
Abstract
Diseases related to smoking are the second leading cause of death in the world. Cigarette smoking is a risk factor for several diseases such as cancer and cardiovascular and respiratory disorders. Despite increasing evidence of genetic determination, the susceptibility genes and loci underlying various aspects of smoking behavior are largely unknown. Moreover, almost all reported genome-wide association studies (GWASs) have been performed on samples of European origin, limiting the applicability of the results to other ethnic populations. In this first GWAS on smoking behavior in an Asian population, after analyzing 8,842 DNA samples from the Korea Association Resource project with 352,228 single nucleotide polymorphisms (SNPs) genotyped for each sample, we identified 8 SNPs significantly associated with smoking initiation (SI) and 4 with nicotine dependence (ND). Because of the current unavailability of an independent Asian smoking sample, we replicated the discoveries in independent samples of European-American and African-American origin. Of the 12 SNPs examined in the replicated samples, we identified two SNPs, in the regulator of G-protein signaling 17 gene (rs7747583, p valuemeta = 6.40 × 10−6; rs2349433, p valuemeta = 5.57 × 10−6), associated with SI. Also, we found two SNPs significantly associated with ND; one in the FERM domain containing 4A (rs4424567, p valuemeta = 2.30 × 10−6) and the other at 7q31.1 (rs848353, p valuemeta = 9.16 × 10−8). These SNPs represent novel targets for examination of smoking behavior and warrant further investigation using independent samples.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cigarette smoking is one of the most significant risk factors for premature death, costing nearly six million human lives per year throughout the world (Warren et al. 2008). According to a recent report from the World Health Organization (WHO), smoking-related diseases rank as the second leading cause of death in the world, and it is expected that the prolonged pattern of current tobacco consumption will double the number of deaths annually by 2020 (WHO; http://www.who.int). Further, cigarette smoking is a well-known risk factor for cardiovascular and respiratory diseases (Giovino 2007; Muller and Wehbe 2008; Vianna et al. 2008).
Many twin studies in the US and other countries have consistently implied that genetic factors contribute to the risk of becoming a regular smoker. Initial evidence for genetic influences on nicotine dependence (ND) came from cross-sectional studies of twins that showed a mean heritability of 0.53 (range 0.28–0.84) for cigarette smoking (for reviews, see Carmelli et al. 1992; Hughes 1986). Our meta-analysis of the genetic parameter estimates for smoking initiation (SI) and ND based on 17 twin studies determined that the weighted mean heritability is 0.50 and 0.59, respectively, for all smokers (Li et al. 2003). Complex segregation analyses of smoking behavior in 493 three-generation families supported a dominant major gene effect with residual familial correlation (Cheng et al. 2000).
Although twin studies provide strong evidence for involvement of genetic factors in various stages of smoking behavior, identifying these susceptibility genes has been a challenge. During the past few years, a significant number of genome-wide association studies (GWAS) have been conducted for various smoking phenotypes, including SI and current smoking (Caporaso et al. 2009; TGC 2010; Thorgeirsson et al. 2010; Vink et al. 2009), age at smoking initiation (Siedlinski et al. 2011), ND (Bierut et al. 2007; Liu et al. 2009; Uhl et al. 2007), smoking quantity (Siedlinski et al. 2011; Berrettini et al. 2008; Caporaso et al. 2009; Liu et al. 2009, 2010; TGC 2010; Thorgeirsson et al. 2010, 2008), and smoking cessation (Siedlinski et al. 2011; TGC 2010; Uhl et al. 2008, 2010). However, most of these studies focused primarily on samples of European origin. Given the distinct differences in linkage disequilibrium (LD) patterns across ethnic populations (Li and Burmeister 2009), a GWAS in other ethnic samples is warranted.
In this study, we performed a GWAS on two smoking-related behaviors, SI and ND, in a cohort from the Korea Association Resource (KARE) project (Cho et al. 2009). A total of 8,842 individuals with 352,228 SNPs genotyped per individual were examined to find any association between smoking behavior and genetic factors. Our initial analysis at the discovery stage identified 12 SNPs (p < 1 × 10−5) significantly associated with smoking behavior. Although they did not reach the genome-wide significance level (p < 1 × 10−7), they represented several genomic regions possibly associated with smoking behavior. Among 12 SNPs, 8 were significantly associated with SI and 4 with ND.
Because of the unavailability of an independent Asian smoking sample, the replication study was performed in a sample of European-American or African-American origin. Of the 12 SNPs examined in independent samples, 4 were replicated. At the 5% significance level, two SNPs, rs7747583 and rs2349433, in RGS17 were replicated in association with SI, and rs4424567 in FRMD4A and rs848353 at 7q31.1 were replicated for association with ND. Finally, we performed meta-analysis on the association results from both the discovery and replication samples.
Materials and methods
Subjects
Discovery sample
All participants provided informed consent on a document that had been approved by all participating Institutional Review Boards. The relevant information has been described in a previous paper (Cho et al. 2009). Briefly, samples from the 10,038 participants in the Korea Association Resource (KARE) project were recruited from two areas, Ansung and Ansan, in South Korea. Participant ages ranged from 40 to 69 years. Two population based cohorts in this study were established as part of Korean Genome and Epidemiology Study (KoGES), which started in 2001, that provides extensive phenotype data over 260 traits through epidemiological surveys, physical examination and clinical examination.
Replication sample
The subjects used in our replication sample were from the Mid-South Tobacco Family (MSTF) study with either African-American (AA) or European-American (EA) origin. They were recruited primarily from Tennessee, Mississippi, and Arkansas during 1999–2004. Proband smokers were required to be at least 21 years of age, to have smoked for at least the last 5 years, and to have consumed an average of 20 cigarettes per day for the last 12 months. Once a proband was identified, siblings and biological parents were recruited full sibs per family. Detailed information of the recruitment and characteristics of study participants have been reported (Li 2008; Li et al. 2005, 2006; Ma et al. 2005). Together, a total of 200 EA and 402 AA families were recruited. Informed consent was obtained from all participants and approval from the appropriate Institutional Review Boards.
DNA extraction and genotyping
Discovery sample
Genomic DNA was isolated from peripheral blood. All DNA samples from the 10,004 participants were genotyped with the Affymetrix Genome-Wide Human SNP Array 5.0. Genotypes were called by Bayesian Robust Linear Modeling using the Mahalanobis Distance (BRLMM) algorithm. After removing samples with low call rates (n = 401), contamination (n = 11), sex inconsistencies (n = 41), cryptic relatedness (n = 608), or serious concomitant illness (n = 101), 8,842 samples remained for use. Similarly, we removed those SNPs with Hardy–Weinberg equilibrium (HWE) p values <10−6, minor allele frequencies >0.01, and genotype call rates >95%, leaving 352,228 SNPs. More detailed statistical analysis of the GWAS data can be found in a previous report (Cho et al. 2009). Considering the samples used for GWAS were recruited from different geographical locations of Korea, we used the magnitude of the genomic inflation factor and multidimensional scaling (MDS)/principal component analysis (PCA) plots to examining the presence of the population stratification, which revealed no evidence of this effect in the KARE samples (Cho et al. 2009).
Replication samples
The DNA was extracted from peripheral blood using a kit from Qiagen Inc. (Valencia, CA). On the basis of the GWAS results from the discovery sample, 12 SNPs were selected for testing in the replication samples. All SNPs were genotyped using the TaqMan SNP Genotyping Assay in a 384-well microplate format (Applied Biosystems, Foster, CA). Briefly, 15 ng of DNA was amplified in a total volume of 7 μl containing an MGB probe and 2.5 μl of TaqMan universal PCR master mix. Allelic discrimination analysis was performed on the ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster CA). To ensure the quality of the genotyping, SNP-specific control samples were added to each 384-well plate.
Definition of smoking-related phenotypes
Discovery sample
Definition of smoking-related phenotypes was essentially the same as in our previous study on the investigation of association of CHRNA5/A3/B4 with smoking in the same sample (Li et al. 2010). Briefly, on the basis of the survey questionnaire, from which information on smoking status (i.e., never smoker, former smoker who quit one year before being enrolled in the study, occasional smoker who smoke irregularly or only on specific occasions such as when drinking, or habitual smoker who smoke regularly on a daily basis) and number of cigarettes smoked per day (CPD) for the habitual smoker was drawn, three smoking-related phenotypes were defined for SI and ND. Regarding SI, the first measure (called SI-1) was defined as a binary trait comparing “never smoked” and “having regular smoking experiences (former, occasional, or habitual smoker),” and the second measure (called SI-2) was defined as an ordinal trait with four categories: never, former, light, and habitual. The ND phenotype was defined as an ordinal trait with five categories according to CPD: non-smoking, <10 CPD, 11–20 CPD, 21–30 CPD, and >31 CPD. Such assessment of ND not only has been commonly used in the literature but also is the most replicative in terms of positive findings across populations (Li 2008; Thorgeirsson et al. 2008). The detailed demographic information as well as smoking-related phenotype information is given in Table 1.
Replication sample
Smoking initiation was defined as the age at which the person began smoking regularly. The presence of ND was ascertained by the three measures most commonly used in the literature: CPD; the Heaviness of Smoking Index (HSI 0–6 scale), which includes CPD and smoking urgency (i.e., how soon after waking up does the subject smoke the first cigarette?); and the FTND (0–10 scale) (Heatherton et al. 1991). CPD in the MSTF sample is categorized CPD, as used in the KARE samples. A detailed description of the demographic and clinical characteristics of the MSTF sample is presented in Table 2.
Statistical analysis
Discovery sample
Statistical analyses were performed using PLINK (Purcell et al. 2007) and R software. A logistic regression model was fit for the binary phenotype, SI-1, adjusting for age, sex, and geographic area under additive, dominant, and recessive models. The cumulative logit model was fit to the ordinal phenotype SI-2 and ND (Bender and Grouven 1997).
To find supporting evidence for the 12 associated SNPs, we performed imputation analysis for untyped SNPs within ±500 kb of these SNPs. Imputation was carried out using the IMPUTE program on the basis of the NCBI (build 36) and dbSNP (build 126) databases. We initially used 90 individuals from JPT and CHB founders in HapMap as a reference panel comprising 3.99 million SNPs (release 22) (Marchini et al. 2007). We removed the imputed SNPs with <0.5 genotype information content, posterior probability scores <0.90, and low call rates (<0.90), MAF < 0.01, and HWE (p < 1 × 10−7).
Replication samples
The PedCheck program was used to determine genotyping consistency for Mendelian inheritance of all 11 SNPs selected for replication. To verify the quality of our genotyping, we also checked the SNP data for any significant departure from HWE. The HWE at each locus was assessed by the χ2 test. The allele frequencies for each genetic marker were calculated using the FREQ program of S.A.G.E. (v. 5.0). Associations between individual SNPs and smoking behavior phenotypes, including the ND measures and age at onset, were determined by the PBAT program using generalized estimating equations (Lange et al. 2003). Three genetic models (additive, dominant, and recessive) were tested, with sex and age as covariates in the AA and EA samples; sex, age, and ethnicity served as covariates for the combined sample. Relations with the three ND measures and age at onset were examined individually.
Meta-analysis
We conducted the meta-analysis using Fisher’s combining p value method (Fisher 1932) in order to combine the results from the discovery and replication samples. Considering that the PBAT approach used in the replication study provides only score statistics with effect directions and p values, we used an equal weight for both studied samples. Prior to conducting meta-analysis, we compared the risk allele of each SNP individually in the two samples and found that all SNPs selected for replication have the same risk allele for the discovery and replication samples. Any variant with discordant direction of effect was not considered as having been replicated.
Results
Description of discovery and replication samples in relation to smoking behaviors
Discovery sample
In total, 8,842 subjects with 352,228 SNPs each were analyzed. The average ages of the participants were 52.22 ± 8.91 (SD) years: 51.78 ± 8.78 for men and 52.61 ± 9.01 for women. Female participants were 52.7% (N = 4,659) of all subjects, however, only 4.93% were smokers, being categorized as former (1.34%), occasional (1.29%), or habitual (2.30%) smokers. In contrast, 80.62% of the male participants smoked. Among them, 31.05% were former smokers, 4.78% occasional smokers, and 44.79% habitual smokers. For habitual smokers, the average number of cigarettes smoked per day (CPD) was 19.51 ± 8.74 for male and 11.93 ± 7.28 for female smokers.
Replication sample
The average age of the subjects was 40.8 ± 14.8 years for EAs and 39.9 ± 14.6 years for AAs. The average family size was 3.16 ± 0.69 for EAs and 3.10 ± 0.75 for AAs. Of these families, 60.5% had at least one biological parent recruited, 50.4% had two full sibs, and 44.6% had three or more full sibs. If a family had no biological parents recruited, we had at least three full sibs. The average Fagerström test for ND (FTND) score was 6.4 ± 2.2 for EA and 6.3 ± 2.1 for AA smokers. The average CPD was 19.8 ± 13.6 for EA and 19.5 ± 13.0 for AA smokers.
GWAS results from the discovery sample
We conducted GWAS for both SI and ND using either total or male samples. We did not perform this analysis on the female sample alone, because there were so few female smokers in the KARE sample.
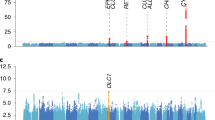
The analysis of the total sample identified 4 SNPs: 1 for SI-1, 1 for SI-2, and 2 for ND, with p values <10−5 (Table 3). The analysis of the male sample identified 10 SNPs: 1 for SI-1, 5 for SI-2, and 4 for ND with p values <10−5 (Table 3). Among the total 14 SNPs identified, two overlapped. For these 12 SNPs brought to replication, the quality of genotyping was investigated through cluster plots, which revealed no evidence of genotyping error. Figure 1 shows the chromosomal plots and quantile–quantile (QQ) plots of the association signals for three smoking-related phenotypes. The values of genomic inflation factor λ (ranging from 0.97 to 1.01) and QQ plot of signals showed no evidence of the presence of population stratification in our discovery sample.


Chromosomal and QQ plots for smoking behaviors. Statistical significance of each SNP is scaled as −log10 (p value). Red dotted line indicates threshold in Discovery sample (p < 10−5), and blue dotted line indicates p value 10−4. We plotted the observed −log10-scaled association results versus −log10-scaled expected association results in QQ plots. Chromosomal and QQ plots for ND phenotype (a), SI-1 phenotype (b), and SI-2 phenotype (c)
Smoking initiation
In the total sample, two SNPs, rs1442815 and rs11134474, located in the gene desert region on chromosomes 15q26 and 5q33, respectively, were associated with SI-1 and SI-2, respectively. Further analysis of the male sample revealed six more SNPs with SI-1 or SI-2 with p values <10−5: rs7761503, rs7747583, and rs2349433, located in RGS17 on chromosome 6; rs10511914 in DCAF12, located on chromosome 9; and rs1385706 and rs12534697, located in an intergenic region at 7q11.
Nicotine dependence
In the total sample, two SNPs, rs1404697 and rs848353, located on chromosome 7q31.1, were associated with ND, with p values <10−5. Further analysis of the male sample revealed four additional SNPs associated with ND with p values <10−5: rs1404697 and rs848353 on chromosome 7, rs12536963 in EXOC4 on chromosome 7, and rs4424567 in FRMD4A on chromosome 10. Two SNPs, rs1404697 and rs848353, were identified in both the total and male samples.
Association analysis results from replication samples
The association analysis of the total sample identified two SNPs for SI and two for ND, and that of the male sample identified six SNPs for SI and four for ND. Among the 14 SNPs, 12 unique ones were selected for the replication study: 8 for SI and 4 for ND.
The replication study was performed for the EA and AA samples separately. Among the eight SNPs for SI, two, rs7747583 and rs2349433 at RGS17, passed the replication significance threshold (p < 0.05) and were replicated in the EA male sample (Table 4). Among the four SNPs for ND, two passed the replication significance threshold: rs4424567 at FRMD4A was replicated in the EA male sample, and rs848353 at 7q31.1, close to the hypothetical protein-coding genes C7orf66 and LOC154907, was replicated in the AA total sample. One SNP, at the intronic region of EXOC4, could not be genotyped in the replication study.
Meta-analysis results of discovery and replication samples
Meta-analysis was performed for the selected 11 SNPs by combining the results from the discovery and replication samples. Three meta-analyses were performed: (1) KARE + EA, (2) KARE + AA, and (3) KARE + EA + AA. Among the 11 replicated SNPs, 10 provided concordant direction of genetic effects, and one (rs10511914) showed discordant direction of effect for SI. All four replicated SNPs provided concordant direction of effects. The results are summarized in Table 4. Through meta-analysis, only 1 SNP rs848353 reached genome-wide significance (p = 9.16 × 10−8). In general, the meta-analysis of KARE + AA decreased the p value for rs848353. For the remaining three SNPs tested, however, meta-analysis did not reduce the p values enough to reach a genome-wide significance level.
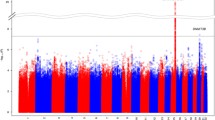
Figure 2 shows the regional association plots for three independent signals near rs4424567 at FRMD4A, rs848353 at 7q31.1, and rs2349433 (rs7747583) at RGS17. We found that two SNPs, rs7747583 and rs2349433 at RGS17, are in complete LD, with r 2 = 0.99. The regional association plots using both genotypes and imputed SNPs support our findings. As shown in Fig. 2a, SNP rs4424567 hovers in the intronic region of FRMD4A. Figure 2b shows that SNP rs848353 is part of a large cluster of associated SNPs near C7orf66. Figure 2c shows the dense cluster of associated SNPs, including the lead SNP rs2349433, in RGS17.

Regional plots of significantly associated SNPs. We show the locus ±500 kb around the lead associated SNP. Statistical significance of each SNP is scaled as −log10 (p value) along the chromosomal position (NCBI build 36). The lead SNP at each locus is shown in a purple diamond. Direct genotyping in SNP genotyping chip is in a circle and imputed SNP in a square. Pairwise correlation between the lead and other SNPs at locus is shown on a scale from minimum (blue) to maximum (red). Estimated recombination rates from HapMap are plotted in aqua. The box below the association signals contains gene information within each locus. The direction of each gene is shown as a left or right arrow, and exon is shown as a filled square, whereas intron is indicated as a line. The LD pattern is at the bottom of each regional plot. A dotted line indicates the ±50-kb boundary from the lead SNP. The black circle stands for the position of the lead associated SNP. We show the LD pattern drawn from HapMap data for the European (CEU), African (YRI), and Asian (CHB/JPT) populations. R 2 of SNPs is drawn on a scale from minimum (white) to maximum (red). Regional association plot for ND-associated SNPs, rs4424567 (a) and rs848353 (b) and SI-associated SNP, rs2349433 (c)
Discussion
This study represents the first large-scale GWAS on smoking-related behaviors, SI and ND, in an Asian population. Among 352,228 genotyped SNPs, our initial GWA analysis identified 12 (p < 1 × 10−5), among which 8 were significantly associated with SI and 4 with ND. In the replication study with American samples of both EA and AA origin, rs7747583 and rs2349433 in RGS17 were replicated in association with SI, and rs4424567 in FRMD4A and rs848353 at 7q31.1 were replicated in association with ND at the 5% significance level. In an additional meta-analysis for these variants, rs848353 reached genome-wide significance (p valuemeta = 9.16 × 10−8), whereas the other three did not.
Three replicated loci have some biologically plausible interpretation supported by previous studies. First, consider FERM domain containing 4A (FRMD4A), which is homologous in mouse EpH4 epithelial cells. This gene encodes a scaffolding protein controlling the activation of Arf6 by connecting Par-3, the Arf6 guanine-nucleotide exchange factor (GEF), and cytohesin-1 (Ikenouchi and Umeda 2010). Previously, a hypermethylation event was detected in FRMD4A in an analysis of DNA methylation of benzopyrene (BaP)-treated MCF-7 cells (Sadikovic et al. 2007). BaP is a common polycyclic aromatic hydrocarbon, an important class of environmental contaminants, that is also present in cigarette smoke. Moreover, Johnson et al. (2008) reported that FRMD4A was associated with substance dependence phenotypes defined by Diagnostic and Statistical Manual (DSM) diagnoses of substance abuse and FTND diagnoses of ND. FRMD4A is responsible for variation of the electrocardiographic RR interval (Marroni et al. 2009).
Second, we should consider the following genes near rs848353 at 7q31.1: C7orf66, LOC154907, and PNPLA8-THAP5-DNAJB9. Although the function of the hypothetical protein-coding genes C7orf66 and LOC154907 remains to be determined, some biological interpretation of PNPLA8-THAP5-DNAJB9 is plausible. Patatin-like phospholipase domain containing 8 (PNPLA8), also known as phospholipase A2, catalyzes cleavage of fatty acids from phospholipids. The encoded product of PNPLA8 regulates membrane physical properties and the release of lipid second messengers and growth factors. THAP5 is a THAP domain containing 5 isoform 2, and DNAJB9 is a DnaJ (Hsp40) homolog, subfamily B, member 9. Numerous previous association findings at 7q31.1 for various phenotypes, such as death among heart failure patients (Morrison et al. 2010), ulcerative colitis (Asano et al. 2009; Barrett et al. 2009; McGovern et al. 2010; Silverberg et al. 2009), cognitive performance (Need et al. 2009), attention deficit hyperactivity disorder (Anney et al. 2008; Lasky-Su et al. 2008), and serum metabolites (Gieger et al. 2008), may imply an important role for this locus in the etiology of these complex disorder/phenotypes.
Third, the encoded protein of RGS17 is a member of the regulator of G-protein signaling family. The gene product of DCAF12, DDB1, and CUL4 associated factor 12 interacts with the COP9 signalosome, a macromolecular complex that interacts with cullin-RING E3 ligases and regulates their activity by hydrolyzing cullin-Nedd8 conjugates. The locus 6q25.2 containing RGS17 was previously reported to be associated with major depressive and bipolar disorders (Ferreira et al. 2008; Liu et al. 2011), general cognitive ability (Davis et al. 2010), and tonometry (Levy et al. 2007).
Finally, a recent GWAS identified FRMD4A associated with substance dependence (Johnson et al. 2008). It is interesting that our result is consistent with that of Johnson et al., as we focused on ND, whereas Johnson et al. focused on substance dependence phenotypes assessed by DSM and FTND. However, Johnson et al. did not report any replication result. As far as we know, the present study is the first GWAS to show that FRMD4A is directly associated with ND with an independent replication.
Interestingly, our replication results appeared to be sex or population specific. For example, the SNPs at FRMD4A and RGS17 were replicated only in the male group of the EA sample. An SNP at 7q31.1 was replicated in the total AA sample but not the EA sample. The analysis result suggests that FRMD4A and RGS17 are associated with ND and SI in a sex-specific manner in conjunction with environmental and epigenetic factors. The complex interaction of genetic, environmental, and epigenetic factors may contribute to the disparity of the results for the total and male samples in the replication study. The discrepancy between the results of the EA and AA samples in the replication samples may be secondary to differences in genetic architecture across populations. For example, two ND-associated SNPs, rs1404697 and rs848353 at 7q31.1, are 4.0 kb apart. The two variants are highly correlated in the Korean population (r 2 = 0.98) and the European population (r 2 = 0.85, HapMap CEU). However, a low correlation was observed in an African population (r 2 = 0.013, HapMap YRI). As shown in Table 2, rs848353 and rs1404697 demonstrate a great difference in allele frequency in the AA sample (0.05 for rs1404697 and 0.32 for rs848353). Also note that rs848353 was replicated in the AA sample, whereas rs1404697 was not.
Of a note, we found that the mode of inheritance that provided the most significant association results also differed from population to population. This is probably attributable to the fact that genetic diversity among populations accounts for the replicated association under different genetic inheritance models in samples with different ancestry. For 7q31.1, the additive model provided the strongest signal in the discovery stage, whereas the dominant model provided the strongest signal in the AA replication sample. For RGS17, the additive model provided the strongest signal in the discovery sample, whereas the recessive model provided the strongest signal in the EA replication sample.
Because of the recruitment criteria used in the MSTF study, which differed from the KARE sample, there were only limited former smokers in the MSTF samples. Thus, we could not find an exactly same measure for both samples on smoking initiation phenotype. The age of onset in the MSTF sample is the age at which regular smoking was started, providing information on smoking initiation. Therefore, we used the age of onset phenotype in the replication study based on the available clinical information in MSTF. Also, the CPD in the MSTF sample is categorized CPD, as used in the KARE samples. In addition, we analyzed HSI and FTND for MSTF sample.
In this study, we used both ordinal phenotype (SI-2) and binary phenotype (SI-1) for smoking initiation. SI-2 is the modified SI-1 (never vs. others) considering the subcategories of “others.” Because the individuals in “former” group were those who smoked in the past but had stopped at the time of recruiting, we placed the “former smoker” between “never” and “occasional” smoker in the context of smoking initiation and current smoking interest. However, there would be an argument for the order of the ordinal phenotypes. Our previous candidate gene-based association study using the same ordinal phenotype definition has replicated the previously well-known smoking-related gene CHRAN5/A3/B5 (Li et al. 2010), which demonstrates the appropriateness of ordinal phenotype definition. In addition, the statistical test for checking the proportional odds assumption with respect to SNP variables showed that the proportionality assumption is reasonable.
For phenotype definition, of course, it would be great if we had biomarker information on those samples, which is impossible for the time being, given the assay cost associated with biomarkers and the unavailability of materials for many of those samples in the Korean population. As far as we know, however, smoking behaviors in almost all reported studies were based on questionnaires, with the number of cigarettes smoked per day being the most commonly used (Li 2008), and only limited studies used the biomarkers. Thus, further GWAS study on ND in an Asian sample with better assessment of smoking behavior, including biomarker information, is warranted.
References
Anney RJ, Lasky-Su J, O’Dushlaine C, Kenny E, Neale BM, Mulligan A, Franke B, Zhou K, Chen W, Christiansen H, Arias-Vasquez A, Banaschewski T, Buitelaar J, Ebstein R, Miranda A, Mulas F, Oades RD, Roeyers H, Rothenberger A, Sergeant J, Sonuga-Barke E, Steinhausen H, Asherson P, Faraone SV, Gill M (2008) Conduct disorder and ADHD: evaluation of conduct problems as a categorical and quantitative trait in the international multicentre ADHD genetics study. Am J Med Genet B Neuropsychiatr Genet 147B(8):1369–1378
Asano K, Matsushita T, Umeno J, Hosono N, Takahashi A, Kawaguchi T, Matsumoto T, Matsui T, Kakuta Y, Kinouchi Y, Shimosegawa T, Hosokawa M, Arimura Y, Shinomura Y, Kiyohara Y, Tsunoda T, Kamatani N, Iida M, Nakamura Y, Kubo M (2009) A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet 41(12):1325–1329
Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA, Phillips A, Wesley E, Parnell K, Zhang H, Drummond H, Nimmo ER, Massey D, Blaszczyk K, Elliott T, Cotterill L, Dallal H, Lobo AJ, Mowat C, Sanderson JD, Jewell DP, Newman WG, Edwards C, Ahmad T, Mansfield JC, Satsangi J, Parkes M, Mathew CG, Donnelly P, Peltonen L, Blackwell JM, Bramon E, Brown MA, Casas JP, Corvin A, Craddock N, Deloukas P, Duncanson A, Jankowski J, Markus HS, McCarthy MI, Palmer CN, Plomin R, Rautanen A, Sawcer SJ, Samani N, Trembath RC, Viswanathan AC, Wood N, Spencer CC, Bellenguez C, Davison D, Freeman C, Strange A, Langford C, Hunt SE, Edkins S, Gwilliam R, Blackburn H, Bumpstead SJ, Dronov S, Gillman M, Gray E, Hammond N, Jayakumar A, McCann OT, Liddle J, Perez ML, Potter SC, Ravindrarajah R, Ricketts M, Waller M, Weston P, Widaa S, Whittaker P, Attwood AP, Stephens J, Sambrook J, Ouwehand WH, McArdle WL, Ring SM, Strachan DP (2009) Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet 41(12):1330–1334
Bender R, Grouven U (1997) Ordinal logistic regression in medical research. J R Coll Physicians Lond 31(5):546–551
Berrettini W, Yuan X, Tozzi F, Song K, Francks C, Chilcoat H, Waterworth D, Muglia P, Mooser V (2008) Alpha-5/alpha-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Mol Psychiatry 13(4):368–373
Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF, Swan GE, Rutter J, Bertelsen S, Fox L, Fugman D, Goate AM, Hinrichs AL, Konvicka K, Martin NG, Montgomery GW, Saccone NL, Saccone SF, Wang JC, Chase GA, Rice JP, Ballinger DG (2007) Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet 16(1):24–35
Caporaso N, Gu F, Chatterjee N, Sheng-Chih J, Yu K, Yeager M, Chen C, Jacobs K, Wheeler W, Landi MT, Ziegler RG, Hunter DJ, Chanock S, Hankinson S, Kraft P, Bergen AW (2009) Genome-wide and candidate gene association study of cigarette smoking behaviors. PLoS ONE 4(2):e4653
Carmelli D, Swan GE, Robinette D, Fabsitz R (1992) Genetic influence on smoking—a study of male twins. N Engl J Med 327(12):829–833
Cheng LS, Swan GE, Carmelli D (2000) A genetic analysis of smoking behavior in family members of older adult males. Addiction 95(3):427–435
Cho YS, Go MJ, Kim YJ, Heo JY, Oh JH, Ban HJ, Yoon D, Lee MH, Kim DJ, Park M, Cha SH, Kim JW, Han BG, Min H, Ahn Y, Park MS, Han HR, Jang HY, Cho EY, Lee JE, Cho NH, Shin C, Park T, Park JW, Lee JK, Cardon L, Clarke G, McCarthy MI, Lee JY, Oh B, Kim HL (2009) A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet 41(5):527–534
Davis OS, Butcher LM, Docherty SJ, Meaburn EL, Curtis CJ, Simpson MA, Schalkwyk LC, Plomin R (2010) A three-stage genome-wide association study of general cognitive ability: hunting the small effects. Behav Genet 40(6):759–767
Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, Smoller JW, Grozeva D, Stone J, Nikolov I, Chambert K, Hamshere ML, Nimgaonkar VL, Moskvina V, Thase ME, Caesar S, Sachs GS, Franklin J, Gordon-Smith K, Ardlie KG, Gabriel SB, Fraser C, Blumenstiel B, Defelice M, Breen G, Gill M, Morris DW, Elkin A, Muir WJ, McGhee KA, Williamson R, MacIntyre DJ, MacLean AW, St CD, Robinson M, Van Beck M, Pereira AC, Kandaswamy R, McQuillin A, Collier DA, Bass NJ, Young AH, Lawrence J, Ferrier IN, Anjorin A, Farmer A, Curtis D, Scolnick EM, McGuffin P, Daly MJ, Corvin AP, Holmans PA, Blackwood DH, Gurling HM, Owen MJ, Purcell SM, Sklar P, Craddock N (2008) Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 40(9):1056–1058
Fisher RA (1932) Statistical methods for research workers, 4th edn. Oliver and Boyd, Edinburgh
Gieger C, Geistlinger L, Altmaier E, Hrabe de Angelis M, Kronenberg F, Meitinger T, Mewes HW, Wichmann HE, Weinberger KM, Adamski J, Illig T, Suhre K (2008) Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet 4(11):e1000282
Giovino GA (2007) The tobacco epidemic in the United States. Am J Prev Med 33(6 Suppl):S318–S326
Heatherton TF, Kozlowski LT, Frecker RC, Fagerstrom KO (1991) The Fagerstrom test for nicotine dependence: a revision of the Fagerstrom Tolerance Questionnaire. Br J Addict 86(9):1119–1127
Hughes JR (1986) Genetics of smoking: a brief review. Behav Ther 17:335–345
Ikenouchi J, Umeda M (2010) FRMD4A regulates epithelial polarity by connecting Arf6 activation with the PAR complex. Proc Natl Acad Sci USA 107(2):748–753
Johnson C, Drgon T, Liu QR, Zhang PW, Walther D, Li CY, Anthony JC, Ding Y, Eaton WW, Uhl GR (2008) Genome wide association for substance dependence: convergent results from epidemiologic and research volunteer samples. BMC Med Genet 9:113
Lange C, Silverman EK, Xu X, Weiss ST, Laird NM (2003) A multivariate family-based association test using generalized estimating equations: FBAT-GEE. Biostatistics 4(2):195–206
Lasky-Su J, Anney RJ, Neale BM, Franke B, Zhou K, Maller JB, Vasquez AA, Chen W, Asherson P, Buitelaar J, Banaschewski T, Ebstein R, Gill M, Miranda A, Mulas F, Oades RD, Roeyers H, Rothenberger A, Sergeant J, Sonuga-Barke E, Steinhausen HC, Taylor E, Daly M, Laird N, Lange C, Faraone SV (2008) Genome-wide association scan of the time to onset of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 147B(8):1355–1358
Levy D, Larson MG, Benjamin EJ, Newton-Cheh C, Wang TJ, Hwang SJ, Vasan RS, Mitchell GF (2007) Framingham Heart Study 100K Project: genome-wide associations for blood pressure and arterial stiffness. BMC Med Genet 8 Suppl 1:S3
Li MD (2008) Identifying susceptibility loci for nicotine dependence: 2008 update based on recent genome-wide linkage analyses. Hum Genet 123(2):119–131
Li MD, Burmeister M (2009) New insights into the genetics of addiction. Nat Rev Genet 10(4):225–231
Li MD, Cheng R, Ma JZ, Swan GE (2003) A meta-analysis of estimated genetic and environmental effects on smoking behavior in male and female adult twins. Addiction 98(1):23–31
Li MD, Beuten J, Ma JZ, Payne TJ, Lou XY, Garcia V, Duenes AS, Crews KM, Elston RC (2005) Ethnic- and gender-specific association of the nicotinic acetylcholine receptor alpha4 subunit gene (CHRNA4) with nicotine dependence. Hum Mol Genet 14(9):1211–1219
Li MD, Payne TJ, Ma JZ, Lou XY, Zhang D, Dupont RT, Crews KM, Somes G, Williams NJ, Elston RC (2006) A genomewide search finds major susceptibility loci for nicotine dependence on chromosome 10 in African Americans. Am J Hum Genet 79(4):745–751
Li MD, Yoon D, Lee JY, Han BG, Niu T, Payne TJ, Ma JZ, Park T (2010) Associations of variants in CHRAN5/A3/B4 gene cluster with smoking behavior in a Korean population. PloS One 16:5(8):e12183
Liu YZ, Pei YF, Guo YF, Wang L, Liu XG, Yan H, Xiong DH, Zhang YP, Levy S, Li J, Haddock CK, Papasian CJ, Xu Q, Ma JZ, Payne TJ, Recker RR, Li MD, Deng HW (2009) Genome-wide association analyses suggested a novel mechanism for smoking behavior regulated by IL15. Mol Psychiatry 14(7):668–680
Liu JZ, Tozzi F, Waterworth DM, Pillai SG, Muglia P, Middleton L, Berrettini W, Knouff CW, Yuan X, Waeber G, Vollenweider P, Preisig M, Wareham NJ, Zhao JH, Loos RJ, Barroso I, Khaw KT, Grundy S, Barter P, Mahley R, Kesaniemi A, McPherson R, Vincent JB, Strauss J, Kennedy JL, Farmer A, McGuffin P, Day R, Matthews K, Bakke P, Gulsvik A, Lucae S, Ising M, Brueckl T, Horstmann S, Wichmann HE, Rawal R, Dahmen N, Lamina C, Polasek O, Zgaga L, Huffman J, Campbell S, Kooner J, Chambers JC, Burnett MS, Devaney JM, Pichard AD, Kent KM, Satler L, Lindsay JM, Waksman R, Epstein S, Wilson JF, Wild SH, Campbell H, Vitart V, Reilly MP, Li M, Qu L, Wilensky R, Matthai W, Hakonarson HH, Rader DJ, Franke A, Wittig M, Schafer A, Uda M, Terracciano A, Xiao X, Busonero F, Scheet P, Schlessinger D, St Clair D, Rujescu D, Abecasis GR, Grabe HJ, Teumer A, Volzke H, Petersmann A, John U, Rudan I, Hayward C, Wright AF, Kolcic I, Wright BJ, Thompson JR, Balmforth AJ, Hall AS, Samani NJ, Anderson CA, Ahmad T, Mathew CG, Parkes M, Satsangi J, Caulfield M, Munroe PB, Farrall M, Dominiczak A, Worthington J, Thomson W, Eyre S, Barton A, Mooser V, Francks C, Marchini J (2010) Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet 42(5):436–440
Liu Y, Blackwood DH, Caesar S, de Geus EJ, Farmer A, Ferreira MA, Ferrier IN, Fraser C, Gordon-Smith K, Green EK, Grozeva D, Gurling HM, Hamshere ML, Heutink P, Holmans PA, Hoogendijk WJ, Hottenga JJ, Jones L, Jones IR, Kirov G, Lin D, McGuffin P, Moskvina V, Nolen WA, Perlis RH, Posthuma D, Scolnick EM, Smit AB, Smit JH, Smoller JW, St Clair D, van Dyck R, Verhage M, Willemsen G, Young AH, Zandbelt T, Boomsma DI, Craddock N, O’Donovan MC, Owen MJ, Penninx BW, Purcell S, Sklar P, Sullivan PF (2011) Meta-analysis of genome-wide association data of bipolar disorder and major depressive disorder. Mol Psychiatry 16(1):2–4
Ma JZ, Beuten J, Payne TJ, Dupont RT, Elston RC, Li MD (2005) Haplotype analysis indicates an association between the DOPA decarboxylase (DDC) gene and nicotine dependence. Hum Mol Genet 14(12):1691–1698
Marchini J, Howie B, Myers S, McVean G, Donnelly P (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39(7):906–913
Marroni F, Pfeufer A, Aulchenko YS, Franklin CS, Isaacs A, Pichler I, Wild SH, Oostra BA, Wright AF, Campbell H, Witteman JC, Kaab S, Hicks AA, Gyllensten U, Rudan I, Meitinger T, Pattaro C, van Duijn CM, Wilson JF, Pramstaller PP (2009) A genome-wide association scan of RR and QT interval duration in 3 European genetically isolated populations: the EUROSPAN project. Circ Cardiovasc Genet 2(4):322–328
McGovern DP, Gardet A, Torkvist L, Goyette P, Essers J, Taylor KD, Neale BM, Ong RT, Lagace C, Li C, Green T, Stevens CR, Beauchamp C, Fleshner PR, Carlson M, D’Amato M, Halfvarson J, Hibberd ML, Lordal M, Padyukov L, Andriulli A, Colombo E, Latiano A, Palmieri O, Bernard EJ, Deslandres C, Hommes DW, de Jong DJ, Stokkers PC, Weersma RK, Sharma Y, Silverberg MS, Cho JH, Wu J, Roeder K, Brant SR, Schumm LP, Duerr RH, Dubinsky MC, Glazer NL, Haritunians T, Ippoliti A, Melmed GY, Siscovick DS, Vasiliauskas EA, Targan SR, Annese V, Wijmenga C, Pettersson S, Rotter JI, Xavier RJ, Daly MJ, Rioux JD, Seielstad M (2010) Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet 42(4):332–337
Morrison AC, Felix JF, Cupples LA, Glazer NL, Loehr LR, Dehghan A, Demissie S, Bis JC, Rosamond WD, Aulchenko YS, Wang YA, Haritunians T, Folsom AR, Rivadeneira F, Benjamin EJ, Lumley T, Couper D, Stricker BH, O’Donnell CJ, Rice KM, Chang PP, Hofman A, Levy D, Rotter JI, Fox ER, Uitterlinden AG, Wang TJ, Psaty BM, Willerson JT, van Duijn CM, Boerwinkle E, Witteman JC, Vasan RS, Smith NL (2010) Genomic variation associated with mortality among adults of European and African ancestry with heart failure: the cohorts for heart and aging research in genomic epidemiology consortium. Circ Cardiovasc Genet 3(3):248–255
Muller F, Wehbe L (2008) Smoking and smoking cessation in Latin America: a review of the current situation and available treatments. Int J Chron Obstruct Pulmon Dis 3(2):285–293
Need AC, Attix DK, McEvoy JM, Cirulli ET, Linney KL, Hunt P, Ge D, Heinzen EL, Maia JM, Shianna KV, Weale ME, Cherkas LF, Clement G, Spector TD, Gibson G, Goldstein DB (2009) A genome-wide study of common SNPs and CNVs in cognitive performance in the CANTAB. Hum Mol Genet 18(23):4650–4661
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575
Sadikovic B, Andrews J, Rodenhiser DI (2007) DNA methylation analysis using CpG microarrays is impaired in benzopyrene exposed cells. Toxicol Appl Pharmacol 225(3):300–309
Siedlinski M, Cho MH, Bakke P, Gulsvik A, Lomas DA, Anderson W, Kong X, Rennard SI, Beaty TH, Hokanson JE, Crapo JD, Silverman EK (2011) Genome-wide association study of smoking behaviours in patients with COPD. Thorax Jun 16 (Epub ahead of print)
Silverberg MS, Cho JH, Rioux JD, McGovern DP, Wu J, Annese V, Achkar JP, Goyette P, Scott R, Xu W, Barmada MM, Klei L, Daly MJ, Abraham C, Bayless TM, Bossa F, Griffiths AM, Ippoliti AF, Lahaie RG, Latiano A, Pare P, Proctor DD, Regueiro MD, Steinhart AH, Targan SR, Schumm LP, Kistner EO, Lee AT, Gregersen PK, Rotter JI, Brant SR, Taylor KD, Roeder K, Duerr RH (2009) Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat Genet 41(2):216–220
TGC (2010) Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet 42(5):441–447
Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, Manolescu A, Thorleifsson G, Stefansson H, Ingason A, Stacey SN, Bergthorsson JT, Thorlacius S, Gudmundsson J, Jonsson T, Jakobsdottir M, Saemundsdottir J, Olafsdottir O, Gudmundsson LJ, Bjornsdottir G, Kristjansson K, Skuladottir H, Isaksson HJ, Gudbjartsson T, Jones GT, Mueller T, Gottsater A, Flex A, Aben KK, de Vegt F, Mulders PF, Isla D, Vidal MJ, Asin L, Saez B, Murillo L, Blondal T, Kolbeinsson H, Stefansson JG, Hansdottir I, Runarsdottir V, Pola R, Lindblad B, van Rij AM, Dieplinger B, Haltmayer M, Mayordomo JI, Kiemeney LA, Matthiasson SE, Oskarsson H, Tyrfingsson T, Gudbjartsson DF, Gulcher JR, Jonsson S, Thorsteinsdottir U, Kong A, Stefansson K (2008) A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 452(7187):638–642
Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, Sulem P, Rafnar T, Esko T, Walter S, Gieger C, Rawal R, Mangino M, Prokopenko I, Magi R, Keskitalo K, Gudjonsdottir IH, Gretarsdottir S, Stefansson H, Thompson JR, Aulchenko YS, Nelis M, Aben KK, den Heijer M, Dirksen A, Ashraf H, Soranzo N, Valdes AM, Steves C, Uitterlinden AG, Hofman A, Tonjes A, Kovacs P, Hottenga JJ, Willemsen G, Vogelzangs N, Doring A, Dahmen N, Nitz B, Pergadia ML, Saez B, De Diego V, Lezcano V, Garcia-Prats MD, Ripatti S, Perola M, Kettunen J, Hartikainen AL, Pouta A, Laitinen J, Isohanni M, Huei-Yi S, Allen M, Krestyaninova M, Hall AS, Jones GT, van Rij AM, Mueller T, Dieplinger B, Haltmayer M, Jonsson S, Matthiasson SE, Oskarsson H, Tyrfingsson T, Kiemeney LA, Mayordomo JI, Lindholt JS, Pedersen JH, Franklin WA, Wolf H, Montgomery GW, Heath AC, Martin NG, Madden PA, Giegling I, Rujescu D, Jarvelin MR, Salomaa V, Stumvoll M, Spector TD, Wichmann HE, Metspalu A, Samani NJ, Penninx BW, Oostra BA, Boomsma DI, Tiemeier H, van Duijn CM, Kaprio J, Gulcher JR, McCarthy MI, Peltonen L, Thorsteinsdottir U, Stefansson K (2010) Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet 42(5):448–453
Uhl GR, Liu QR, Drgon T, Johnson C, Walther D, Rose JE (2007) Molecular genetics of nicotine dependence and abstinence: whole genome association using 520, 000 SNPs. BMC Genet 8:10
Uhl GR, Liu QR, Drgon T, Johnson C, Walther D, Rose JE, David SP, Niaura R, Lerman C (2008) Molecular genetics of successful smoking cessation: convergent genome-wide association study results. Arch Gen Psychiatry 65(6):683–693
Uhl GR, Drgon T, Johnson C, Ramoni MF, Behm FM, Rose JE (2010) Genome-wide association for smoking cessation success in a trial of precessation nicotine replacement. Mol Med 16(11–12):513–526
Vianna EO, Gutierrez MR, Barbieri MA, Caldeira RD, Bettiol H, Da Silva AA (2008) Respiratory effects of tobacco smoking among young adults. Am J Med Sci 336(1):44–49
Vink JM, Smit AB, de Geus EJ, Sullivan P, Willemsen G, Hottenga JJ, Smit JH, Hoogendijk WJ, Zitman FG, Peltonen L, Kaprio J, Pedersen NL, Magnusson PK, Spector TD, Kyvik KO, Morley KI, Heath AC, Martin NG, Westendorp RG, Slagboom PE, Tiemeier H, Hofman A, Uitterlinden AG, Aulchenko YS, Amin N, van Duijn C, Penninx BW, Boomsma DI (2009) Genome-wide association study of smoking initiation and current smoking. Am J Hum Genet 84(3):367–379
Warren CW, Jones NR, Peruga A, Chauvin J, Baptiste JP, Costa de Silva V, el Awa F, Tsouros A, Rahman K, Fishburn B, Bettcher DW, Asma S (2008) Global youth tobacco surveillance, 2000–2007. MMWR Surveill Summ 57(1):1–28
Acknowledgments
This work was supported by a grant from the National Institute of Health of Korea 2009-E73007-00 and National Research Foundation NRF-2008-313-C00146 to TP; and National Institutes of Health grant DA-12844 to MDL. The KARE data were obtained from the Korean Genome Analysis Project 4845-301, which was funded by a grant from the Korea National Institute of Health (Korea Center for Disease Control, Ministry for Health, Welfare and Family Affairs), Republic of Korea. We are grateful for the invaluable contributions of clinical information and tissue samples by the participants in this study, as well as for the dedicated work of the research staff at the clinical sites and molecular research laboratory. We thank Nam H. Cho (Department of Preventive Medicine, Ajou University, South Korea) and Chol Shin (Department of Internal Medicine, Korea University Ansan Hospital, South Korea) for their great efforts in generating and providing these valuable data. We also thank Dr. David L. Bronson for his excellent editing of this manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Yoon, D., Kim, YJ., Cui, WY. et al. Large-scale genome-wide association study of Asian population reveals genetic factors in FRMD4A and other loci influencing smoking initiation and nicotine dependence. Hum Genet 131, 1009–1021 (2012). https://doi.org/10.1007/s00439-011-1102-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-1102-x