J 21(21):5853–5863. doi:10.1093/emboj/cdf585
Meister G, Bühler D, Laggerbauer B, Zobawa M, Lottspeich F, Fischer U (2000) Characterization of a nuclear 20S complex containing the survival of motor neurons (SMN) protein and a specific subset of spliceosomal Sm proteins. Hum Mol Genet 9(13):1977–1986
Meister G, Bühler D, Pillai R, Lottspeich F, Fischer U (2001) A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat Cell Biol 3(11):945–949. doi:10.1038/ncb1101-945
Miller N, Shi H, Zelikovich AS, Ma YC (2016) Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum Mol Genet 25(16):3395–3406. doi:10.1093/hmg/ddw262
Misteli T, Cáceres JF, Clement JQ, Krainer AR, Wilkinson MF, Spector DL (1998) Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo. J Cell Biol 143(2):297–307
Monani UR (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 8(7):1177–1183. doi:10.1093/hmg/8.7.1177
Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S et al (2000) The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet 9(3):333–339
Monani UR, Pastore MT, Gavrilina TO, Jablonka S, Le TT, Andreassi C, DiCocco JM et al (2003) A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol 160(1):41–52. doi:10.1083/jcb.200208079
Murray LM, Beauvais A, Gibeault S, Courtney NL, Kothary R (2015) Transcriptional profiling of differentially vulnerable motor neurons at pre-symptomatic stage in the Smn (2b/−) mouse model of spinal muscular atrophy. Acta Neuropathol Commun 3(1):55. doi:10.1186/s40478-015-0231-1
Nash LA, Burns JK, Chardon JW, Kothary R, Parks RJ (2016) Spinal muscular atrophy: more than a disease of motor neurons? Curr Mol Med 16(9):779–792. doi:10.2174/1566524016666161128113338
Neuenkirchen N, Englbrecht C, Ohmer J, Ziegenhals T, Chari A, Fischer U (2015) Reconstitution of the human U snRNP assembly machinery reveals stepwise Sm protein organization. EMBO J 34(14):1925–1941. doi:10.15252/embj.201490350
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133
Niedzielska E, Smaga I, Gawlik M, Moniczewski A, Stankowicz P, Pera J, Filip M (2016) Oxidative stress in neurodegenerative diseases. Mol Neurobiol 53(6):4094–4125. doi:10.1007/s12035-015-9337-5
Nilsen TW, Graveley BR (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature 463(7280):457–463. doi:10.1038/nature08909
Nobutoki T, Ihara T (2015) Early disruption of neurovascular units and microcirculatory dysfunction in the spinal cord in spinal muscular atrophy type I. Med Hypotheses 85(6):842–845. doi:10.1016/j.mehy.2015.09.028
Obata T, Brown GE, Yaffe MB (2000) MAP kinase pathways activated by stress: the P38 MAPK pathway. Crit Care Med 28(4 Suppl):N67–N77
Ogawa C, Usui K, Aoki M, Ito F, Itoh M, Kai C, Kanamori-Katayama M, Hayashizaki Y, Suzuki H (2007) Gemin2 plays an important role in stabilizing the survival of motor neuron complex. J Biol Chem 282(15):11122–11134. doi:10.1074/jbc.M609297200
Ogawa C, Usui K, Ito F, Itoh M, Hayashizaki Y, Suzuki H (2009) Role of survival motor neuron complex components in small nuclear ribonucleoprotein assembly. J Biol Chem 284(21):14609–14617. doi:10.1074/jbc.M809031200
Otter S, Grimmler M, Neuenkirchen N, Chari A, Sickmann A, Fischer U (2007) A comprehensive interaction map of the human survival of motor neuron (SMN) complex. J Biol Chem 282(8):5825–5833. doi:10.1074/jbc.M608528200
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40(12):1413–1415. doi:10.1038/ng.259
Panas MD, Ivanov P, Anderson P (2016) Mechanistic insights into mammalian stress granule dynamics. J Cell Biol 215(3):313–323
Papp LV, Holmgren A, Khanna KK (2010) Selenium and selenoproteins in health and disease. Antioxid Redox Signal 12(7):793–795. doi:10.1089/ars.2009.2973
Parker SJ, Meyerowitz J, James JL, Liddell JR, Crouch PJ, Kanninen KM, White AR (2012) Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem Int 60(4):415–424. doi:10.1016/j.neuint.2012.01.019
Paronetto MP, Zalfa F, Botti F, Geremia R, Bagni C, Sette C (2006) The nuclear RNA-binding protein Sam68 translocates to the cytoplasm and associates with the polysomes in mouse spermatocytes. Mol Biol Cell 17(1):14–24. doi:10.1091/mbc.E05-06-0548
Parsons DW, McAndrew PE, Iannaccone ST, Mendell JR, Burghes AH, Prior TW (1998) Intragenic telSMN mutations: frequency, distribution, evidence of a founder effect, and modification of the spinal muscular atrophy phenotype by cenSMN copy number. Am J Hum Genet 63(6):1712–1723. doi:10.1086/302160
Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, Hua Y et al (2011) Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med 3(72):72ra18. doi:10.1126/scitranslmed.3001777
Patitucci TN, Ebert AD (2016) SMN deficiency does not induce oxidative stress in SMA iPSC-derived astrocytes or motor neurons. Hum Mol Genet 25(3):514–523. doi:10.1093/hmg/ddv489
Pedrotti S, Bielli P, Paronetto MP, Ciccosanti F, Fimia GM, Stamm S, Manley JL, Sette C (2010) The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J 29(7):1235–1247. doi:10.1038/emboj.2010.19
Pellizzoni L (2007) Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep 8(4):340–345. doi:10.1038/sj.embor.7400941
Pellizzoni L, Charroux B, Dreyfuss G (1999) SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc Natl Acad Sci USA 96(20):11167–11172
Pellizzoni L, Yong J, Dreyfuss G (2002) Essential role for the SMN complex in the specificity of snRNP assembly. Science 298(5599):1775–1779. doi:10.1126/science.1074962
Perera ND, Sheean RK, Crouch PJ, White AR, Horne MK, Turner BJ (2016) Enhancing survival motor neuron expression extends lifespan and attenuates neurodegeneration in mutant TDP-43 mice. Hum Mol Genet 25(18):4080–4093. doi:10.1093/hmg/ddw247
Praveen K, Wen Y, Matera AG (2012) A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. Cell Rep 1(6):624–631. doi:10.1016/j.celrep.2012.05.014
Prior TW (2007) Spinal muscular atrophy diagnostics. J Child Neurol 22(8):952–956. doi:10.1177/0883073807305668
Prior TW, Krainer AR, Hua Y, Swoboda KJ, Snyder PC, Bridgeman SJ, Burghes AHM, Kissel JT (2009) A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet 85(3):408–413. doi:10.1016/j.ajhg.2009.08.002
Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, Tang AA, Oldham MC, Wang H, Shorter J, Filiano AJ, Roberson ED, Tourtellotte WG, Chen B, Tsai LH, Huang EJ (2014) ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Investig 124(3):981–999. doi:10.1172/JCI72723
Rajendra TK, Gonsalvez GB, Walker MP, Shpargel KB, Salz HK, Matera AG (2007) A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J Cell Biol 176(6):831–841. doi:10.1083/jcb.200610053
Raker VA, Plessel G, Lührmann R (1996) The snRNP core assembly pathway: identification of stable core protein heteromeric complexes and an snRNP subcore particle in vitro. EMBO J 15(9):2256–2269
Raker VA, Hartmuth K, Kastner B, Lührmann R (1999) Spliceosomal U snRNP core assembly: Sm proteins assemble onto an Sm site RNA nonanucleotide in a specific and thermodynamically stable manner. Mol Cell Biol 19(10):6554–6565
Ratti A, Buratti E (2016) Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem 138(Suppl 1):95–111. doi:10.1111/jnc.13625
Reber S, Stettler J, Filosa G, Colombo M, Jutzi D, Lenzken SC, Schweingruber C, Bruggmann R, Bachi A, Barabino SM, Mühlemann O, Ruepp MD (2016) Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. EMBO J 35(14):1504–1521. doi:10.15252/embj.201593791
Richert JR, Antel JP, Canary JJ, Maxted WC, Groothuis D (1986) Adult onset spinal muscular atrophy with atrophic testes: report of two cases. J Neurol Neurosurg Psychiatry 49(5):606–608
Riessland M, Ackermann B, Förster A, Jakubik M, Hauke J, Garbes L, FritzscheI Mende Y, Blumcke I, Hahnen E, Wirth B (2010) SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet 19(8):1492–1506. doi:10.1093/hmg/ddq023
Robbins KL, Glascock JJ, Osman EY, Miller MR, Lorson CL (2014) Defining the therapeutic window in a severe animal model of spinal muscular atrophy. Hum Mol Genet 23(17):4559–4568. doi:10.1093/hmg/ddu169
Rochette CF, Surh LC, Ray PN, McAndrew PE, Prior TW, Burghes AH, Vanasse M, Simard LR (1997) Molecular diagnosis of non-deletion SMA patients using quantitative PCR of SMN exon 7. Neurogenetics 1(2):141–147
Rossoll W, Jablonka S, Andreassi C, Kröning A-K, Karle K, Monani UR, Sendtner M (2003) Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol 163(4):801–812. doi:10.1083/jcb.200304128
Sahashi K, Hua Y, Ling KKY, Hung G, Rigo F, Horev G, Katsuno M et al (2012) TSUNAMI: an antisense method to phenocopy splicing-associated diseases in animals. Genes Dev 26(16):1874–1884. doi:10.1101/gad.197418.112
Saltzman AL, Pan Q, Blencowe BJ (2011) Regulation of alternative splicing by the core spliceosomal machinery. Genes Dev 25(4):373–384. doi:10.1101/gad.2004811
Sanchez G, Dury AY, Murray LM, Biondi O, Tadesse H, El Fatimy R, Kothary R, Charbonnier F, Khandjian EW, Côté J (2013) A novel function for the survival motoneuron protein as a translational regulator. Hum Mol Genet 22(4):668–684. doi:10.1093/hmg/dds474
Saxena S, Caroni P (2011) Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71(1):35–48. doi:10.1016/j.neuron.2011.06.031
Schrank B, Götz R, Gunnersen JM, Ure JM, Toyka KV, Smith AG, Sendtner M (1997) Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA 94(18):9920–9925
Scotti MM, Swanson MS (2016) RNA mis-splicing in disease. Nat Rev Genet 17(1):19–32. doi:10.1038/nrg.2015.3
Seng CO, Magee C, Young PJ, Lorson CL, Allen JP (2015) The SMN structure reveals its crucial role in snRNP assembly. Hum Mol Genet 24(8):2138–2146. doi:10.1093/hmg/ddu734
Seo J, Singh NN, Ottesen EW, Sivanesan S, Shishimorova M, Singh RN (2016) Oxidative stress triggers body-wide skipping of multiple exons of the spinal muscular atrophy gene. PLoS One 11(4):e0154390. doi:10.1371/journal.pone.0154390
Shababi M, Lorson CL, Rudnik-Schöneborn SS (2014) Spinal muscular atrophy: a motor neuron disorder or a multi-organ disease? J Anat 224(1):15–28. doi:10.1111/joa.12083
Sharma A, Lambrechts A, Hao LT, Le TT, Sewry CA, Ampe C, Burghes AHM, Morris GE (2005) A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp Cell Res 309(1):185–197. doi:10.1016/j.yexcr.2005.05.014
Shpargel KB, Matera AG (2005) Gemin proteins are required for efficient assembly of Sm-class ribonucleoproteins. Proc Natl Acad Sci USA 102(48):17372–17377. doi:10.1073/pnas.0508947102
Singh NK, Singh NN, Androphy EJ, Singh RN (2006a) Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol 26(4):1333–1346. doi:10.1128/MCB.26.4.1333-1346.2006
Singh NK, Singh NN, Androphy EJ, Singh RN (2006b) Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol 26(4):1333–1346. doi:10.1128/MCB.26.4.1333-1346.2006
Singh NN, Lee BM, Singh RN (2015) Splicing regulation in spinal muscular atrophy by an RNA structure formed by long-distance interactions. Ann NY Acad Sci 1341:176–187. doi:10.1111/nyas.12727
Singh RN, Howell MD, Ottesen EW, Singh NN (2017a) Diverse role of survival motor neuron protein. Biochim Biophys Acta 1860(3):299–315. doi:10.1016/j.bbagrm.2016.12.008
Singh NN, Howell MD, Singh RN (2017b) Transcriptional and splicing regulation of spinal muscular atrophy genes. In: Sumner CJ, Paushkin S, Ko C-P (eds) Spinal muscular atrophy. Academic, Cambridge, Massachusetts, pp 75–97
Somers E, Stencel Z, Wishart TM, Gillingwater TH, Parson SH (2012) Density, calibre and ramification of muscle capillaries are altered in a mouse model of severe spinal muscular atrophy. Neuromuscul Disord NMD 22(5):435–442. doi:10.1016/j.nmd.2011.10.021
Somers E, Lees RD, Hoban K, Sleigh JN, Zhou H, Muntoni F, Talbot K, Gillingwater TH, Parson SH (2016) Vascular defects and spinal cord hypoxia in spinal muscular atrophy. Ann Neurol 79(2):217–230. doi:10.1002/ana.24549
Speert DP (2006) Bacterial infections of the lung in normal and immunodeficient patients. Novartis Found Symp 279:42–51 (disussion 51–5–216–9)
Stamm S (2008) Regulation of alternative splicing by reversible protein phosphorylation. J Biol Chem 283(3):1223–1227. doi:10.1074/jbc.R700034200
Stoilov P, Daoud R, Nayler O, Stamm S (2004) Human Tra2-Beta1 autoregulates its protein concentration by influencing alternative splicing of its pre-mRNA. Hum Mol Genet 13(5):509–524. doi:10.1093/hmg/ddh051
Sumpter V, Kahrs A, Fischer U, Kornstädt U, Lührmann R (1992) In vitro reconstitution of U1 and U2 snRNPs from isolated proteins and snRNA. Mol Biol Rep 16(4):229–240
Sun Y, Grimmler M, Schwarzer V, Schoenen F, Fischer U, Wirth B (2005) Molecular and functional analysis of intragenic SMN1 mutations in patients with spinal muscular atrophy. Hum Mutat 25(1):64–71. doi:10.1002/humu.20111
Sun S, Ling SC, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, Li H, Qiu H, Bui A, Yeo GW, Huang EJ, Eggan K, Zhou H, Fu XD, Lagier-Tourenne C, Cleveland DW (2015) ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun 6:6171. doi:10.1038/ncomms7171
Talbot K, Ponting CP, Theodosiou AM, Rodrigues NR, Surtees R, Mountford R, Davies KE (1997) Missense mutation clustering in the survival motor neuron gene: a role for a conserved tyrosine and glycine rich region of the protein in RNA metabolism? Hum Mol Genet 6(3):497–500
Temsamani J, Rhoadhouse M, Pederson T (1991) The U2 small nuclear ribonucleoprotein particle associates with nuclear factors in a pre-mRNA independent reaction. J Biol Chem 266(30):20356–20362
Tsai L-K, Chen C-L, Tsai Y-C, Ting C-H, Chien Y-H, Lee N-C, Hwu W-L (2016) Hypothermia improves disease manifestations in SMA mice via SMN augmentation. Hum Mol Genet 25(4):631–641. doi:10.1093/hmg/ddv500
Tsalikis J, Tattoli I, Ling A, Sorbara MT, Croitoru DO, Philpott DJ, Girardin SE (2015) Intracellular bacterial pathogens trigger the formation of U small nuclear RNA bodies (U bodies) through metabolic stress induction. J Biol Chem 290(34):20904–20918. doi:10.1074/jbc.M115.659466
Turner BJ, Parkinson NJ, Davies KE, Talbot K (2009) Survival motor neuron deficiency enhances progression in an amyotrophic lateral sclerosis mouse model. Neurobiol Dis 34(3):511–517. doi:10.1016/j.nbd.2009.03.005
Turunen JJ, Niemelä EH, Verma B, Frilander MJ (2013) The significant other: splicing by the minor spliceosome. Wiley Interdiscip Rev RNA 4(1):61–76. doi:10.1002/wrna.1141
van der Houven van Oordt W, Diaz-Meco MT, Lozano J, Krainer AR, Moscat J, Cáceres JF (2000) The MKK(3/6)-P38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J Cell Biol 149(2):307–316
Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211. doi:10.1126/science.1165942
Veldink JH, Kalmijn S, Van der Hout AH, Lemmink HH, Groeneveld GJ, Lummen C, Scheffer H, Wokke JH, Van den Berg LH (2005) SMN genotypes producing less SMN protein increase susceptibility to and severity of sporadic ALS. Neurology 65(6):820–825
Vezain M, Saugier-Veber P, Goina E, Touraine R, Manel V, Toutain A, Fehrenbach S et al (2010) A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum Mutat 31(1):E1110–E1125. doi:10.1002/humu.21173
Wan L, Battle DJ, Yong J, Gubitz AK, Kolb SJ, Wang J, Dreyfuss G (2005) The survival of motor neurons protein determines the capacity for snRNP assembly: biochemical deficiency in spinal muscular atrophy. Mol Cell Biol 25(13):5543–5551. doi:10.1128/MCB.25.13.5543-5551.2005
Wan L, Ottinger E, Cho S, Dreyfuss G (2008) Inactivation of the SMN complex by oxidative stress. Mol Cell 31(2):244–254. doi:10.1016/j.molcel.2008.06.004
Wang CH, Papendick BD, Bruinsma P, Day JK (1998) Identification of a novel missense mutation of the SMN(T) gene in two siblings with spinal muscular atrophy. Neurogenetics 1(4):273–276
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456(7221):470–476. doi:10.1038/nature07509
Wang ZB, Zhang X, Li XJ (2013) Recapitulation of spinal motor neuron-specific disease phenotypes in a human cell model of spinal muscular atrophy. Cell Res 23(3):378–393. doi:10.1038/cr.2012.166
Winkler C, Eggert C, Gradl D, Meister G, Giegerich M, Wedlich D, Laggerbauer B, Fischer U (2005) Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev 19(19):2320–2330. doi:10.1101/gad.342005
Workman E, Saieva L, Carrel TL, Crawford TO, Liu D, Lutz C, Beattie CE, Pellizzoni L, Burghes AHM (2009) A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum Mol Genet 18(12):2215–2229. doi:10.1093/hmg/ddp157
Xu CC, Denton KR, Wang ZB, Zhang X, Li XJ (2016) Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis Model Mech 9(1):39–49. doi:10.1242/dmm.021766
Yamazaki T, Chen S, Yu Y, Yan B, Haertlein TC, Carrasco MA, Tapia JC, Zhai B, Das R, Lalancette-Hebert M, Sharma A, Chandran S, Sullivan G, Nishimura AL, Shaw CE, Gygi SP, Shneider NA, Maniatis T, Reed R (2012) FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep 2(4):799–806. doi:10.1016/j.celrep.2012.08.025
Yasuda K, Clatterbuck-Soper SF, Jackrel ME, Shorter J, Mili S (2017) FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J Cell Biol 216(4):1015–1034. doi:10.1083/jcb.201608022
Young PJ, Didonato CJ, Hu D, Kothary R, Androphy EJ, Lorson CL (2002) SRp30c-dependent stimulation of survival motor neuron (SMN) exon 7 inclusion is facilitated by a direct interaction with hTra2 beta 1. Hum Mol Genet 11(5):577–587
Zhang H, Xing L, Rossoll W, Wichterle H, Singer RH, Bassell GJ (2006) Multiprotein complexes of the survival of motor neuron protein SMN with Gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci 26(33):8622–8632. doi:10.1523/JNEUROSCI.3967-05.2006
Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G (2008) SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133(4):585–600. doi:10.1016/j.cell.2008.03.031
Zou T, Yang X, Pan D, Huang J, Sahin M, Zhou J (2011) SMN deficiency reduces cellular ability to form stress granules, sensitizing cells to stress. Cell Mol Neurobiol 31(4):541–550. doi:10.1007/s10571-011-9647-8
Acknowledgements
Work in the authors’ lab has been funded by National Institute of Neurological Diseases and Stroke (R21-NS084187 to DSC and F31-NS079032 to CED). The authors would like to thank Ashlee Smith for topic discussions, critical reading of the manuscript draft and help with reference management during the writing process.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Dominguez, C.E., Cunningham, D. & Chandler, D.S. SMN regulation in SMA and in response to stress: new paradigms and therapeutic possibilities. Hum Genet 136, 1173–1191 (2017). https://doi.org/10.1007/s00439-017-1835-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-017-1835-2