
Abstract
While hundreds of genes have recently been implicated in an organism’s response to thermal stress, our insight into the cellular and physiological mechanisms affected by these genes has advanced to a lesser extent. We focus on an enigmatic Drosophila heat stress RNA gene, hsr-omega, which encodes two RNA transcripts that are constitutively expressed in almost all developing and adult tissues, omega-n in the nucleus and omega-c in the cytoplasm; both being readily induced to high levels by mild heat stress. We derived three hsr-omega mutant lines via imprecise P-element excision and characterised them for changes in expression, in both the presence and absence of heat stress. Viability estimates indicate that a low level of omega-n is required for normal development. Consistent with the model of omega-n as a negative regulator of intron-processed mRNA levels the mutants displayed a 1.5-fold increase in rates of protein synthesis measured in ovarian tissue in the absence of heat stress, a result suggesting that an important function of hsr-omega is the modulation of general protein synthesis. The mutants had little effect on two measures commonly used to assess heat tolerance, heat-knockdown time and heat hardening ability, suggesting that more subtle heat-related fitness components need to be examined for effects of these mutations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The genetic and cellular mechanisms that underlie heat-stress tolerance have recently been the focus of considerable research in diverse organisms (Larkindale et al. 2005; Collier et al. 2008; Healy et al. 2010), as has been the mechanisms that underlie heat acclimation (Horowitz 2001; Bernabucci et al. 2010). In this era of global warming these mechanisms are likely to be subject to adaptive evolutionary change (Deutsch et al. 2008; Hoffmann 2010; Thomas et al. 2004), and from conservational and agricultural perspectives, we need to understand the processes involved. Drosophila provides a useful model organism to study such mechanisms and processes (Lindquist 1980; Sgrò et al. 2010) and recent technological advances have revealed hundreds of new Drosophila genes that are likely to be involved (Sørensen et al. 2005; Laayouni et al. 2007). However, these genetic data have not yet led to equivalent advances in elucidating the cellular mechanisms affected by these genes. One enigmatic heat-inducible gene that is likely to affect Drosophila heat tolerance and heat-acclimation ability is hsr-omega (Pardue et al. 1990; McKechnie et al. 1998), despite the fact that just how this gene protects the cells has remained elusive (Lakhotia et al. 1999; Weeks et al. 2002). To address this issue we derive some new mutations in hsr-omega using imprecise P-element excision. We characterise the mutant strains for changes in gene transcript level, with and without heat stress, we test a current hypothesis about the cellular function of hsr-omega, and we look for effects of the mutations on two common measures of Drosophila heat tolerance.
While no protein product is known for hsr-omega its two RNA transcripts are constitutively expressed in nearly all tissues, omega-n in the nucleus and omega-c in the cytoplasm, and both are rapidly and significantly up-regulated in response to thermal and other stresses (Pardue et al. 1990; Lakhotia et al. 1999). Growing evidence indicates that natural variation in genotype and expression of hsr-omega influence heat tolerance (McColl et al. 1996; Anderson et al. 2003; Rako et al. 2007; Collinge et al. 2008). The gene is particularly interesting in the context of heat acclimation or heat hardening as divergent changes occurred in the resting level of its transcripts in heat resistant populations, depending on whether they were heat-hardened or not prior to laboratory selection for heat resistance (McKechnie et al. 1998). The roles of the hsr-omega transcripts, however, and how they influence whole organism fitness under heat stress, is poorly understood, especially when compared to our understanding of the function of the heat stress proteins and the heat shock transcription factor (HSF).
Indications that hsr-omega affects whole organism heat tolerance come from early study of genomic deficiencies that uncover the gene (Lakhotia 1987; Mohler and Pardue 1984). However the insight provided has been limited because the genetic lesions also removed adjacent chromosomal genes (Gerasimova et al. 1995) rendering the causes of trait changes ambiguous, or because low viability of the lines precluded measurements of ecologically relevant traits such as adult heat-knockdown time (Hoffmann et al. 2002). Nonetheless, these aberrations have been useful in suggesting an important role for omega-n during heat stress. Omega-n regulates availability of nuclear factors known primarily to be involved in pre-mRNA processing (Lakhotia et al. 1999; Prasanth et al. 2000). Thus, we might expect lines differing in the levels of the nuclear transcript to have altered levels of general protein synthesis, and recent evidence supports this idea (Johnson et al. 2009a).
Levels of cellular protein synthesis are profoundly changed following heat shock (Ashburner and Bonner 1979; Lindquist 1980). In the nucleus, repression of general gene transcription (Spradling et al. 1975) and arrest of nascent transcript processing occurs (Mayrand and Pederson 1983) and these changes are paralleled in the cytoplasm by programmed shutdown of general protein synthesis and the induction of the protective heat shock proteins (Yost and Lindquist 1986). Both the levels of heat shock proteins and the extent of shutdown of general protein synthesis following heat stress changed in lines successfully selected for increased or decreased heat tolerance, demonstrating a link between these processes and heat-stress adaptation (Stephanou et al. 1983). Further, levels of protein synthesis following heat stress have been associated with the capacity of a line to increase knockdown time when heat hardened (Johnson et al. 2009b). If hsr-omega is able to modulate levels of protein synthesis, it may influence variation in thermal tolerance via an effect on protein synthesis.
Here, we generate three new alleles of the hsr-omega gene by excision of a P-element insertion (EP3128) in close proximity to the hsr-omega transcription start site. Lines homozygous for these mutations have been characterised for hsr-omega expression with and without heat-induction, for rates of protein synthesis, for viability and for heat tolerance measured as adult knockdown time following two different levels of heat stress. We report that a markedly reduced level of both of the main hsr-omega transcripts is not necessarily lethal, but it disrupts development, affects rates of protein synthesis and has little effect on adult heat-knockdown time.
Materials and methods
Fly maintenance and lines used
Flies were maintained at 25°C under a 12 h light regime in 42 ml vials or 250 ml bottles containing potato-dextrose medium. Flies were raised under constant density for testing (Clancy and Kennington 2001). P-element insertion line EP3128 (Rørth et al. 1998) was sourced from Szeged stock centre (Hungary). The transposase source used was w 1118; Sb, P[ry +, ∆2–3]/TM6.
Derivation of mutant lines
For P-element excision mutagenesis, line EP3128 was selected due to the elements close proximity to the hsr-omega transcriptional start site. Firstly, the transposase source (∆2–3) was crossed to EP3128/TM6B en masse and the progeny containing both the P-element and transposase chromosomes crossed again to the transposase source. Individual excision progeny were crossed back to the transposase source to generate balanced (TM6B) lines. Once 200 independent excision lines had been established, DNA was isolated using a standard phenol–chloroform method and each line was subject to PCR with primers designed to detect deletions not likely to affect the upstream gene (Fig. 1a, F-5′ TGT AGG TCC TTG TTG CCA AAC, R-5′ GGA GAG AGA GAA CGA GCC AGT). Amplicons smaller than that expected from wild-type (3.3 kb) were gel-extracted (Geneclean II, Qbiogene) and sequenced (BigDye v3.1 mix, Applied Biosystems 3730S Genetic analyser) to determine the nature of excision events. For precise excisions, additional sequence was obtained using an extra reverse primer (R-5′ TCC GAG GGA CTA TTC TCG TAA) to survey the region for evidence of other local sequence aberrations. All sequences were analysed for the creation of a new transcription start site for hsr-omega using McPromoter for Drosophila (Ohler 2006). Imprecise excision lines (56, 66) and a precise excision line (29) for use as a control, were maintained with a floating TM6B balancer chromosome. Homozygous mutant lines were derived at least two generations prior to use in all tests.

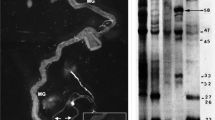
The hsr-omega gene and nature of excision alleles derived via P-element mobilisation. Relative position of the P-element insertion in line EP3128 (triangle) and structure of the hsr-omega gene including the 5′ promoter region (left of arrow, green), exons (right of arrow, blue) and intron (right of arrow, pale-blue) are displayed (TSS transcription start site). Small arrows represent primer-binding sites used for PCR screen of P-element excision events (a). Alleles derived from a precise excision event (hsr-omega 29) and imprecise excisions events hsr-omega 56 (∆1,853 bp) and hsr-omega 66 (∆1,598 bp) are detailed with their respective genomic lesions indicated by dashed lines (b). The sequences below (unhighlighted) are the sequences of fragments found to be inserted in place of deletions. Agarose gel (1%) separation of duplicate PCR products from excision line homozygotes using aforementioned primers (c). Wild-type expected product size is ~3.3 kb. DNA marker is 1 kb (Promega); NTC no template control
Quantification of hsr-omega transcript levels
Levels of omega-n and omega-c were estimated by real time RT-PCR as previously described (Collinge et al. 2008; Johnson et al. 2009a). Transcript specificity was assured by primer placement; for omega-n both primers were downstream of the full omega-c template (and upstream of the repeat region), and for omega-c the forward primer spanned the intron splice junction while the reverse primer was close-by (near the 5′ end of the omega-c template, as depicted in Johnson et al. 2009a). Three replicate RNA extractions were made for each line for both basal and heat-shocked treatments. Replicates consisted of twenty adult females reared in separate culture vials and aged 5–7 days. The heat shock was performed by placing flies in capped 10 ml centrifuge tubes, before submerging in a circulating water-bath set to 36.0°C for 1 h. RNA was isolated from replicate groups using TriSure reagent (Bioline) and DNase treated (New England Biolabs) for 40 min at 37°C prior to cDNA conversion. For realtime RT-PCR, each run consisted of one replicate from each line for the two treatments with omega-n and omega-c run separately in addition to the reference gene, Cyclin K. Amplification conditions and primer sets for all genes were validated across a range of cDNA concentrations, and melt curves analysed prior to transcript quantification.
Viability
Homozygous adults from each line and line w 1118 were allowed to mate and lay overnight on culture media containing food dye for ease in embryo picking, and three times the standard agar amount to keep eggs above the media surface. Ten replicate groups of twenty eggs each were picked and carefully transferred to 42 ml culture vials containing fresh media supplemented with ampicillin (50 mg/ml) to minimise infection. Vials were placed at 25°C and scored only for both the number individuals surviving to pupate and the number of adults to emerge.
Rates of protein synthesis
Protein synthesis rates were assessed by measuring the amount of 35 S-methionine incorporated into dissected ovarian tissue in 1 h, as previously described (Johnson et al. 2009a). For each line (w 1118 and lines homozygous for 29, EP1328 and 56) rearing occurred in parallel at 25°C at controlled densities in five separate cultures vials in which adults were allowed to mate until 5–7 days old. For each line, between seven and nine replicates of four females (up to two replications per culture vial) were lightly anaesthetised under CO2, and their intact ovaries immediately dissected for pulse-labelling. Immediately following the 1 h labelling reaction samples were thoroughly washed and centrifuged (three times on ice) to remove unincorporated 35 S-methionine (Johnson et al. 2009a). Each block of replication included all lines and the experiment was complete within 12 h. Counts per minute (CPM) of radioactive emissions were corrected for block effects by multiplying each by grand mean/block mean. Incorporation of label into protein and its successful recovery were confirmed using autoradiography by the detection of labelled protein bands from the supernatant run on electrophoretic gels. In addition, a control assay in which ovaries were pre-incubated with 3 mM cycloheximide (a protein synthesis inhibitor; Sigma-Aldrich Co, St Louis, C-7698; Bendena et al. 1989; Marcos et al. 1982) at 25°C for 20 min prior to and during labelled methionine incorporation confirmed that >75% of recovered label was due to protein synthesis. To account for line differences in ovary mass, radiolabel incorporation was standardised against total ovary protein measured in a separate set of females at the same age and raised under identical culture and density conditions as those used for radio-labelling. Three replicates per line each consisting of ovaries from ten dissected females were added to 100 μl of phosphate buffered saline, supplemented with anti-protease agents (2 mM phenyl methyl sulphonyl fluoride, and 1% (v/v) anti-protease cocktail (100 μg/ml pepstatin A, 50 μg/ml leupeptin, 10 mM benzamidine, 10 mM sodium metabisulphite)) in a centrifuge tube. Each replicate was thoroughly homogenised then centrifuged. The protein content of each replicate was assessed using the bicinchoninic acid (BCA) protein assay (Pierce Biochemicals). Lines differed in total ovary protein mass (F 3,8 = 13.8, p < 0.02), so rates of protein synthesis were expressed as counts per minute per microgram of ovarian protein.
Heat tolerance
Heat tolerance assays, using w 1118 and homozygous derived lines, were conducted in a similar manner to Hoffmann et al. (2002) but at both a mild and a severe stress temperature (37 and 39°C, respectively) and both with and without a hardening pre-treatment. For each heat-stress assay, all lines were reared in parallel at controlled densities. Adults aged 5–7 days were used with sexes separated under light CO2 at least 2 days prior to testing. For the hardening treatment, flies were exposed to 37°C for 1 h in 10 ml centrifuge tubes, then transferred to food vials and left to recover for 6 h before testing. Both untreated and hardened flies were tested within a single run in both assays and sexes run separately. For the 39°C assay, four flies of a single sex (either all hardened or not) were placed in a 33 ml sealed glass vial and submerged in a clear glass tank set to 39°C. Two runs for each sex were necessary to assay five replicate vials of hardened and untreated flies for all lines, with line, treatment and vial position randomised across all runs. The time taken for each fly to reach heat-induced paralysis was recorded. Similar randomisation of line, replicate and treatment occurred for the 37°C assay, except one fly was tested per vial and runs were repeated until approximately ten flies had been tested from each line and for each treatment. Sexes were run separately.
Statistical analyses
Effects of heat hardening, temperature and hsr-omega expression disruption on thermal tolerance were assessed using univariate one-way and two-way ANOVA functions in SPSS 17.0 for Windows. Sexes were analysed separately. Differences between lines for transcript levels were detected by Dunnett’s multiple comparison t tests, and effects on viability and heat tolerance were assessed using unpaired t tests comparing mutant lines individually to the precise excision line 29.
Results
By mobilising the EP3128 P-element three independent hsr-omega excision lines were selected from about 200 lines for further characterisation, one (line 29) with a precise excision and two (lines 56 and 66) that had sizeable genomic lesions as detected by the size of a PCR product generated from the proximal region of the promoter (Fig. 1c). Sequencing revealed that the deletion in line 56 included 1,853 bp of upstream 5′ regulatory sequences (from −13 bp), whereas the 1,598 bp deletion of upstream regulatory sequences in line 66 removed 9 bp of transcribed sequence including the transcriptional start site (TSS) (Fig. 1b). Sequence analysis indicated deletions of the TATA box (−31 bp), initiator sequence, and known heat-shock and GAGA elements in both lines (Mutsuddi and Lakhotia 1995; Ryseck et al. 1987). Additionally, short sequences of 41 and 24 bp were found inserted in place of the 56 and 66 deletions, respectively (Fig. 1b), likely resulting from the incorporation of excised fragments during DNA break repair. Promoter prediction analysis of the regions upstream of hsr-omega including these short insertions in the deletion lines suggested no new TSS was created in either excision event (data not shown). For the precise excision event, sequencing that extended from the gene located upstream until the beginning of the hsr-omega tandem repeat region confirmed the restoration of an intact promoter and 5′ gene region.
Expression levels of omega-n and omega-c, with and without heat exposure, were markedly affected in homozygous lines EP3128, 56 and 66 (Fig. 2). Precise excision of the P-element (line 29) restored near normal levels of both transcripts under basal and heat-shock conditions as assessed by comparison to w 1118 . The EP element itself (8 kb) inserted 5 bp upstream from the hsr-omega TSS (line EP3128) reduced basal omega-n and omega-c to 10–15% of normal. Notably, omega-c in EP3128 was still heat-inducible to roughly double that of basal levels (t 4 = 3.19, p = 0.017, see inset Fig. 2b), however, not close to the 16-fold induction observed for the control line w 1118. Heat-induction of omega-n was not significant for EP3128 (t 4 = 1.06, p = 0.17). The deletion in line 66 completely abolished expression of both transcripts as their amplification levels were not different from the background levels observed in no-template control reactions. While no omega-c was detected in line 56 (see insets of Fig. 2b), low levels of omega-n were consistently observed (insets of Fig. 2a). Omega-n in line 56 compared to control line w 1118 had about 1% of basal levels and 0.1% of heat-shocked levels as no increase of omega-n following heat treatment was detected (t 4 = 0.06, p = 0.478).

Disruption and rescue of expression levels of the heat-inducible transcripts omega-n (a) and omega-c (b) determined by qRT-PCR. Both basal (white) and heat-induced (36°C 1 h, grey) transcript levels in adult females are shown. ^Indicates not significantly different from zero levels as determined by comparison to negative control reactions (data not shown). Significant differences between w 1118 (control) and other lines within each treatment (ANOVA, Dunnett’s test, *p < 0.05, **p < 0.01, ***p < 0.001) are indicated above columns. Error bars represent +1 standard error, n = 3
Balanced cultures of line 66 (on TM6B) displayed noticeable levels of recessive lethality. Nonetheless, surviving homozygotes were fertile and able to be cultured. Viability tests confirmed semi-lethality in line 66 (Fig. 3) where almost half of line 66 individuals died at pre-pupal stages, and of those that survived to pupate, only 67% emerged as adults compared to 93% and 98% for lines 29 and w 1118, respectively. Line 66 was difficult to work with as very few healthy individuals could be obtained; adults were slow moving and cultures were susceptible to bacterial infection. In contrast, line 56 homozygotes showed no obvious affects of the deletion, as there was no difference in pupa-to-adult viability (Fig. 3). Cultures of line 56 were healthy and fertile. Examination of line 56 ovaries from several females confirmed all stages of oogenesis to be present and in ‘normal’ proportions (data not shown).

The affect of genomic deletions at the hsr-omega locus on viability. Viability was assessed at 25°C from egg to pupa (a), pupa to adult (b) and egg to adult (c). Comparisons are made between excision lines (grey) and control line 29 (two-tailed t test, **p < 0.01, ***p < 0.001). Error bars represent +1 standard error, n > 8 in all cases
Protein synthesis rates were determined for two lines with reduced hsr-omega expression, homozygous lines EP3128 and 56, and two control lines, w 1118 and homozygous line 29, and significant differences were detected (Fig. 4) as assessed by one-way ANOVA (F 3,31 = 12.00, p < 0.001). A Tukey post hoc test clearly identified two homogeneous subsets of two lines each, the control lines (w 1118, 29) and the mutant lines (EP3128, 56). Each line was significantly different to both lines in the alternate subgroup (all p < 0.01). The marked down-regulation of hsr-omega expression in the mutated lines has increased the levels of protein synthesis in ovarian tissues by about 1.5-fold.

Protein synthesis rates of control lines (w 1118 and 29) and lines with altered hsr-omega expression (EP3128 and 56). Dissected ovaries were metabolically labelled with 35 S-methionine for 1 h. Rates are measured as counts per minute (CPM) per microgram ovarian protein. Error bars represent ±1 standard error
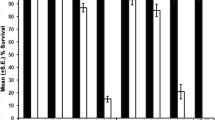
Effects of the homozygous hsr-omega mutations on heat-knockdown time were measured at 37°C and 39°C and analysed separately, as were the sexes. Note that average knockdown times were reduced from about 80 min when exposed to 37°C down to 20 min at 39°C. When tests were conducted at the milder temperature our hardening treatment did not significantly increase knockdown time, in both females (ANOVA, F 1,160 = 3.51, p = 0.063) and males (F 1,136 = 0.006, p = 0.937), so the treatment data were pooled to assess the effect of disrupting hsr-omega. Females from line 56 had a slightly reduced tolerance to 37°C compared to control lines, and while EP3128 also knocked down sooner, the result was not significant (Fig. 5a). In males, however, mutant lines were not significantly different from controls (Fig. 5b). In contrast, for the severe heat stress tests at 39°C, heat hardening caused a significant increase in knockdown time in both females and males (Fig. 6; females: F 1,36 = 13.86, p = 0.001; males: F 1,36 = 23.55, p < 0.001). For both unhardened and hardened treatments, however, the effects of the mutations on knockdown time were not evident. Consideration of the two hsr-omega disrupted lines only, EP3128 and line 56, left no doubt that the heat hardening treatment of the 39°C test was effective at increasing knockdown time in the disrupted lines (F 1,16 = 14.70, p < 0.001 for males and F 1,16 = 12.98, p < 0.01 for females).

Mild-temperature heat tolerances in hsr-omega mutant lines. Heat tolerance was measured by the heat-knockdown test at 37°C in females (a) and males (b). No affect of hardening treatment was detected for females (two-way ANOVA, F 1,160 = 3.51, p = 0.063) or males (F 1,136 = 0.006, p = 0.937), so treatment data were pooled to test genotype. A significant difference between control line 29 and hsr-omega mutant line 56 is denoted above column (one-tailed t test *p < 0.05). Error bars represent +1 standard error

High-temperature heat tolerances in hsr-omega mutant lines. Heat tolerance was tested at 39°C in females (a, b) and males (c, d). Basal (a, c) and hardened (1 h at 37°C followed by 6 h recovery, b, d) heat tolerance is shown. No significant differences between control (line 29) and hsr-omega mutant lines (EP3128 and line 56, grey) were detected. Error bars represent +1 standard error
Discussion
A high rate of P-element transposition into the hsr-omega gene in screens following P-element mutagenesis has provided numerous lines that could potentially disrupt expression of hsr-omega. We worked with line EP3128 that had potential to severely affect hsr-omega function due to the proximity of the insertion to the transcription start site (TSS). Homozygotes for EP3128 were healthy and fertile, unlike those previously described to lack hsr-omega expression (Mohler and Pardue 1984), suggesting that hsr-omega was still functional. This proved to be the case as expression of both omega-c and omega-n in EP3128 were not abolished, although they were markedly reduced. In addition, omega-c, but not omega-n, remained heat-inducible, suggesting the heat shock regulatory elements remain intact despite being shifted ~8 kb (the size if the P-element) upstream. The hsr-omega gene appears to have a striking capacity to remain functional even with large insertions. This is relevant given the high frequency of P transposable element insertions observed in the hsr-omega promoter in natural populations (Walser et al. 2006) and suggests that these insertions could result in significant natural variation in hsr-omega expression levels. Fine control of hsr-omega transcript levels in Drosophila adults appears not to be important for ‘normal’ development and viability.
To increase the chance of recovering deletions that included both a substantial portion of the hsr-omega regulatory region and of transcribed sequence a large number of independent excision lines (>200) were derived from EP3128. The deletions in lines 56 and 66 that removed core and responsive hsr-omega 5′ regulatory sequences (Garbe and Pardue 1986; Garbe et al. 1989) achieved this end. In line 66, the TSS was also deleted and no expression of omega-n nor omega-c was detected. However, line 56 while having no detectable omega-c had low but detectable levels of omega-n. How does line 56 achieve omega-n transcription given almost complete lack of promoter sequence? The presence of effective regulatory signals within the transcribed region of hsr-omega cannot be ruled out, although no consensus downstream promoter element (DPE) or motif ten element (MTE) appear to be present (Lim et al. 2004). In addition, it is possible that such a signal occurs within the short sequence found in place of the genomic deletion (Fig. 1b), however promoter prediction analysis suggests this is unlikely. The non-inducibility of omega-n in line 56 following heat shock is consistent with the absence of heat-shock elements in this line (Pelham 1982).
Gross differences in fitness occurred between homozygous cultures of lines 56, that was healthy, and 66 that was not. Explanations other than hsr-omega expression differences, such as a variable genetic background or multiple P-element insertions in EP3128, are unlikely since all lines derived in this manner retain w 1118 chromosomes and few EP lines have more than one insert (Berkeley Drosophila Genome Project). The development characteristics of these hsr-omega mutations are notably less severe than those described by Mohler and Pardue (1984) for their eGp4/GC14 deficiency trans-heterozygotes, and suggest that the additional lethality of their trans-heterozygotes is likely to be associated with other genes that were also disrupted by these chromosomal deficiencies. Our data indicate that only low concentrations of omega-n are enough for successful larval viability, pupation, and reproductive performance, as occurred in line 56 that had no detectable omega-c. While complete loss of both transcripts, as occurred in line 66, was not necessarily lethal and did not remove reproductive ability, surviving adults were generally weak unfit individuals.
The pre-adult fitness loss of line 66 that had no detectable omega-n could be due to the absence of this transcript in the prothoracic gland of larvae where it occurs at high levels (Mutsuddi and Lakhotia 1995). During larval development, this is the site of production for ecdysone, the major Drosophila larval moulting hormone that is required for developmental growth transitions (Berreur et al. 1979). It is possible that the previously described indirect interactions between omega-n and ecdysone production (Ashburner 1967; Lakhotia and Singh 1982) are crucial for normal pre-adult development, particularly for the transition between pre-adult stages—this interaction being missing in line 66.
Since recent data suggest that natural variation in levels of omega-n transcript influence basal rates of general protein synthesis (Johnson et al. 2009a), we examined protein synthesis in lines with disrupted hsr-omega expression. While we used ovaries from gravid females for convenience, and for tissue uniformity, transcript levels were measured in RNA extracts from whole adult females. We thought this not unreasonable since a large percentage of RNA extracted from gravid females will be ovarian derived and high levels of omega-n normally occur in ovarian nurse and follicle cells (Mutsuddi and Lakhotia 1995). We were also concerned that the ‘normal’ progression of oogenesis in the mutants could be different than the controls and affect levels of total protein synthesis measured at any particular age, despite the fact that these mutant females were healthy and fertile. To minimise such an effect we adjusted our measure of protein synthesis for differences in ovary size across lines. Thus, our test of this cellular model of omega-n function has been indirect; we have not studied a uniform tissue or cell system. Nonetheless, a reduction in hsr-omega expression led to the predicted increase in rates of ovarian protein synthesis. The result is consistent with the working model of omega-n function, whereby omega-n sequesters pre-messenger RNA splicing factors away from pre-mRNA processing activity (Prasanth et al. 2000) thus delaying production of mature mRNA for export and translation, and limiting levels of protein synthesis (Johnson et al. 2009a). Since omega-n shows a widespread and general tissue distribution (Lakhotia et al. 2001) we postulate that constitutive levels of omega-n may set an upper limit to protein synthesis that occurs in most adult tissues of unstressed Drosophila—a hypothesis that needs to be tested at both the cellular and tissue levels.
While we were only effective in testing heat hardening at the higher stress temperature it is clear that the deleted lines successfully heat hardened. Therefore, ‘normal’ expression levels of hsr-omega appear not to be an essential part of the heat hardening mechanism. Heat hardening capacity in D. melanogaster has recently been negatively associated with protein synthesis rates measured immediately following a mild heat shock (Johnson et al. 2009b), suggesting a link between the control of protein synthesis and heat hardening. While these hsr-omega mutations have had an effect on basal rates of protein synthesis in ovaries we have not detected an effect on adult fly hardening capacity.
The current data indicate that any effect of hsr-omega on adult heat-knockdown time, a specific measure of heat tolerance, is subtle and/or sex-specific. A small affect of hsr-omega in protection from heat paralysis was suggested by the increased knockdown time detected in mutant females in the less severe 37°C test, but not in mutant males. An overlapping but unique genetic architecture in each sex has been previously reported to underlie resistance to high temperature extremes in D. melanogaster (Morgan and Mackay 2006). Also, different measures of heat resistance can be surprisingly unrelated, both across strains and between sexes (Hoffmann et al. 1997), so a difference between sexes in this measure is not surprising.
In our more severe heat-knockdown tests at 39°C, no effects of the mutations were detected. Could hsr-omega affect heat-knockdown tolerance only at lower heat-shock temperatures? A functional hsr-omega could be more relevant in the 37°C test when individuals took about 80 min to knockdown (compared to ~20 min in the 39°C test). If body temperature is increased quickly, there may be insufficient time for a protective cellular heat-stress response to be implemented, as has been established for Drosophila cells in cultures above 37°C (Lindquist 1980). Small differences in exposure conditions can alter relevant underlying gene expression patterns (Krebs and Feder 1997; Sørensen et al. 2005; Stephanou et al. 1983) and result in different effects on any particular measure of heat tolerance. Slower heating conditions would be more relevant in natural populations on hot days when ambient temperatures increase slowly to stressful levels (Sgrò et al. 2010). However, it is possible that hsr-omega has no effect on heat-knockdown time, and the genotype associations detected in similar knockdown tests (Anderson et al. 2003; Rako et al. 2007) are due to affects of genes in linkage disequilibrium with hsr-omega, such as those within In ( 3R)P (although the tolerance associations in Rako et al. (2007) were independent of In(3R)P).
What heat-related fitness components might be affected by hsr-omega expression? The strongest evidence for a role of hsr-omega in heat tolerance comes from work on heat selected lines (McColl et al. 1996; McKechnie et al. 1998) where selection involved fast increase in body temperature to 39°C (resulting in a 5 min knockdown time) followed by a recovery period and breeding each generation. In these heat-tolerant lines, a protective role of hsr-omega could have been during recovery rather than during the heat stress itself. Perhaps hsr-omega acted to delay post-heat-stress mortality, not measured in the current study. In addition, given the high abundance of hsr-omega transcripts in the ovaries and testes (Mutsuddi and Lakhotia 1995) it is possible that hsr-omega contributes to a more efficient post-stress reproductive success, such as higher fertility or fecundity. These and other more subtle heat-related fitness tests need to be examined in the mutant strains.
The hsr-omega gene appears to be essential for full-fitness in an optimal environment since removal of both major transcripts resulted in a ‘sick’ strain. However, it is not a lethal gene since surviving homozygous males and females are viable and reproductively capable. It is likely that hsr-omega is important for a healthy and competitive organism in nature where daily and sometimes harsh temperature changes occur along with other environmental stresses. Our data suggest that hsr-omega is not a major gene that affects two commonly used laboratory measures of heat tolerance, heat-knockdown time when abrupt changes to high body temperature are applied, and heat hardening ability. Nonetheless, the expression responses of the gene following heat stress, and the repeated spatial and environmental genotype associations, are consistent with the idea that hsr-omega plays a role in coping with thermal stress in natural populations. Further research should elucidate whether or not these hsr-omega mutants influence heat-knockdown tolerance when slow natural rates of body-temperature increase are involved, or when measurements are made on post-heat-stress survival or reproductive performance. Perhaps of greater significance is our data suggesting that one hsr-omega transcript acts as a negative regulator of general protein synthesis. This idea needs to be tested directly at the molecular/cellular level, and if it proves to be correct hsr-omega modulation of protein synthesis could help facilitate changes in thermal tolerance.
References
Anderson AR, Collinge JE, Hoffmann AA, Kellett M, McKechnie SW (2003) Thermal tolerance trade-offs associated with the right arm of chromosome 3 and marked by the hsr-omega gene in Drosophila melanogaster. Heredity 90:195–202
Ashburner M (1967) Patterns of puffing activity in the salivary gland chromosomes of Drosophila. 1. Autosomal puffing patterns in a laboratory stock of Drosophila melanogaster. Chromosoma 21:398–428
Ashburner M, Bonner JJ (1979) The induction of gene activity in Drosophila by heat shock. Cell 17:241–254
Bendena W, Garbe J, Traverse K, Lakhotia S, Pardue M-L (1989) Multiple inducers of the Drosophila heat shock locus 93D (hsr omega): inducer-specific patterns of the three transcripts. J Cell Biol 108:2017–2028
Bernabucci U, Lacetera N, Baumgard LH, Rhoads RP, Ronchi B, Nardone A (2010) Metabolic and hormonal acclimation to heat stress in domesticated ruminants. Animal 4:1167–1183
Berreur P, Porcheron P, Berreur-Bonnenfant J, Simpson P (1979) Ecdysteroid levels and pupariation in Drosophila melanogaster. J Exp Zool 210:347–352
Clancy DJ, Kennington JW (2001) A simple method to achieve consistent larval density in bottle cultures. Dros Inf Serv 84:168–169
Collier RJ, Collier JL, Rhoads RP, Baumgard LH (2008) Invited review: genes involved in the bovine heat stress response. J Dairy Sci 91:445–454
Collinge JE, Anderson AR, Weeks AR, Johnson TK, McKechnie SW (2008) Latitudinal and cold-tolerance variation associate with DNA repeat-number variation in the hsr-omega RNA gene of Drosophila melanogaster. Heredity 101:260–270
Deutsch CA, Tewksbury JJ, Huey RB, Sheldon KS, Ghalambor CK, Haak DC, Martin PR (2008) Impacts of climate warming on terrestrial ectotherms across latitude. Proc Natl Acad Sci USA 105:6668–6672
Garbe JC, Pardue M-L (1986) Heat shock locus 93D of Drosophila melanogaster: a spliced RNA most strongly conserved in the intron sequence. Proc Natl Acad Sci USA 83:1812–1816
Garbe JC, Bendena WG, Pardue M-L (1989) Sequence evolution of the Drosophila heat shock locus hsrw. I. The nonrepeated portion of the gene. Genetics 122:403–415
Gerasimova TI, Gdula DA, Gerasimov DV, Simonova O, Corces VG (1995) A Drosophila protein that imparts directionality on a chromatin insulator is an enhancer of position-effect variegation. Cell 82:587–597
Healy TM, Tymchuk WE, Osborne EJ, Schulte PM (2010) Heat shock response of killifish (Fundulus heteroclitus): candidate gene and heterologous microarray approaches. Physiol Genomics 41:171–184
Hoffmann AA (2010) Physiological climatic limits in Drosophila: patterns and implications. J Exp Biol 213:870–880
Hoffmann AA, Dagher H, Hercus M, Berrigan D (1997) Comparing different measures of heat resistance in selected lines of Drosophila melanogaster. J Insect Physiol 43:393–405
Hoffmann AA, Anderson AR, Hallas R (2002) Opposing clines for high and low temperature resistance in Drosophila melanogaster. Ecol Lett 5:614–618
Horowitz M (2001) Heat acclimation: phenotypic plasticity and cues to the underlying molecular mechanisms. J Therm Biol 25:357–363
Johnson TK, Carrington LB, Hallas RJ, McKechnie SW (2009a) Protein synthesis rates in Drosophila associate with levels of the hsr-omega nuclear transcript. Cell Stress and Chaperones 14:569–677
Johnson TK, Cockerell FE, Carrington LB, Rako L, Hoffmann AA, McKechnie SW (2009b) The capacity of Drosophila to heat harden associates with low rates of heat-shocked protein synthesis. J Therm Biol 34:327–331
Krebs RA, Feder ME (1997) Natural variation in the expression of the heat-shock protein HSP70 in a population of Drosophila melanogaster and its correlation with tolerance of ecologically relevant thermal stress. Evolution 51:173–179
Laayouni H, Garcia-Franco F, Chavez-Sandoval BE, Trotta V, Beltan S, Corominas M, Santos M (2007) Thermal evolution of gene expression profiles in Drosophila subobscura. BMC Evol Biol 7:42–56
Lakhotia SC (1987) The 93D heat shock locus in Drosophila: a review. J Genet 66:139–157
Lakhotia SC, Singh AK (1982) Conservation of the 93D puff of Drosophila melanogaster in different species of Drosophila. Chromosoma 86:265–278
Lakhotia SC, Ray P, Rajendra TK, Prasanth KV (1999) The non-coding transcripts of hsr-omega gene in Drosophila: do they regulate trafficking and availability of nuclear RNA-processing factors? Curr Sci 77:553–563
Lakhotia SC, Rajendra TK, Prasanth KV (2001) Developmental regulation and complex organization of the promoter of the non-coding hsrω gene of Drosophila melanogaster. J Biosci 26:25–38
Larkindale J, Hall JD, Knight MR, Vierling E (2005) Heat stress phenotypes of Arabidopsis mutants implicate multiple signaling pathways in the acquisition of thermotolerance. Plant Physiol 138:882–897
Lim CY, Santoso B, Boulay T, Dong E, Ohler U, Kadonaga JT (2004) The MTE, a new core promoter element for transcription by RNA polymerase II. Genes Dev 18:1606–1617
Lindquist S (1980) Varying patterns of protein synthesis in Drosophila during heat shock: implications for regulation. Dev Biol 77:463–479
Marcos R, Lloberas J, Creus A, Xamena N, Cabré O (1982) Effect of cycloheximide on different stages of Drosophila melanogaster. Toxicol Lett 13:105–112
Mayrand S, Pederson T (1983) Heat shock alters nuclear ribonucleoprotein assembly in Drosophila cells. Mol Cell Biol 3:161–171
McColl G, Hoffmann AA, McKechnie SW (1996) Response of two heat shock genes to selection for knockdown heat resistance in Drosophila melanogaster. Genetics 143:1615–1627
McKechnie SW, Halford MM, McColl G, Hoffmann AA (1998) Both allelic variation and expression of nuclear and cytoplasmic transcripts of hsr-omega are closely associated with thermal phenotype in Drosophila. Proc Natl Acad Sci USA 95:2423–2428
Mohler J, Pardue M-L (1984) Mutational analysis of the region surrounding the 93D heat shock locus of Drosophila melanogaster. Genetics 106:249–265
Morgan TJ, Mackay TFC (2006) Quantitative trait loci for thermotolerance phenotypes in Drosophila melanogaster. Heredity 96:232–242
Mutsuddi M, Lakhotia SC (1995) Spatial expression of the hsr-omega (93D) gene in different tissues of Drosophila melanogaster and identification of promoter elements controlling its developmental expression. Dev Genet 17:303–311
Ohler U (2006) Identification of core promoter modules in Drosophila and their application in improved promoter prediction. Nucleic Acids Res 34:5943–5950
Pardue M-L, Bendena WG, Fini ME, Garbe JC, Hogan NC, Traverse KL (1990) Hsr-omega, a novel gene encoded by a Drosophila heat shock puff. Biol Bull 179:77–86
Pelham HRB (1982) A regulatory upstream promoter element in the Drosophila hsp70 heat shock gene. Cell 30:517–528
Prasanth KV, Rajendra TK, Lal AK, Lakhotia SC (2000) Omega speckles—a novel class of nuclear speckles containing hnRNPs associated with non-coding hsr-omega RNA in Drosophila. J Cell Sci 113:3485–3497
Rako L, Blacket MJ, McKechnie SW, Hoffmann AA (2007) Candidate genes and thermal phenotypes: identifying ecologically important genetic variation for thermotolerance in the Australian Drosophila melanogaster cline. Mol Ecol 16:2948–2957
Rørth P, Szabo K, Bailey A, Laverty T, Rehm J, Rubin GM, Weigmann K, Milan M et al (1998) Systematic gain-of-function genetics in Drosophila. Development 125:1049–1057
Ryseck R-P, Walldorf U, Hoffmann T, Hovemann B (1987) Heat shock loci 93D of Drosophila melanogaster and 48B of Drosophila hydei exhibit a common structural and transcriptional pattern. Nucleic Acids Res 15:3317–3333
Sgrò CM, Overgaard J, Kristensen TN, Mitchell K, Cockerell FE, Hoffmann AA (2010) A comprehensive assessment of geographic variation in heat tolerance and hardening capacity in populations of Drosophila melanogaster from eastern Australia. J Evol Biol 23:2484–2493
Sørensen JG, Nielsen MM, Kruhøffer M, Justesen J, Loeschcke V (2005) Full genome gene expression analysis of the heat stress response in Drosophila melanogaster. Cell Stress and Chaperones 10:312–328
Spradling A, Penman S, Pardue M-L (1975) Analysis of Drosophila mRNA by in situ hybridization: sequences transcribed in normal and heat shocked cultured cells. Cell 4:395–404
Stephanou G, Alahiotis SN, Christodoulou C, Marmaras VJ (1983) Adaptation of Drosophila to temperature: heat-shock proteins and survival in Drosophila melanogaster. Dev Genet 3:299–308
Thomas CD, Cameron A, Green RE, Bakkenes M, Beaumont LJ, Collingham YC, Erasmus BFN, Ferreira de Siqueira M et al (2004) Extinction risk from climate change. Nature 427:145–148
Walser J-C, Chen B, Feder ME (2006) Heat-shock promoters: targets for evolution by P transposable elements in Drosophila. PLoS Genet 2:1541–1555
Weeks AR, McKechnie SW, Hoffmann AA (2002) Dissecting adaptive clinal variation: markers, inversions and size/stress associations in Drosophila melanogaster from a central field population. Ecol Lett 5:756–763
Yost JH, Lindquist S (1986) RNA splicing is interrupted by heat shock and is rescued by heat shock protein synthesis. Cell 45:185–193
Acknowledgments
We are grateful to Winston Yee and Nicole Derycke for excellent technical help, to two anonymous reviewers for helpful comments on the manuscript, and to the Australian Research Council for financial support through their Special Research Centre scheme.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T. Clandinin.
Rights and permissions
About this article
Cite this article
Johnson, T.K., Cockerell, F.E. & McKechnie, S.W. Transcripts from the Drosophila heat-shock gene hsr-omega influence rates of protein synthesis but hardly affect resistance to heat knockdown. Mol Genet Genomics 285, 313–323 (2011). https://doi.org/10.1007/s00438-011-0610-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-011-0610-7