
Abstract
Erythrokeratodermia variabilis 3 (Kamouraska type) or EKV3 is a newly described autosomal recessive disorder observed in patients from the Bas St-Laurent region of Quebec. It has similar skin lesions as observed for EKV, including congenital hyperkeratosis and red patches of variable sizes, shapes, and duration. EKV3 is also characterized by ichthyosis, sensorineural hearing loss, peripheral neuropathy, psychomotor retardation, congenital chronic diarrhea, and an elevation of very long chain fatty acids (VLCFAs). To map the disease locus, we performed candidate gene analysis and a genomewide scan to identify a common homozygous region in affected individuals from three non-consanguineous families. Mutations in connexin 31 (GJB3) and connexin 30.3 (GJB4), implicated in previous reports of EKV, and connexin 26 (GJB2), implicated in palmoplantar keratoderma, were unlikely given the lack of shared homozygous haplotypes in the regions surrounding these genes. The most promising region of common homozygosity observed in a 4,600 single-nucleotide polymorphism genome scan was further characterized by using microsatellites. A 6.8-Mb region on chromosome 7 between D7S2539 and rs727708 was found to be homozygous for the same haplotype in all affected individuals but not in the parents or an unaffected sibling. This region contains connexin 31.3 (GJE1), and although no mutation have been observed in the coding region of this gene, further analyses are required in order to exclude it. Identification of the gene responsible for this disorder will provide insights into the etiology of this multisystemic disorder.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Erythrokeratodermia variabilis (EKV; MIM 133200) is a congenital disease of the skin and causes hyperkeratosis and red patches of variable sizes, shapes, and duration (Richard et al. 2000). EKV is usually inherited as an autosomal dominant disease resulting from mutations in connexin genes. Connexins are a group of approximately 20 genes that encode gap junctions channels allowing intercellular communication (Sohl and Willecke 2004). Mutations in connexin 31 (GJB3) and connexin 30.3 (GJB4), located within a region containing four connexin genes on chromosome 1p34-35, have been linked to the dominant form of EKV. GJB3 has also been implicated in peripheral neuropathy and sensorineural hearing impairment (Lopez-Bigas et al. 2001; Macari et al. 2000; Plantard et al. 2003). A recent study suggests that a specific mutation in connexin 31, whereby a leucine residue is replaced with a proline, may also cause a recessive form of EKV (Gottfried et al. 2002). Mutations in connexin 26 (GJB2) on chromosome 13 cause palmoplantar keratoderma and syndromic hearing impairment (Leonard and Freedberg 2003; Rabionet et al. 2000). There are also several forms of EKV or EKV-like syndromes of unknown genetic etiology (Itin et al. 2003; Levy et al. 2003).
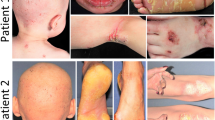
Five children from three families originating from the Kamouraska region of the province of Quebec have been diagnosed with an atypical form of EKV; this form is similar to a disorder previously described by Beare (1972) and includes sensorineural deafness, peripheral neuropathy and psychomotor retardation (Fig. 1). In addition to these symptoms, the Kamouraska patients have congenital diarrhea, an elevation of very long chain fatty acids (VLCFAs), and a recessive mode of inheritance (see Table 1). Two of the children died at an early age from severe congenital diarrhea. This unique and atypical form of EKV in the Kamouraska region is thus a novel disorder, termed erythrokeratodermia variabilis 3 (Kamouraska type) or EKV3.

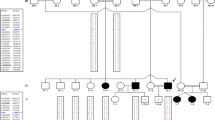
Pedigrees of three families of the Kamouraska region with EKV3
All three families are likely to share common ancestors, as they live in a relatively isolated population descended from founders of French origin who settled the region in the seventeenth and eighteenth centuries. Given the rarity of the disorder across the world, one can assume that the parents are carriers of the same mutation, which is present on a common extended haplotype. We decided to investigate the genetic cause of this new disease by homozygosity mapping. Homozygosity mapping with a single-nucleotide polymorphism (SNP)-based genomewide scan and microsatellite markers has allowed us to reject GJB3 on chromosome 1 as a candidate gene and to identify a large region on chromosome 7 that is identical by descent in all affected individuals but not in an unaffected first-degree relative.
Materials and methods
Ten individuals were available for the genetic study of EKV3: three sets of parents, three affected children, and one unaffected sibling (Fig. 1). Peripheral blood was taken at Le Service de Dermatologie du CHRGP de Rivière-du-Loup and sent to the Hôpital St-François d’Assise. Lymphocytes were isolated, and lymphoblastic cell lines were produced by using Epstein-Barr virus (Tremblay and Khandjian 1998). Genetic material of affected individuals and unaffected siblings and parents was isolated from the lymphoblasts. DNA was purified by the usual phenol-chloroform procedure (Drouin et al. 2001). The study was approved by the Institutional Review Board of the Hôpital St-François d’Assise, and informed consent was obtained from all family members.
Microsatellite marker information was obtained from the July 2003 assembly of the UCSC Genome Browser. Polymerase chain reactions (PCR) were performed under standard conditions on a GeneAmp PCR System 9700 (Applied Biosystems), and products were separated on 3700 or 3730 DNA Analysers (Applied Biosystems). Genotypes were determined by using Genescan, Genotyper and GeneMapper software.
The genomewide scan was performed with the Illumina linkage set III, which consists of 4,637 SNP markers distributed evenly throughout the genome. All reactions were carried out by using the Illumina BeadArray System according to the manufacturers protocol (Oliphant et al. 2002).
Primer design for sequencing was performed by using the Primer3 program (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). Primers used for sequencing GJE1 and for confirming SNP heterozygosity are shown in Table 2. Sequencing was carried out on the ABI 3700 and ABI 3730 DNA analyser platforms as described elsewhere (Engert et al. 2002). Sequences were analyzed by using Sequencing Analysis software version 3.6 (Applied BioSystems, Foster City, Calif., USA) and were aligned with Autoassembler 2.1.1 (Applied BioSystems) and the Phred/Phrap/Consed System (http://www.phrap.org/).
Results
The involvement of GJB3 in a recessive form of EKV with sensorineural deafness and peripheral neuropathy implicates this gene as a prime candidate. However, the genotypes of three microsatellite markers, D1S2783, D1S2613, and D1S3463, located in the 1p34-35 region, revealed no common shared haplotype between affected individuals (data not shown). Furthermore, an unaffected sibling (KEKV02-04) inherited the same two chromosomes as his affected sister; this would be incompatible with fully penetrant recessive mutations at this locus. Therefore, GJB3, and the three other connexin genes in the region, GJB4, GJB5 and GJA4, were excluded as the cause of EKV3.
We subsequently decided to perform a genomewide scan to identify a homozygous region shared by all three affected individuals. The Illumina linkage set consists of 4,637 polymorphic SNPs, covering the human genome with an average inter-SNP distance of 0.78 cM. Kruglyak (1999) has previously estimated that a 1-cM to 2-cM bi-allelic map of polymorphic markers can extract most of the inheritance information. Furthermore, the average information content of the set is 95%, which is higher than standard microsatellite-based genome scan panels. Using this set, we obtained a genotyping call rate of 99.5%, and the reproducibility and Mendelian inheritance error rates were both below 0.1%. The SNP genotyping data at 1p34-35 supported the microsatellite analysis in excluding this locus. No homozygous SNP alleles and haplotypes were shared among affected individuals in the vicinity of GJB3. In addition, there was no evidence of shared homozygosity near GJB2 on chromosome 13, a gene implicated in palmoplantar keratoderma and hearing loss.
The global analysis of the genome scan data involved a search for regions of complete shared homozygosity consisting of at least three consecutive informative SNPs covering more than 1 Mb. Only five such regions were observed on the studied autosomal chromosomes: chromosome 3q22 between markers rs1502186 and rs768496 (four SNPs; 2 Mb); chromosome 5p12 between markers rs10522 and rs726941 (three SNPs; 1.2 Mb); chromosome 10p14 between markers rs942434 and rs1623807 (three SNPs; 1.8 Mb); and two adjacent regions on chromosome 7q22 of six and four consecutive SNPs spanning 1.5 and 2.2 Mb, respectively (Fig. 2). In the first four regions, the unaffected sibling was homozygous for the same alleles as his affected sibling. In addition, for all patients, except for patient 10 (KEKV01-03) at the chromosome 5p12 locus, the stretch of homozygosity in single individuals did not extend further than the shared common region of homozygosity observed in the three patients (data not shown). In contrast, the 2.2-Mb region on chromosome 7 was a result of the intersection of large homozygous regions observed in each patient, spanning 10, 4, and 12 Mb, respectively (Fig. 2).

Affected individuals share a region of homozygosity spanning 6.8 Mb. The disease chromosome is shaded. The region of shared homozygosity in the three affected children is framed with a solid line. A second region of homozygosity, which was initially observed with SNP marker from the genome scan, is framed with a dotted line.
To refine the region and confirm shared homozygosity, we added 17 microsatellite markers on chromosome 7 (Fig. 2). We determined the boundaries of the region of shared homozygosity to lie between rs219798 and rs727708 spanning a distance of 4.6 Mb. The presence of a heterozygous SNP (rs219798) at the border of the common shared haplotype in individual KEKV01-03 was surprising. The heterozygous genotype divided what would otherwise be a large 20-Mb homozygous stretch. We hypothesized that this could correspond to a genotyping error. However, the observation was confirmed by sequencing the SNP. In this process, another heterozygous SNP (rs219797) was identified 48 bp from rs219798. Moreover, the mother (KEKV01-02) was homozygous for the allele that differed from the common shared haplotype, supporting the findings of heterozygozity at these markers. The mother otherwise appeared to have the EKV3 chromosome on both sides of these SNPs. Given the possibility of a gene conversion for this marker that could be unrelated to EKV3 (see below), we defined a centromeric border of the shared region of homozygosity at marker D7S2539, for a total length of the shared interval identical-by-descent estimated to be 6.8 Mb.
This 6.8-Mb region of chromosome 7 contains approximately 100 genes. Interestingly, one of these genes is connexin 31.3 (GJE1). Because of the role of connexins in several skin and hearing disorders, this gene is a likely candidate. We sequenced the two known coding exons of GJE1 and a possible downstream connexin-like pseudogene in all samples. However, no missense mutation was observed. Two SNPs in the 3′ untranslated region were found only in parents and the unaffected sibling.
Discussion
A 6.8-Mb haplotype on chromosome 7 has been observed to be homozygous in three affected individuals from three non-consanguineous families derived from a young founder population of Quebec. The presence of a common shared haplotype in these individuals is consistent with the hypothesis that they share the same mutation identical by descent. The 6.8-Mb interval defined by 12 SNPs and eight microsatellites is thus likely to harbor the disease mutation. The presence of a small heterozygous interval in an otherwise long homozygous stretch in patient KEKV01-03 is consistent with a gene conversion event that occurred in the recent ancestry of the haplotype inherited from the mother. Given that this phenomenon is not observed in the two other families, we believe that this observation is not relevant to the EKV3 mutation. A problem in the genome assembly in this region has been considered. However, by comparisons with the sequences of the mouse and the rat, we could not find any evidence of misassembly (data not shown).
GJE1 has not previously been reported as a cause of EKV or any other disease. In mice, GJE1 is expressed in myelinating glial cells in the peripheral and central nervous systems but has not been investigated in keratinocytes (Altevogt et al. 2002). The sequence of the two known coding exons of the GJE1 gene and a downstream connexin-like pseudogene has revealed no missense mutations. Further work requires the analysis of regulatory regions of the gene and a search for alternate exons, prior to an analysis of other genes found in the 6.8-Mb disease interval.
Although many of the symptoms of EKV3 are strikingly similar to those of peroxisomal biogenesis disorders, such as Refsum’s disease or Zellweger syndrome (see Table 1), several lines of evidence suggest that they are separate diseases. In patients with Zellweger syndrome, deafness is observed in only 40% of the patients (Naidu and Moser 1994). In another study, Baumgartner et al. (1998) have observed seven Refsum’s disease patients with digestive problems, sensorineural hearing loss, elevated VLCFAs, and peripheral neuropathy. However, in contrast with EKV3 patients, deafness is less marked and not congenital. Furthermore, no anomaly in peroxisomal enzyme levels has been observed in EKV3 patients (data not shown). It is expected that peroxisomal and connexin-induced diseases exhibit similar symptoms: both induce a disturbance in the transport of electrical current, the first by its action on the production of myelin and the second by interfering with ion transportation during intercellular communication (Sohl and Willecke 2004). Finally, no obvious peroxisomal gene is present in the candidate region on chromosome 7.
In conclusion, we have identified a 6.8-Mb region on chromosome 7 that harbors a disease gene causing EKV3, an atypical form of EKV specific to the Kamouraska region of the province of Quebec.
References
Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL (2002) Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci 22:6458–6470
Baumgartner MR, Poll-The BT, Verhoeven NM, Jakobs C, Espeel M, Roels F, Rabier D, Levade T, Rolland MO, Martinez M, Wanders RJ, Saudubray JM (1998) Clinical approach to inherited peroxisomal disorders: a series of 27 patients. Ann Neurol 44:720–730
Beare JM, Nevin NC, Froggatt P, Kernohan DC, Allen IV (1972) Atypical erythrokeratoderma with deafness, physical retardation and peripheral neuropathy. Br J Dermatol 87:308–314
Drouin R, Therrien J-P, Angers M, Ouellet S (2001) In vivo DNA analysis. Methods Mol Biol 148:175–219
Engert JC, Vohl MC, Williams SM, Lepage P, Loredo-Osti JC, Faith J, Dore C, Renaud Y, Burtt NP, Villeneuve A, Hirschhorn JN, Altshuler D, Groop LC, Despres JP, Gaudet D, Hudson TJ (2002) 5′ Flanking variants of resistin are associated with obesity. Diabetes 51:1629–1634
Gottfried I, Landau M, Glaser F, Di WL, Ophir J, Mevorah B, Ben-Tal N, Kelsell DP, Avraham KB (2002) A mutation in GJB3 is associated with recessive erythrokeratodermia variabilis (EKV) and leads to defective trafficking of the connexin 31 protein. Hum Mol Genet 11:1311–1316
Itin PH, Moschopulos M, Richard G (2003) Reticular erythrokeratoderma: a new disorder of cornification. Am J Med Genet 120A:237–240
Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 22:139–144
Leonard AL, Freedberg IM (2003) Palmoplantar keratoderma of Sybert. Dermatol Online J 9:30
Levy J, Chung W, Garzon M, Gallagher MP, Oberfield SE, Lieber E, Anyane-Yeboa K (2003) Congenital myopathy, recurrent secretory diarrhea, bullous eruption of skin, microcephaly, and deafness: a new genetic syndrome? Am J Med Genet 116A:20–25
Lopez-Bigas N, Olive M, Rabionet R, Ben-David O, Martinez-Matos JA, Bravo O, Banchs I, Volpini V, Gasparini P, Avraham KB, Ferrer I, Arbones ML, Estivill X (2001) Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment. Hum Mol Genet 10:947–952
Macari F, Landau M, Cousin P, Mevorah B, Brenner S, Panizzon R, Schorderet DF, Hohl D, Huber M (2000) Mutation in the gene for connexin 30.3 in a family with erythrokeratodermia variabilis. Am J Hum Genet 67:1296–1301
Naidu S, Moser HW (1994) Peroxisomal disorders. Neurol Clin 12:727–739
Oliphant A, Barker DL, Stuelpnagel JR, Chee MS (2002) BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques 32:S56–S61
Plantard L, Huber M, Macari F, Meda P, Hohl D (2003) Molecular interaction of connexin 30.3 and connexin 31 suggests a dominant-negative mechanism associated with erythrokeratodermia variabilis. Hum Mol Genet 12:3287–3294
Rabionet R, Gasparini P, Estivill X (2000) Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Hum Mutat 16:190–202
Richard G, Brown N, Smith LE, Terrinoni A, Melino G, Mackie RM, Bale SJ, Uitto J (2000) The spectrum of mutations in erythrokeratodermias–novel and de novo mutations in GJB3. Hum Genet 106:321–329
Sohl G, Willecke K (2004) Gap junctions and the connexin protein family. Cardiovasc Res 62:228–232
Tremblay S, Khandjian EW (1998) Successful use of long-term frozen lymphocytes for the establishment of lymphoblastoid cell lines. Clin Biochem 31:555–556
Acknowledgements
The authors thank David Rosenblatt for useful advice and are grateful for the technical help given by members of the McGill University and Genome Quebec Innovation Centre, including Corinne Darmond-Zwaig and Carole Doré. The authors are also grateful to Nancy Dallaire, Isabelle Paradis, and Sandra Tremblay for their excellent technical support. This project was supported by the Canadian Genetics and Diseases Network (to T.J.H. and R.D.). R.D. holds the Canada Research Chair in “Genetics, Mutagenesis, and Cancer”. A.M. is supported by a fellowship from the Canadian Institute of Health Research (CIHR).
Author information
Authors and Affiliations
Corresponding author
Additional information
T.G. Saba and A. Montpetit have contributed equally to this work
Rights and permissions
About this article
Cite this article
Saba, T.G., Montpetit, A., Verner, A. et al. An atypical form of erythrokeratodermia variabilis maps to chromosome 7q22. Hum Genet 116, 167–171 (2005). https://doi.org/10.1007/s00439-004-1193-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-004-1193-8