
Abstract
Primary ovarian insufficiency (POI) affects ~ 1–3, 7% of women under forty and is a public health problem. Most causes are unknown, but an increasing number of genetic causes have been identified recently. The identification of such causes is essential for genetic and therapeutic counseling in patients and their families. We performed whole exome sequencing in two Caucasian sisters displaying non syndromic POI and their unaffected mother. We identified two novel pathogenic variants in STAG3 encoding a meiosis-specific subunit of the cohesin ring, which ensures correct sister chromatid cohesion: a c.3052delC truncating mutation in exon 28 yielding p.Arg1018Aspfs*14, and a c.659T > G substitution in exon seven yielding p.Leu220Arg. Leu220, highly conserved throughout species, belongs to the STAG domain conserved with other mitotic subunits of the cohesion complex STAG1 and 2. In silico analysis reveals that this substitution markedly impacts the structure of this domain. The truncation removes the last 206 C-terminal residues, not conserved in STAG1 and 2, supporting an important specific role in STAG3, especially meiosis. This is the first occurrence of STAG3 mutations in a Caucasian family. Very little is known about the function of STAG proteins domains. The “knock out-like” phenotype described here supports the crucial role of a single residue in the STAG domain and of the C-terminal region in STAG3 function. In conclusion, this observation shows the necessity to perform the genetic study of POI worldwide including STAG3. This could lead to appropriate genetic counseling and long term follow-up since these patients may develop ovarian tumors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary ovarian insufficiency (POI) is defined by amenorrhea or oligomenorrhea over 4 months with elevated follicle stimulating hormone (FSH) ≥ 25 UI/l in women under 40 years (Huhtaniemi et al. 2018). A very recent meta-analysis revealed that the prevalence of POI is 3.7% worldwide (Golezar et al. 2019). POI is associated with a significant morbidity due in part to steroid hormones deficiency: an increased risk of osteoporosis and fractures, overall cardiovascular diseases, type 2 diabetes and total mortality has been described. A premature decline in cognitive function and mood disorders have also been described (Huhtaniemi et al. 2018; Golezar et al. 2019). This makes this condition a public health problem (Golezar et al. 2019). POI is often diagnosed too late, generating severe irreversible consequences for fertility and well-being of the affected women. Other extra-ovarian morbidities are direct consequences of specific causes of syndromic POI (Huhtaniemi et al. 2018). About one-third of POI is considered to be of genetic origin and there is a high genetic heterogeneity. Most causes are unknown but recently an increased number of genetic causes have been identified especially by whole exome sequencing (WES). The genetic diagnosis of POI is critical not only to establish the cause of patients’ infertility and to perform genetic and therapeutic counseling within the family but also to search for co-morbidities associated with specific gene defects. Recently, genes involved in DNA repair and meiosis have been shown to cause POI (Smirin-Yosef et al. 2017; Huhtaniemi et al. 2018; Zhou et al. 2018; Zhang et al. 2018). It has been shown that tumor or cancer predispositions are associated with mutations of such genes (Caburet et al. 2014; Wood-Trageser et al. 2014; AlAsiri et al. 2015; Fouquet et al. 2017; Weinberg-Shukron et al. 2018).
Up to now, more than 60 genes have been reported to cause POI (Huhtaniemi et al. 2018). However, they have been found at most in one or two families, and in specific populations. They require replication in independent studies (Smirin-Yosef et al. 2017; Huhtaniemi et al. 2018; Zhou et al. 2018; Zhang et al. 2018). As mutations of meiosis/DNA repair genes may also be implicated in tumor/cancer, it is, therefore, very important to confirm their involvement in the etiology of POI and to know the prerequisite that should lead to their study.
Patients with POI may have primary amenorrhea (PA) in combination with delayed puberty, or secondary amenorrhea (SA) occurring after regular menarche for a variable period of time. Although many iatrogenic (chemotherapy, surgery, or irradiation), autoimmune, or viral factors can cause POI, more than 70% of cases remain idiopathic (Huhtaniemi et al. 2018).
Mutations of STAG3 (stromal antigen 3), which encodes a meiosis-specific subunit of the cohesin complex involved in sister chromatids pairing, were identified in rare Middle-eastern and Asian POI (Caburet et al. 2014; Le Quesne Stabej et al. 2016; Colombo et al. 2017; He et al. 2018). Here, we report the first Caucasian family with two sisters presenting a severe phenotype of non-syndromic POI including PA, lack of puberty and streak ovaries, with novel biallelic mutations in STAG3. This observation further supports the crucial role of meiotic genes in ovarian pathophysiology. STAG3 has to be studied in all unexplained POI with PA worldwide for genetic diagnosis and appropriate monitoring in the long term since such patients could also develop ovarian tumors, as shown by a previous POI case and by the rodent knock out model. The novel mutations identified highlight for the first time the crucial role of a residue within the STAG domain and of the C-terminus in STAG3 function.
Patients and methods
Medical history and clinical data
Informed consent was obtained from the patients and all institutions involved in the study. The pedigree of this Caucasian family is presented in Fig. 1. The proposita was referred at 13 years for PA and lack of pubertal development. She measured 1 m 46 (− 1.5 DS) and weighed 35 kg (− 2 DS). Her body mass index (BMI) was 16.4 kg/m2. Physical examination revealed an absence of breast development and no other clinical sign. Hormonal assays confirmed POI with high FSH (86 UI/l) and luteinizing hormone (LH) (27.5 UI/l) and low estradiol (E2) (< 3 pg/ml) plasma levels. Ovarian antibodies were undetectable, and karyotype was 46, XX. FMR1 premutation screening was negative. Pelvic ultrasonography (US) showed a small uterus with streak ovaries. Hormone replacement therapy was started. The last clinical examination at 29 years old in another center confirmed POI without any other clinical sign. She measured 1 m 68 and weighted 49 kg (BMI = 17.6 kg/m2).

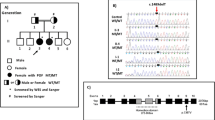
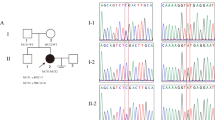
Pedigree of the family and molecular analysis. a Pedigree of the family with two POI sisters and electropherograms of sanger sequencing. WES whole exome sequencing. b Visualization of exome data in IGV (integrative genomics viewer). The coverage for each variant is reported. Both variants have coverage of at least 50× for the three individuals. c Structure of the STAG3 gene and protein, and position of the causal variants. STAG3 stromal antigen 3. STAG domain is represented in green and the armadillo (ARM)-type domain, which is predicted to interact with a nucleic acid or another protein, in purple. The diagrams of STAG proteins are adapted from (Pezzi et al. 2000) and from PFAM (http://pfam.xfam.org/family/PF08514). WT wild type
Her younger sister was seen at 17 years of age in another center for lack of pubertal development and PA. Hormonal assays confirmed the diagnosis of POI. Hormone replacement therapy was started. The last clinical examination at the age 25 years does not reveal any particular complication. She measured 1.66 cm and weighted 55 kg (BMI = 19.9 kg/m2). There was no familial history of POI, autoimmune, or cancer diseases especially ovarian tumors. The mother had menopause at the age of 51 years.
Exome and sanger sequencing
Exome sequencing was performed on genomic DNA extracted from the peripheral blood of patients I.2 (mother), II.1 (proposita), and II.2 (sister) (Fig. 1b). Library preparation, exome capture, sequencing, and data processing were performed by IntegraGen SA (Evry, France) according to their in-house procedures. Data analysis was performed as described in (Fouquet et al. 2017). Briefly, exon enrichment was performed on 600 ng of DNA, using the Agilent SureSelect Human All Exons kit version CRE (Agilent Technologies, Santa Clara, USA). Exon-enriched libraries were subjected to a 75 bp paired-end sequencing on an HiSeq2500, according to the manufacturer’s protocol. Reads alignment to the human reference genome (GRCh38) and variant calling were performed using the Illumina pipeline (CASAVA 1.8.2). Variant annotation was performed using variant effect predictor tools. The variants were filtered using SIRIUS, an IntegraGen in-house pipeline platform.
Each exome was processed using the following bioinformatic filters: (i) variants with a read coverage of less than 5× and a Qscore of below 20 were filtered out; (ii) we filtered the variations against the 1000 Genomes Project data set (May 2011, 20101123 release, http://www.1000genomes.org), the ESP cohort data set (ESP6500, http://evs.gs.washington.edu/EVS/) and GnomAD (exome and genome; http://gnomad.broadinstitute.org/) using an allelic frequency filter of 0.01. In the absence of consanguinity in this Caucasian family, we tested the hypothesis of compound heterozygous mutations transmitted in a recessive model. Two pathogenic heterozygous variants in the same gene, transmitted by both parents, would be present in the POI patients. For missense variants, we used the following in silico prediction tools: SIFT, Polyphen, CADD and FATHMM-MKL throw Varsome website (https://varsome.com/) and M-CAP (http://bejerano.stanford.edu/mcap/). The latter is a variant pathogenicity classifier of rare missense variants that combines previous pathogenicity scores with higher sensitivity (Jagadeesh et al. 2016).
Relevant pathogenic variants detected were confirmed by direct genomic sanger sequencing of STAG3 using the followings primers pairs:
In silico protein 3D structure analysis
For structural analysis of the missense variant, we used MODELLER (v9.19) software to build a 3D model of STAG3 wild-type and mutant as described (Sali and Blundell 1993; Webb and Sali 2014). This software is based on the satisfaction of spatial restraints of atoms and the comparison between a given protein sequence (as target) with one or more related proteins with known structures (templates). It requires first alignments of target to templates sequences. In our case, since the 3D model of STAG3 is not known, we first performed the sequence alignment of STAG3 and STAG2 using EMBOSS program based on Smith and Waterman algorithm (Rice et al. 2000). STAG2 has two known 3D structures (PDB: 4PJU and 4PK7). Regions 154–1048 in STAG2 and 112–998 in STAG3 shared 56.8% identity (75.2% similarity). Then, we generated the STAG3 3D model for the wild-type and the mutant proteins using the STAG2 PDB structure 4PJU as a template (Hara et al. 2014). The ten best models were selected according to the DOPE score (Shen and Sali 2006). Results for the wild-type and the mutant STAG3 proteins are shown in Fig. 3.
Results
Exome sequencing allowed to detect two exonic variants shared between patient II.1 and patient II.2 (Supplementary Tables 1 and 2). The first missense variant, also found in the mother, is located in exon seven of STAG3 (NM_001282716.1: c.659T > G; p.Leu220Arg) (Fig. 1b). This residue is located in the conserved domain among all stromalin antigen (STAG) proteins (STAG1, 2 and 3) (Figs. 1c, 3) (Pezzi et al. 2000, p. 3). This residue is remarkably conserved in all species throughout the evolution in all STAG proteins (Fig. 3). The change is predicted pathogenic by different predicting softwares (Table 3 in Supplementary data). The second pathogenic variant is a 1 bp-deletion in exon 28 of STAG3 (c.3052delC; p.Arg1018AspfsTer14) yielding a truncated protein (Fig. 1c) devoid of the last 206 C-terminal residues.
The missense mutation was reported in dbSNP as rs1289429589 with minor allele frequency (MAF) of 3.23e-05 in the GnomAD genome database, which includes 15.496 whole genome sequences, and is absent from the 123,136 exome sequences of this database. The truncating mutation has a MAF of 4.06e-06 in the Exome GnomAD database and is absent from all other human variants databases.
Sanger sequencing confirmed the presence of the missense mutation in both patients and their mother (Fig. 1a). The truncated mutation was transmitted from the father. The brother inherited the two reference alleles (Fig. 1a).
In silico analysis revealed that STAG3 adopts a conformation very close to that of the dragon-shaped crystal structure of STAG2 (Hara et al. 2014) (Fig. 2A). Leu 220 localized on helix nine at the N-terminal region is mainly involved in hydrophobic interactions. The ten best models built with MODELLER are superimposed in Fig. 2 (B and C). For the wild-type STAG3 models, the positions of the Leu 220 side chain are conserved in the ten models and this side chain is in close contact with Leu 216 and Leu 242. When Leu 220 is substituted by an Arginine, disruption of interaction with other hydrophobic residues in the vicinity is observed. Indeed, according to the ten models, either Arg 220 side chain is excluded from the helix loop and the two Leu 216 and 242 stay nearby (Fig. 2C-a) or the side chain points to the two leucines that deviate (Fig. 2C-b).

Models of wild-type and mutant STAG3 proteins. A Cartoon diagram of the wild-type (WT) STAG3 model on the left and the same view at 180° on the right. The N-terminal region is circled in blue. Leu 220 is labelled and represented in green sticks. B–C Zoomed-in view of the N-terminal region of the WT STAG3 model (left) and the mutant (Mut) STAG3 model (right). In B, the ten best models for WT (left) and Mut (right) are superimposed. Leu 220 and Arg 220 are represented in sticks, respectively, in green for WT and in red for Mut. In C hydrophobic residues Leu 216 and Leu 242 are shown in sticks and are in close contact with Leu 220 in WT models. These contacts are disrupted in the presence of Arg 220 whose side chain can adopt two distinct conformations: a directed to the other side of the two leucines that remain close or b directed to the two leucines that deviate (in Mut models)
Discussion
STAG3 encodes the meiosis-specific subunit of the cohesin complex that also contain SMC1α/SMC1β and SMC3 in addition to RAD21/REC8 or RAD21L (Nishiyama 2018). The multiprotein cohesin complex forms a ring around sister chromatids to hold them together during mitosis and meiosis. Whereas SMC1α and RAD21 are ubiquitous, SMC1β, REC8, and RAD21L are present only in meiotic cells (Ward et al. 2016). STAG1/2 are present in mitotic cells (Carramolino et al. 1997) whereas STAG3 is present only in ovaries and testes (Pezzi et al. 2000; Caburet et al. 2014). It is worth noting that the impact of STAG proteins mutations is different in meiotic and mitotic cells. Indeed, while mono-allelic inactivation of STAG3 appears to be tolerated by meiotic cells, heterozygous loss of function mutations of STAG1/2 have been linked to cohesinopathies characterized by syndromic developmental delay (Mullegama et al. 2017; Lehalle et al. 2017).
Until now, only two consanguineous Asian and two Middle-eastern families (Caburet et al. 2014; Le Quesne Stabej et al. 2016; Colombo et al. 2017; He et al. 2018) with STAG3 defects have been reported. All these patients display isolated POI with PA, lack of pubertal development, infantile uterus and streak gonads as in our patient. This phenotype is in line with that observed in the mouse model. Histological analysis revealed that stag3-/- mice lack ovarian follicles (Caburet et al. 2014). Our observation of the first Caucasian family supports the study of STAG3 in all unexplained POI with PA worldwide for genetic diagnosis and counselling. Stag3-/- mouse females developed ovarian tumors (Caburet et al. 2014; Fukuda et al. 2014; Hopkins et al. 2014). Caburet et al. reported the presence of an ovarian tumor in relation with this biallelic inactivation of STAG3 in a Palestinian family (Caburet et al. 2014). A regular ovarian monitoring by the US should be recommended for these patients.
All previous mutations of STAG3 were large homozygous truncating mutations. Therefore, no information on the structure–function correlation of STAG3 could be established. Here, using WES, we identified compound heterozygous mutations in STAG3. The first mutation resulted in the replacement of a hydrophobic leucine (L), by a basic arginine (R), at position 220 (L220R). This residue is highly conserved in STAG3 during the evolution and between STAG1, 2. This conservation, together with the “knock out-like” phenotype observed in our patients, supports the functional importance of this residue in all meiotic STAG3 and also mitotic STAG proteins.
The second pathogenic variant was a 1 bp deletion p.Arg1018Aspfs*14 resulting in a premature stop codon with a deduced truncated protein devoid of the last 206 C-terminal residues (Fig. 1c). This region of STAG3 is not conserved with STAG 1 and 2 (Fig. 3). This supports an important specific functional role of this region in STAG3, especially meiosis.

Comparison between STAG3 and STAG½. a Comparison between STAG3 and STAG1 and 2. respectively. STAG3 is shown in the middle, STAG2 on top and STAG1 below. The two mutations are indicated by a red line. Between STAG3 and STAG 1 and 2, respectively, the identity percentages of the sliding window of 11 AA are represented between white (0%) and black (100%). STAG stromal antigene. STAG domain is represented in green and armadillo (ARM)-type domain, which is predicted to interact with a nucleic acid or another protein, in purple. The diagrams of STAG are adapted from (Pezzi et al. 2000) and from PFAM (http://pfam.xfam.org/family/PF08514). b Alignment of the three STAG (1, 2, 3) (on top) and of STAG3 proteins in different species (below) focused in the STAG domain using SeaView software (Gouy et al. 2010). The position of the amino acid (Leu 220) mutated in our patients is showed by an arrow
Recently, two truncating mutations of STAG3 were reported in a Brazilian POI (França et al. 2018). However, the authors were unable to confirm the compound heterozygous status of these variants in the absence of parent’s DNA. Therefore, they could not demonstrate the implication of STAG3 in the pathogenesis of the POI.
All Stag3-/- male mice described to date are infertile (Caburet et al. 2014; Fukuda et al. 2014; Llano et al. 2014). Very recently the first mutation of STAG3 was described in a man with azoospermia (Riera-Escamilla et al. 2019). This observation is consistent with the mouse model and highlights the role of STAG3 in meiosis in both sexes.
In conclusion, we report the first implication of STAG3 in a Caucasian family with POI. This observation has wide impact in the genetic counseling and management of patients with POI; (i) Screening of this gene and all meiosis/DNA repair genes involved in POI should be performed in all unexplained POI worldwide with PA. (ii) Prolonged monitoring of patients with STAG3 mutations should be performed considering the risk of ovarian tumors. Furthermore, the two novel mutations of STAG3 identified here support the critical role of Leu220 and of the C-terminal part of STAG3 in cohesion function. Altogether, our work supports the crucial role of the cohesin complex in ovarian physiology and female reproduction worldwide. Larger studies and long term follow-up of POI patients harboring biallelic mutations of STAG3 will allow us to better evaluate the risk of tumors in these patients.
References
AlAsiri S, Basit S, Wood-Trageser MA, Yatsenko SA, Jeffries EP, Surti U, Ketterer DM, Afzal S, Ramzan K, Faiyaz-Ul Haque M, Jiang H, Trakselis MA, Rajkovic A (2015) Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. J Clin Invest 125:258–262
Caburet S, Arboleda VA, Llano E, Overbeek PA, Barbero JL, Oka K, Harrison W, Vaiman D, Ben-Neriah Z, García-Tuñón I, Fellous M, Pendás AM, Veitia RA, Vilain E (2014) Mutant cohesin in premature ovarian failure. N Engl J Med 370:943–949
Carramolino L, Lee BC, Zaballos A, Peled A, Barthelemy I, Shav-Tal Y, Prieto I, Carmi P, Gothelf Y, González de Buitrago G, Aracil M, Márquez G, Barbero JL, Zipori D (1997) SA-1, a nuclear protein encoded by one member of a novel gene family: molecular cloning and detection in hemopoietic organs. Gene 195:151–159
Colombo R, Pontoglio A, Bini M (2017) A STAG3 missense mutation in two sisters with primary ovarian insufficiency. Eur J Obstet Gynecol Reprod Biol 216:269–271
Fouquet B, Pawlikowska P, Caburet S, Guigon C, Mäkinen M, Tanner L, Hietala M, Urbanska K, Bellutti L, Legois B, Bessieres B, Gougeon A, Benachi A, Livera G, Rosselli F, Veitia RA, Misrahi M (2017) A homozygous FANCM mutation underlies a familial case of non-syndromic primary ovarian insufficiency. Elife. https://doi.org/10.7554/elife.30490
França MM, Nishi MY, Funari MFA, Lerario AM, Baracat EC, Hayashida SAY, Maciel GAR, Jorge AAL, Mendonca BB (2018) Two rare loss-of-function variants in the STAG3 gene leading to primary ovarian insufficiency. Eur J Med Genet. https://doi.org/10.1016/j.ejmg.2018.07.008
Fukuda T, Fukuda N, Agostinho A, Hernández-Hernández A, Kouznetsova A, Höög C (2014) STAG3-mediated stabilization of REC8 cohesin complexes promotes chromosome synapsis during meiosis. EMBO J 33:1243–1255
Golezar S, Ramezani Tehrani F, Khazaei S, Ebadi A, Keshavarz Z (2019) The global prevalence of primary ovarian insufficiency and early menopause: a meta-analysis. Climacteric 22:403–411
Gouy M, Guindon S, Gascuel O (2010) SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224
Hara K, Zheng G, Qu Q, Liu H, Ouyang Z, Chen Z, Tomchick DR, Yu H (2014) Structure of cohesin subcomplex pinpoints direct shugoshin-Wapl antagonism in centromeric cohesion. Nat Struct Mol Biol 21:864–870
He W-B, Banerjee S, Meng L-L, Du J, Gong F, Huang H, Zhang X-X, Wang Y-Y, Lu G-X, Lin G, Tan Y-Q (2018) Whole-exome sequencing identifies a homozygous donor splice-site mutation in STAG3 that causes primary ovarian insufficiency. Clin Genet 93:340–344
Hopkins J, Hwang G, Jacob J, Sapp N, Bedigian R, Oka K, Overbeek P, Murray S, Jordan PW (2014) Meiosis-specific cohesin component, STAG3 is essential for maintaining centromere chromatid cohesion, and required for DNA repair and synapsis between homologous chromosomes. PLoS Genet 10:e1004413. https://doi.org/10.1371/journal.pgen.1004413
Huhtaniemi I, Hovatta O, La Marca A, Livera G, Monniaux D, Persani L, Heddar A, Jarzabek K, Laisk-Podar T, Salumets A, Tapanainen JS, Veitia RA, Visser JA, Wieacker P, Wolczynski S, Misrahi M (2018) Advances in the molecular pathophysiology, genetics, and treatment of primary ovarian insufficiency. Trends Endocrinol Metab 29:400–419
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, Bernstein JA, Bejerano G (2016) M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 48:1581–1586
Le Quesne Stabej P, Williams HJ, James C, Tekman M, Stanescu HC, Kleta R, Ocaka L, Lescai F, Storr HL, Bitner-Glindzicz M, Bacchelli C, Conway GS (2016) STAG3 truncating variant as the cause of primary ovarian insufficiency. Eur J Hum Genet 24:135–138
Lehalle D, Mosca-Boidron A-L, Begtrup A, Boute-Benejean O, Charles P, Cho MT, Clarkson A, Devinsky O, Duffourd Y, Duplomb-Jego L, Gérard B, Jacquette A, Kuentz P, Masurel-Paulet A, McDougall C, Moutton S, Olivié H, Park S-M, Rauch A, Revencu N, Rivière J-B, Rubin K, Simonic I, Shears DJ, Smol T, Taylor Tavares AL, Terhal P, Thevenon J, Van Gassen K, Vincent-Delorme C, Willemsen MH, Wilson GN, Zackai E, Zweier C, Callier P, Thauvin-Robinet C, Faivre L (2017) STAG1 mutations cause a novel cohesinopathy characterised by unspecific syndromic intellectual disability. J Med Genet 54:479–488
Llano E, Gomez-H L, García-Tuñón I, Sánchez-Martín M, Caburet S, Barbero JL, Schimenti JC, Veitia RA, Pendas AM (2014) STAG3 is a strong candidate gene for male infertility. Hum Mol Genet 23:3421–3431
Mullegama SV, Klein SD, Mulatinho MV, Senaratne TN, Singh K, Clinical Genomics Center UCLA, Nguyen DC, Gallant NM, Strom SP, Ghahremani S, Rao NP, Martinez-Agosto JA (2017) De novo loss-of-function variants in STAG2 are associated with developmental delay, microcephaly, and congenital anomalies. Am J Med Genet A 173:1319–1327
Nishiyama T (2018) Cohesion and cohesin-dependent chromatin organization. Curr Opin Cell Biol 58:8–14
Pezzi N, Prieto I, Kremer L, Pérez Jurado LA, Valero C, Del Mazo J, Martínez-A C, Barbero JL (2000) STAG3, a novel gene encoding a protein involved in meiotic chromosome pairing and location of STAG3-related genes flanking the Williams–Beuren syndrome deletion. FASEB J 14:581–592
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277
Riera-Escamilla A, Enguita-Marruedo A, Moreno-Mendoza D, Chianese C, Sleddens-Linkels E, Contini E, Benelli M, Natali A, Colpi GM, Ruiz-Castañé E, Maggi M, Baarends WM, Krausz C (2019) Sequencing of a “mouse azoospermia” gene panel in azoospermic men: identification of RNF212 and STAG3 mutations as novel genetic causes of meiotic arrest. Hum Reprod 34:978–988
Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815
Shen M-Y, Sali A (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci 15:2507–2524
Smirin-Yosef P, Zuckerman-Levin N, Tzur S, Granot Y, Cohen L, Sachsenweger J, Borck G, Lagovsky I, Salmon-Divon M, Wiesmüller L, Basel-Vanagaite L (2017) A biallelic mutation in the homologous recombination repair gene SPIDR is associated with human gonadal dysgenesis. J Clin Endocrinol Metab 102:681–688
Ward A, Hopkins J, Mckay M, Murray S, Jordan PW (2016) Genetic interactions between the meiosis-specific cohesin components, STAG3, REC8, and RAD21L. G3 (Bethesda) 6:1713–1724
Webb B, Sali A (2014) Comparative protein structure modeling using MODELLER. Curr Protoc Bioinform 47:5.6.1–5.6.32
Weinberg-Shukron A, Rachmiel M, Renbaum P, Gulsuner S, Walsh T, Lobel O, Dreifuss A, Ben-Moshe A, Zeligson S, Segel R, Shore T, Kalifa R, Goldberg M, King M-C, Gerlitz O, Levy-Lahad E, Zangen D (2018) Essential role of BRCA2 in ovarian development and function. N Engl J Med 379:1042–1049
Wood-Trageser MA, Gurbuz F, Yatsenko SA, Jeffries EP, Kotan LD, Surti U, Ketterer DM, Matic J, Chipkin J, Jiang H, Trakselis MA, Topaloglu AK, Rajkovic A (2014) MCM9 mutations are associated with ovarian failure, short stature, and chromosomal instability. Am J Hum Genet 95:754–762
Zhang D, Liu Yifeng, Zhang Z, Lv P, Liu Yun, Li J, Wu Y, Zhang R, Huang Y, Xu G, Qian Yeqing, Qian Yuli, Chen S, Xu C, Shen J, Zhu L, Chen K, Zhu B, Ye X, Mao Y, Bo X, Zhou C, Wang T, Chen D, Yang W, Tan Y, Song Y, Zhou D, Sheng J, Gao H, Zhu Y, Li M, Wu L, He L, Huang H (2018) Basonuclin 1 deficiency is a cause of primary ovarian insufficiency. Hum Mol Genet 27:3787–3800
Zhou Y, Qin Yan, Qin Yingying, Xu B, Guo T, Ke H, Chen M, Zhang L, Han F, Li Y, Chen M, Behrens A, Wang Y, Xu Z, Chen Z-J, Gao F (2018) Wdr62 is involved in meiotic initiation via activating JNK signaling and associated with POI in humans. PLoS Genet 14:e1007463. https://doi.org/10.1371/journal.pgen.1007463
Acknowledgement
We thank Prof. Reiner Veitia and Dr Sandrine Caburet (Jacques Monod Institute, Paris) for their support and helpful discussions.
Funding
This work was supported by the Universities Paris South-Paris Saclay and Paris Diderot and by The French National Biomedical Agency (ABM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Communicated by Stefan Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Heddar, A., Dessen, P., Flatters, D. et al. Novel STAG3 mutations in a Caucasian family with primary ovarian insufficiency. Mol Genet Genomics 294, 1527–1534 (2019). https://doi.org/10.1007/s00438-019-01594-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-019-01594-4