
Abstract
In this study, two melon bacterial artificial chromosome (BAC) clones have been sequenced and annotated. BAC 1-21-10 spans 92 kb and contains the nsv locus conferring resistance to the Melon Necrotic Spot Virus (MNSV) in melon linkage group 11. BAC 13J4 spans 98 kb and belongs to a BAC contig containing resistance gene homologues, extending a previous sequenced region of 117 kb in linkage group 4. Both regions have microsyntenic relationships to the model plant species Arabidopsis thaliana, and to Medicago truncatula and Populus trichocarpa. The network of synteny found between melon and each of the sequenced genomes reflects the polyploid structure of Arabidopsis, Populus, and Medicago genomes due to whole genome duplications (WGD). A detailed analysis revealed that both melon regions have a lower relative syntenic quality with Arabidopsis (eurosid II) than when compared to Populus and Medicago (eurosid I). Although phylogenetically Cucurbitales seem to be closer to Fabales than to Malphigiales, synteny was higher between both melon regions and Populus. Presented data imply that the recently completed Populus genome sequence could preferentially be used to obtain positional information in melon, based on microsynteny.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Melon (Cucumis melo L.) is a major horticultural crop worldwide. It belongs to the Cucurbitaceae family, which is only second to the Solanaceae as the most economically important vegetable crop. This diploid species (2n = 2x = 24) has a relatively small genome size of 4.5 × 108 bp (Arumuganathan and Earle 1991), being in the same range as the genome of rice and approximately three-times that of the Arabidopsis genome. Several melon genetic maps are available, consisting of molecular markers and a few important agronomic traits (Périn et al. 2002; Gonzalo et al. 2005). These studies have shown that melon is a highly diverse crop with wide variation in plant growth habit and morphology, as well as having diverse agronomical traits involved in fruit ripening, flowering time, and tolerance to biotic and abiotic stress.
Although important agronomic traits in crop species can be isolated by a map-based cloning approach [e.g., the Pto disease resistance gene in tomato (Martin et al. 1993) and the fw2.2 QTL in tomato (Frary et al. 2000)], the procedure is time consuming and expensive in crops without high-density maps, or with large genomes. Over the last few years, whole genome sequences of plant species such as Arabidopsis thaliana (The Arabidopsis Genome Initiative 2000), rice (International Rice Genome Sequencing Project 2005), and recently, black cottonwood (Populus trichocarpa) (Tuskan et al. 2006) have become available. These offer an efficient alternative for the isolation of agronomically important genes by making use of large blocks of conserved gene order between model plant species and the crop plant of interest (Schmidt 2002). By combining the information of annotated genome sequences with fine mapping strategies, the number of putative candidate genes in a delimited genomic region, harboring the trait of interest, can be reduced (Cnops et al. 1996; Schwarz et al. 1999). Microsynteny has been reported between Arabidopsis and tomato, near the ovate, and lateral suppressor regions (Ku et al. 2000; Rossberg et al. 2001) among others, leading in some cases to the identification of a candidate gene from the corresponding Arabidopsis region (Liu et al. 2002).
In melon, this strategy has resulted in the cloning of the nsv locus that confers resistance to the Melon necrotic spot virus (MNSV), a major viral disease (Nieto et al. 2006). When comparing bacterial artificial chromosome (BAC) end sequences from a 500 kb BAC contig near the melon nsv locus and the Arabidopsis genome, a syntenic region in Arabidopsis chromosome 4 was found (Nieto et al. 2006). Among the annotated genes in this region of the Arabidopsis genome, the eukaryotic translation initiation factor 4E (eIF4E) was identified as a good candidate for the nsv gene, which was confirmed after transient complementation experiments (Nieto et al. 2006). Previous work has also described sequence colinearity between two duplicated regions in A. thaliana and a 117 kb region in melon linkage group 4, showing that microsynteny may be found in short intervals between the two species, although important local genome rearrangements are frequently found (van Leeuwen et al. 2003). In order to get further insight into the genome organization around the nsv locus and microsynteny with closely related species, we have sequenced a melon 92 kb BAC clone containing the eIF4E region in linkage group 11 and another BAC clone of 98 kb from a contig in linkage group 4 that contains a cluster of disease resistance genes, extending the previously annotated BAC 60K17 (van Leeuwen et al. 2003). Both regions (92 and 215 kb) have been compared with the A. thaliana, Medicago truncatula, and P. trichocarpa genomes.
Materials and methods
BAC isolation and DNA preparation
The BAC clone 1-21-10, encompassing the nsv locus, was isolated from a melon genomic BAC library constructed from the MNSV susceptible WMR29 genotype (Nsv/Nsv) (Morales et al. 2005). BAC clone 13J4 belongs to the BAC contig MRGH63, a region in linkage group 4 that contains a cluster of resistance gene homologues (van Leeuwen et al. 2003, 2005). A PCR screening for BAC clones located downstream the 3′-end of the already sequenced melon BAC 60K17 was performed using DNA pools of the PIT92 BAC library (van Leeuwen et al. 2003), using BAC 60K17 3′-end specific primers. Isolated positive clones were analyzed for the size of the inserts using pulse-field electrophoresis, as described in van Leeuwen et al. (2003). BAC ends from the selected clones were sequenced in order to establish the relative position of the genomic fragments. Prior to the PCR sequence reaction, 10 μl DNA samples were mixed with 10 μl H2O and then sheared with a cut-off 0.8 mm syringe and incubated 30 min at 65°C. One μl DMSO, 2 μl SP6 or D primers, 8 μl premix, and 8 μl (Terminator cycle sequencing ready reaction kit, Applied Biosystems, Warrington, UK) were then added and the sequencing reaction performed as follows: 95°C 5′, 60× (95°C 60″, 50°C 50″, 60°C 4′). BAC 13J4 was then selected for shotgun sequencing based in a compromise between maximum insert length and minimum length of the overlapping region with BAC 60K17. BAC DNA was prepared as described by van Leeuwen et al. (2005).
Subcloning, sequencing, and assembly
A shotgun subcloning sequencing strategy was used to obtain the full-length insert sequence of BACs 1-21-10 and 13J4. Subclone libraries of BAC 1-21-10 and 13J4 consisting of 1,152 and 1,104 clones with average insert sizes of 1,500 and 2,000 bp, respectively, and plasmid DNA from these clones were provided by GATC Biotech, Konstanz, Germany. Insert fragments were cloned into the pCR® 4Blunt-TOPO® (1-21-10) and the pSAMRT-HCkan (13J4) vectors. Seven hundred and sixty-eight clones were sequenced for BAC 1-21-10 using the M13 forward primer and the BigDye™ Terminal Cycle DNA sequencing kit 1.1 (Applied Biosystems) with average reads of about 700 bp, so representing a fivefold coverage of the 92 kb BAC insert. For BAC 13J4, 858 independent readings using primers SL1 (5′CAGTCCAGTTACGCTGGAGTC) and SR2 (5′GGTCAGGTATGATTTAAATGGTCAGT) produced a sixfold coverage of the 98 kb BAC insert. The sequences were then assembled with the Pregap4 and Gap4 programs from the STADEN package (Staden 1996). Remaining gaps in the contigs were resolved by sequencing additional clones using the reverse-oriented primers.
Sequence analysis, gene prediction, and annotation
The nucleotide sequences of BACs 1-21-10 and 13J4 were initially analyzed using the TBLASTX program (Altschul et al. 1990) and subsequently with multiple ab initio gene prediction programs (van Leeuwen et al. 2003). The entire sequence of the BAC inserts was analyzed and then parsed into 3 and 6 kb pieces for similar analysis. Additional gene prediction programs available on the web were included for comparison, and to search for additional open reading frames that might not have been detected by the previous algorithms, such as ORNL GRAIL (Version 1.3) (http://compbio.ornl.gov/Grail-1.3/), FgenesH and BestORF (http://sun1.softberry.com), and AUGUSTUS (Stanke et al. 2006). Refinement of ab initio predicted genes was performed with web-based gene prediction programs that allow the incorporation of homologous NCBI-annotated gene structures to infer improved predictions, such as Genomescan (Yeh et al. 2001), GeneBuilder (http://l25.itba.mi.cnr.it/∼webgene/genebuilder.html) and Geneid (Parra et al. 2000). Further analysis of putative proteins was performed using TBLASTN and BLASTP (Altschul et al. 1997), SMART (http://smart.embl-heidelberg.de; Letunic et al. 2006), Blast2Sequences (http://genopole.toulouse.inra.fr/blast/wblast2.html), and InterProScan (Quevillon et al. 2005; http://www.ebi.ac.uk/InterProScan). The BAC sequences containing the predicted melon proteins were further analyzed with TBLASTX and TBLASTN at the MELOGEN melon database (http://www.melogen.upv.es) to look for the presence of ESTs for the predicted genes. Repetitive sequences were searched using STADEN (Staden 1996), Sputnik (Abajian 1994), Webtroll (Castelo et al. 2002), and Palindrome (http://bioweb.pasteur.fr/seqanal/interfaces/palindrome; Rice et al. 2000) programs.
Analysis of microsynteny
Predicted melon proteins were analyzed for synteny with A. thaliana at the NCBI Blast site (http://www.ncbi.nlm.nih.gov/blast/) with the program TBLASTN. The BLAST parameters were modified to restrict the search to Arabidopsis sequences. In addition, this process was repeated by limiting the search to nucleotide entries with lengths between 100 and 6,000 bases to select for cDNA sequences. Syntenic regions were defined as contiguous regions containing two or more homologous genes in A. thaliana and C. melo, irrespective of the orientation and exact order of the genes. These regions were investigated within the ‘Genomic Content’ and ‘MapViewer’ sections at NCBI to look at all the genes. Synteny between C. melo and M. truncatula was identified using the TBLASTN program under standard conditions at http://www.medicago.org/genome. The syntenic gene sequences in the annotated Medicago BAC clones were retrieved from NCBI. Synteny between C. melo and P. trichocarpa was also analyzed at the Populus genomeDB (http://www.genome.jgi-psf.org/Poptr1). Levels of identity and similarity between the amino acid sequences were determined with the BLASTP program of BLAST 2 Sequences.
The relative syntenic quality in a region, expressed as a percentage, was calculated by dividing the sum of the conserved genes in both syntenic regions by the sum of the total number of genes in both regions, excluding retroelements and transposons, and collapsing tandem duplications (Cannon et al. 2006).
For phylogenetical analysis of paralogous genes, sequences were aligned using the MEGA3 package (Kumar et al. 2004).
Results
BACs 1-21-10 and 13J4 shotgun sequence and annotation
The sequence of the BAC clones 1-21-10 and 13J4 was obtained using a shotgun sequencing approach after sequencing 768 and 858 shotgun clones, respectively. The size of the BAC inserts was 92,343 and 98,716 bp, with GC% contents of 33.0 and 33.9% (Table 1). Sequence annotation revealed 13 genes in BAC 1-21-10 and 16 genes and in BAC 13J4 (including putative transposable elements) (Table 1). Gene densities within BACs 1-21-10 and 13J4 were similar with one gene per 7.7 and 6.8 kb, respectively, considering the partial genes at the 5′ and 3′ BAC ends as half genes.
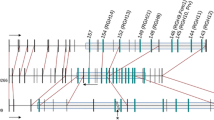
Thirteen genes were annotated in BAC 1-21-10, with two partial genes in the 5′ and 3′ borders (Fig. 1, Table 2). Eight of the predicted genes are supported by an EST from the MELOGEN database (www.melogen.upv.es). The nsv gene, which was cloned using a positional cloning and microsynteny approach (Nieto et al. 2006), is located from 23,049 to 25,863 bp in BAC 1-21-10. This gene encodes a eukaryotic transcription initiation factor (eIF) with high homology to eIF4E from Arabidopsis. 15d_03-C10 and 15d_01-G03 genes were predicted from the presence of matching ESTs in the melon database. These putative novel genes do not show any similarity with sequences in the NCBI or in the Populus genome database. The SH3P3 gene, which is located in position 35,930–51,616 bp, contains a last intron of 7 kb in length, increasing the overall length of the gene drastically when compared to its Arabidopsis homolog. Sequence analysis showed that the intron is longer largely because of the insertion of a retroelement, although the intron/exon splice site and donor site of the melon SH3P3 gene are intact. The retroelement is homologous to the Ty3-gypsy class of transposable elements (see below) previously described in melon by van Leeuwen et al. (2003). A transposase belonging to the DDE superfamily is also found in position 72,923–77,435 bp. This transposase belongs to a possibly autonomous mobile genetic element related to the insertional sequence 4 (IS4) (Klaer et al. 1981), which is required for the excision and insertion of the mobile element.

Overview of microsynteny between melon BAC 1-21-10 and regions in the Arabidopsis thaliana, Medicago truncatula, and Populus trichocarpa genomes. Genes are represented by arrows, with the gene name, number or ID above or below it. Homologous genes are illustrated with the same color and indicated by narrow connecting lines of the corresponding color. Arrows representing genes that have one or more ESTs are spotted. Genes without homologs are black. Transposable elements are in gray and indicated by Tn. Mt4 consists in BAC clones AC137837, AC153460, and AC144608. At1g, At2g, At4g Arabidopsis thaliana chromosomes 1, 2, and 4, respectively, Cm11 Cucumis melo linkage group 11, Pt_XI Populus trichocarpa linkage group XI, Pt_204 Populus unmapped scaffold 204, Mt4 Medicago truncatula chromosome 4. * = End of scaffold. Figure not drawn to scale
Sixteen genes were annotated in BAC 13J4, with two partial genes in the 5′ and 3′ borders (Table 2). Five of the predicted genes are supported by an EST from the MELOGEN database (www.melogen.upv.es). The Mki1 gene in the 5′ border of BAC 14J4 corresponds to the 3′ of the Mki1 gene that was annotated in BAC 60K17 (van Leeuwen et al. 2003) (Fig. 2). The gene is interrupted by the insertion of a residual retroelement of the Ty3-gypsy class. BACs 60K17 and 13J4 are not contiguous and they are separated by an unsequenced region of around 3 kb that may contain two exons of the Mki1 gene that are missing in both BAC ends (Fig. 2). 15d_15-H04 gene was predicted by the presence of an EST in the MELOGEN database, and it shows no homology with sequences present in public databases. A second retrotransposon is found in the 3′ region, containing regions with homology to a reverse transcriptase and an integrase of the copia-type. A partial resistance gene (R-gene) homolog (MRGH) is found in the 3′ of BAC 13J4, confirming the presence of a cluster of disease resistance gene homologues in this region (van Leeuwen et al. 2003).

Overview of microsynteny between BAC contig in melon linkage group 4 (60K17 and 13J4) and regions in the Arabidopsis thaliana, Medicago truncatula, and Populus trichocarpa genomes. Genes are represented by arrows, with the gene name, number or ID above or below it. Homologous genes are illustrated with the same color and indicated by narrow connecting lines of the corresponding color. Arrows representing genes that have one or more ESTs are spotted. Genes without homologs are black. Transposable elements are in gray and indicated by Tn. At1g, At3g, At5g Arabidopsis thaliana chromosomes 1, 3, and 5, respectively, Cm4 Cucumis melo linkage group 4, Pt_XIII Populus trichocarpa linkage group XIII, Pt_70 Populus unmapped scaffold 70, Mt1 Medicago truncatula chromosome 1, Mt7 Medicago truncatula chromosome 7. Bars in Cm4 indicate a 3 kb gap of sequence between 60K17 and 13J4. About 40 kb in the 5′ of Cm4 corresponding to BAC 60K17 are not represented. Figure not drawn to scale
Repetitive sequences in BACs 1-21-10 and 13J4
CURE retrotransposons (Cucumis retrotransposable element, van Leeuwen et al. 2003) were discovered into the last intron of the melon SH3P3 gene (BAC 1-21-10), and into the Mki1 gene (BAC 13J4). The CURE retrotransposons, identified based on strong homology to previously isolated retroelement polyproteins from Arabidopsis and rice, were established as Ty3-Gypsy retroelements due to the protein domains found and their distribution. The total length of the retroelements was 5,413 and 2,862 bp, the second one probably representing a residual retroelement with homology to a reverse transcriptase.
Simple sequence repeats (SSRs) were identified from BACs 1-21-10 and 13J4 using identical parameters to those described by van Leeuwen et al. (2003, 2005) (Table 1). The average frequency of occurrence in 1-21-10 was one SSR per 0.37 kb (245 SSRs in 92.3 kb), and in 13J4 it was one SSR per 0.97 kb (101 SSRs in 98.7 kb). For 1-21-10, it corresponds to the same value found in melon BAC 60K17 (1 SSR per 0.35 kb; van Leeuwen et al. 2003).
Two large palindromic sequences were identified using the program Palindrome in BAC 1-21-10. The first sequence was over 62 bp from 59,791 to 59,852 bp, and its palindrome was located between 60,193 and 60,254 bp. It contained only 6 mismatches in total. The second palindromic sequence was interspersed with the first one, running from 59,882 to 59,968 bp, with the palindromic sequence between 60,068 and 60,154 bp. This latter palindromic region was 88 bp in length and had five mismatches. The palindromic sequences were located in a region containing a large number of SSRs.
Synteny between A. thaliana, M. truncatula, P. trichocarpa, and the melon nsv region
Microsynteny between the region surrounding the nsv locus in melon and a 128 kb region on chromosome 4 of A. thaliana was previously described by Nieto et al. (2006). To further analyze the presence and nature of the microsynteny between melon and Arabidopsis, we compared the amino acid sequences of the predicted melon proteins in BAC 1-21-10 with the NCBI database using TBLASTN and BLASTP. Two additional Arabidopsis genomic regions were identified on chromosomes 1 and 2 that harbored multiple genes with similarity to those predicted in BAC 1-21-10 (Fig. 1).
Four melon genes are found in a syntenic region in Arabidopsis At4g <20 kb in length (Fig. 1, Table 3). These genes are not part of multigene families, but are either single copy genes or have one to three paralogous genes in Arabidopsis. Among the syntenic genes, the nsv locus in melon was 78% identical to At4g18040. This 20 kb region in Arabidopsis chromosome 4 contains eight genes in total, leading to a gene density of 1 gene per 2.9 kb. Beyond this 20 kb region, we screened for further homologies, in both directions, by browsing the Arabidopsis MapViewer (http://www.ncbi.nlm.nih.gov/mapview/maps.cgi?), but no further genes were found with synteny to melon. The resulting relative syntenic quality between the nsv region in melon and the corresponding region in Arabidopsis on chromosome 4 is 44.4% (Table 4).
We also found several syntenic genes on At1g, with a syntenic quality of 50% after collapsing all tandem duplications on Arabidopsis and excluding the transposable elements in the melon nsv region (Table 4). The gene density of this 32.8 kb Arabidopsis region, with 13 genes, is one gene per 2.7 kb. The melon nsv gene was homologous to two genes in this region, At1g29550 and At1g29590 (Table 3), suggesting a duplication of the ancestral eIF4E gene during evolution in the lineage that led to Arabidopsis. Both Arabidopsis proteins showed high similarity to the melon eIF4E using BLASTP (65% identity), as well as to each other (95% identity). This duplication event is also suggested from the duplicated genes At1g29610 and At1g29600, which with the eIF4E gene At1g29590, form a tandem duplication block with At1g29570, At1g29560, and At1g29550.
Further synteny in the nsv region between melon and Arabidopsis was revealed on At2g with a syntenic quality of 25% (Table 4). With one gene every 3.2 kb, gene density in this 15.9 kb region is lower than the in the above mentioned Arabidopsis syntenic regions, although it is still more than double the density of the melon nsv region. An eIF4E paralog is absent in Atg2.
To uncover higher degrees of synteny with the region harboring the nsv locus, we analyzed the partial sequence of the M. truncatula genome. Medicago has been phylogenetically grouped among the eurosids I, along with the Cucurbitaceae, whereas Arabidopsis, as a member of the Brassicaceae is clustered within the clade of the eurosids II (Hilu et al. 2003).
The nsv region in melon showed synteny to a single contiguous region in M. truncatula consisting of BAC clones AC137837, AC153460, and AC144608 (Fig. 1). Regions flanking the contiguous region containing the synteny were analyzed, but additional homologs of the nsv region in melon were not identified. The Medicago syntenic region between TPS1 and SH3P3 spans 51.5 kb and consists of nine predicted genes, giving an average of one gene per 6.4 kb. The low number of syntenic genes between melon and Medicago leads to a rather low-relative syntenic quality of 30.0% (Table 4).
With the publication of the black cottonwood (P. trichocarpa) genome sequence (Tuskan et al. 2006), another member of the eurosid I clade of higher eudicots was analyzed for putative synteny with melon. The melon BAC 1-21-10 has homology to two syntenic regions in the Populus genome (Table 3). The first region is found on chromosome XI, whereas the other is the unmapped scaffold 204. The region on chromosome XI is a 205.3 kb region containing 17 predicted genes, with seven of them showing synteny with the nsv region resulting in a syntenic quality of 53.8% (Table 4). The syntenic genes have conserved the order and orientation, with the exception of the exonuclease gene.
The scaffold 204 is an 88.8 kb segment of the Populus genome that stops abruptly upstream from an ABC transporter (ID 100357) (Fig. 1). Synteny to melon was found in four genes, with a syntenic quality between scaffold 204 and the melon nsv region of 40.9% (Table 4). Additionally, a metallophosphoesterase gene (ID 674792) is highly conserved in the syntenic region on Populus chromosome XI (ID 568470), and the pentatricopeptide (PPR) gene (ID 100353) is conserved in the Medicago syntenic region (gene21).
Synteny between A. thaliana, M. truncatula, P. trichocarpa, and the melon region in linkage group 4
In order to confirm the synteny found around the nsv locus and Arabidopsis, Medicago, and Populus, a second melon genomic region containing a cluster of disease resistance genes was also compared. The region of 215 kb spans a contig of two BACs, 60K17 (117 kb, van Leeuwen et al. 2003) and 13J4 (98 kb) that are separated by a gap of 3 kb (Fig. 2). High microsynteny between 60K17 and two Arabidopsis regions in At3g and At5g was previously described, but 60K17 was not compared with Populus and Medicago (van Leeuwen et al. 2003). The 5′ 40 kb region of 60K17 was not included in Fig. 2 because it did not show synteny with any of the three sequenced species.
Again, syntenic regions were found between melon and three segments of Arabidopsis (At1g, At3g, and At5g), two segments of Populus (Pt_XIII and scaffold Pt_70), and two segments of Medicago (Mt1 and Mt7). The highest syntenic quality values were obtained between melon and Pt_70 (59.6%) and Pt_XIII (54.2%). The lower syntenic values were obtained with the three Arabidopsis regions (from 25.6 to 47.1%). The At1g region was not found in van Leeuwen et al. (2003) because synteny is higher in the region of the newly described BAC 13J4.
Discussion
The melon BAC clone 1-21-10 was sequenced to obtain information on gene distribution around the nsv locus in melon linkage group 11. Its physical structure was analyzed in detail and the predicted genes were annotated. A second genomic region in linkage group 4 consisting in two BAC clones (60K17 an 13J4) spanning 215 kb was also analyzed. BAC 13J4 was sequenced, whereas BAC 60K17 sequence was already available (van Leeuwen et al. 2003). Subsequently, the microsynteny between the melon genes in both regions to other eudicotyledonous model plant species was established.
The BACs 1-21-10 and 13J4 contain a total of 13 and 16 predicted genes, respectively. Among them are homologs of transposable elements belonging to different classes: two Ty3-gypsy retrotransposons, a transposase sequence, and a retrotransposon with homology to copia-like elements. Transposable elements have been shown to be important for the physical structure of many plant genomes (Schmidt 2002). Amplification of these elements plays a large part in the shaping of plant genomes, causing insertions, deletions, and even translocations of genes to new locations in the genome (Bennetzen 2000). Integration of transposons into the genome often occurs in gene poor areas, leading to an increase in genome size over time (Bennetzen et al. 2005; Biemont and Vieira 2006). Transposon integration within the coding sequence of a gene frequently leads to a knock-out allele of that gene. In BAC 1-21-10, however, a retrotransposon was found within the intron sequence of the melon SH3P3 gene. This event apparently left the coding region of the gene intact, while significantly increasing the total size of the gene.
Three novel genes 15d_03-C10, 15d_01-G03, and 15d_15-H04 were found that did not show similarity to any gene in the sequence databases. They were predicted due to the isolation of corresponding EST sequences in the melon EST database. This shows that the melon EST database could be a useful tool for gene discovery, annotation, and description, as other species such as Arabidopsis (The Arabidopsis Genome Initiative 2000), rice (International Rice Genome Sequencing Project 2005), and Populus (Tuskan et al. 2006).
The rest of the predicted genes in BACs 1-21-10 and 13J4 are homologous to single or low-copy number genes of known function, with ESTs supporting the expression of most of them (Table 2). The values of 33.0 and 33.9% GC content in 1-21-10 and 13J6, respectively, are similar to values measured in two previously annotated melon BACs, 60K17 (32.4%) and 31O16 (33.9%) (van Leeuwen et al. 2003, 2005). Gene densities for the new BACs were also similar to melon BACs 60K17 and 31O16, with one gene per 7.7 kb and 6,8 kb as opposed to one gene per 9.0 and 8.0 kb, respectively (van Leeuwen et al. 2003, 2005). These values are higher than in the Populus genome (1 gene per 11.1 kb), but lower than in Arabidopsis and Medicago (1 gene per 4.5 and 6.0 kb, respectively) (Tuskan et al. 2006; The Arabidopsis Genome Initiative 2000; Mudge et al. 2005). It has been suggested that gene density is primarily related to the average length of introns and non-coding intergenic DNA (van Leeuwen et al. 2003; Kevei et al. 2005; Schmidt 2002). The differences in gene spacing might be influenced by local genomic arrangements. Arabidopsis and Populus gene densities are based on the availability of the whole genome sequence, whereas in Medicago a 3 Mb section of the genome was used for the calculation. In melon, however, the estimation of gene density was based on four BAC sequences, which may not be representative for the overall gene density in the species.
Microsynteny has been previously described among many monocot and eudicot species (Ku et al. 2000; Schmidt 2002; Yan et al. 2003). Unraveling microsynteny between phylogenetically related genomes has allowed the isolation of genes encoding agronomically important traits (Ku et al. 2000; Rossberg et al. 2001; Ramakrishna and Bennetzen 2003). Melon is an important crop species that lacks genome sequence information. The isolation of genes responsible for agronomically important traits could be speeded up by analyzing genomic regions of closely related model species, as has been shown by cloning the nsv resistance gene to MNSV using data based on Arabidopsis (Nieto et al. 2006). In this report, we have analyzed the corresponding syntenic regions in two additional dicot species, M. truncatula and P. trichocarpa. A second unrelated region in the melon genome has also been analyzed for comparison.
Phylogenetically, Medicago and Populus are more closely related to melon than Arabidopsis; however, their exact relationships remain unclearly defined within the eurosids I clade (Hilu et al. 2003; Judd and Olmstead 2004). Arabidopsis is a eurosid II member species, being more distantly related to the other three species. This taxonomical classification has been mirrored in our microsyntenic analysis (Figs. 1, 2). Both melon regions are scattered over three Arabidopsis chromosomes, with some chromosomal regions having retained more genes arranged syntenically than others. Synteny, measured as syntenic quality, was usually lower between melon and Arabidopsis than with the eurosids I species (Table 4). In contrast, synteny of the melon region in linkage group 11 with Populus was highest on linkage group XI (53.8%), and still fairly high on scaffold 204 (40.9%). A similar result was obtained when comparing the region in linkage group 4 and the two corresponding syntenic regions in Populus. The level of synteny between Populus chromosome XI and melon might be even higher, as several genes in Populus may be incorrectly annotated, since they are not supported by ESTs and do not have homologies with known genes. The Populus scaffold 204 also ends upstream from the ABC transporter gene (ID100357), preventing further insight into a putative extension of synteny in this direction. Synteny values between melon in linkage group 4 and Medicago were also high, whereas low-syntenic values were found between the melon nsv region and Medicago (30%).
Synteny was previously demonstrated between Arabidopsis chromosome 4 and the nsv BAC contig containing BAC 1-21-10 (Nieto et al. 2006). Three BAC ends in the contig containing BAC 1-21-10 had high homology with genes in the chromosome 4 region (At4g18260, At4g18810, and At4g20140). We searched for this synteny in Populus, Medicago, and Arabidopsis chromosomes 1 and 2. Although synteny was absent in Medicago and A. thaliana chromosomes 1 and 2, the Populus protein ProtID 660544 on linkage group XI had homology to BAC end 41I23sp6 (63% identity). In addition, internal sequences from BAC 7K20 are syntenic with the LRR protein kinase RFK1 (At1g29750; 50% identity) and the Populus homolog ProtID 82916 (identity 63%) on chromosome XI, showing that microsynteny of the nsv region may extend beyond BAC 1-21-10.
Independent whole genome duplications (WGD) have been suggested for Arabidopsis, Medicago, and Populus after separation of the eurosids I and II and within eurosids I after the separation of Salicaceae and Fabaceae (Tuskan et al. 2006; Cannon el al. 2006), followed by selective gene divergence and gene loss (Ku et al. 2000). In Populus a recent duplication affected ∼92% of the genome and a more ancient duplication may have happened in an ancestral eurosid lineage, probably shared by Populus and Arabidopsis (Tuskan et al. 2006). We have found two syntenic regions for each melon region in Populus and three in Arabidopsis. In Medicago, two syntenic regions with melon linkage group 4 have been found and only one with melon linkage group 11, which might be attributed to the sequence of the Medicago genome only being partial (149 Mbp available from the estimated 475 Mbp in Cannon et al. 2006). Our findings would reflect the polyploidy of the three genomes used for comparison with melon, and for Arabidopsis even the existence of a more ancient WGD event, as we found three syntenic regions for each melon segment. A comparison of a 105-kb region from the ovate-containing region of tomato with Arabidopsis revealed four syntenic segments probably originated after two genome duplication events in Arabidopsis (Ku et al. 2000). The same pattern of synteny has also been obtained between the coffee SH3 region and Arabidopsis (Mahé et al. 2007). Both tomato and coffee are asterids, more distant phylogenetically to Arabidopsis than melon. The Drzf gene in melon linkage group 4 is found in the syntenic regions of At3g, At5g, Pt_70, Pt_XIII, Mt1, and Mt7 (Fig. 2). A phylogenetical analysis of the Drzf paralogous sequences has shown that each pair of genes from Arabidopsis, Populus, and Medicago is located in the same cluster, respectively. The same result is obtained for other paralogous genes that are found in two syntenic regions from the same species in Figs. 1 and 2 (SH3P3, Mki1, Gpi1/2, nGTP, GTPb, Gtf, NPK1, and Tangled from Populus; HLH, Gpi1/2, Gtf, and Tangled from Medicago; Spp from Arabidopsis), probably indicating that each gene pair is the result of the recent genome duplications that happened independently in Populus, Medicago, and Arabidopsis after they diverged.
The higher microsynteny found between both melon regions and Populus may be attributed to the reduced rate of evolution in Populus since the ancient eurosid whole-genome duplication (Tuskan et al. 2006). A second possibility is that Cucurbitaceae are phylogenetically closely related to Salicaceae than to Fabaceae. Hilu et al. (2003) studied the angiosperm phylogeny using the plastid matK gene and classified Cucurbitales and Fabales in a different clade than Malphigiales, among eurosids I. We have compared the paralogous genes among melon syntenic regions and Arabidopsis, Medicago, and Populus. From the 14 genes tested, half of them were closer phylogenetically to Populus, whereas the other half were closer to Medicago. In general, Arabidopsis genes were always more distant to the other three species, as expected. With the data presented here it is not possible to resolve the phylogenetical relationships between Cucurbitaceae, Salicaceae, and Fabaceae. It should be pointed out that we have only compared two independent melon regions spanning around 300 kb, 0.06% of the melon genome. More melon sequences are necessary in order to verify if the higher mycrosynteny with Populus is consistent and due to closer phylogenetical relationships between both species. Also, additional comparisons should be made when the complete Medicago genome is available.
The data presented may suggest that the Populus genome is a useful dicot genome to obtain positional information on candidate genes for crop species in the Cucurbitaceae family, thereby facilitating their identification. Research into dicot crop species will benefit over the next few years with the advent of new plant genome sequences (e.g., tomato, Lotus japonicus, grape) and the completion of the Medicago genome.
References
Abajian C (1994) SPUTNIK. Bioinformatics 20:1475–1476
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Arumuganathan K, Earle ED (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Rep 9:208–218
Biemont C, Vieira C (2006) Junk DNA as an evolutionary force. Nature 443:521–524
Bennetzen JL (2000) Transposable element contributions to plant gene and genome evolution. Plant Mol Biol 42:251–269
Bennetzen JL, Ma J, Devos KM (2005) Mechanisms of recent genome size variation in flowering plants. Ann Bot 95:127–132
Cannon SB, Sterck L, Rombauts S, Sato S, Cheung F, Gouzy G, Wang X, Mudge J, Vasdewani J, Scheix T, Spannagl M, Monaghan E, Nicholson C, Humphray SJ, Schoof H, Mayer KFX, Rogers J, Quetier F, Oldroyd GE, Debelle F, Cook DR, Retzel EF, Roe BA, Town CD, Tabata S, Van de Peer Y, Young ND (2006) Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc Natl Acad Sci USA 103:14959–14964
Castelo A, Martins W, Gao G (2002) TROLL–tandem repeat occurrence locator. Bioinformatics J 18:634–636
Cnops G, den Boer B, Gerats A, Van Montagu M, Van Lijsebettens M (1996) Chromosome landing at the Arabidopsis TORNADO1 locus using an AFLP-based strategy. Mol Gen Genet 253:32–41
Frary A, Nesbitt TC, Frary A, Grandillo S, van der Knaap E, Cong B, Liu J, Meller J, Elber R, Alpert K, Tanksley S (2000) Cloning and transgenic expression of fw2.2: a quantitative trait locus key to the evolution of tomato fruit. Science 289:85–87
Gonzalo MJ, Oliver M, Garcia-Mas J, Monfort A, Dolcet-Sanjuan R, Katzir N, Arus P, Monforte AJ (2005) Simple-sequence repeat markers used in merging linkage maps of melon (Cucumis melo L). Theor Appl Genet 110:802–811
Hilu KW, Borsch T, Müller K, Soltis DE, Soltis PS, Savolainen V, Chase MW, Powell M, Alice LA, Rodger Evans R, Sauquet H, Neinhuis C, Slotta TA, Rohwer JG, Campbell CS, Chatrou L (2003) Angiosperm phylogeny based on matK sequence information. Am J Bot 90:1758–1776
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
Judd WS, Olmstead RG (2004) A survey of tricolpate (eudicot) phylogenetic relationships. Am J Bot 91:1627–1644
Kevei Z, Seres A, Kereszt A, Kalo P, Kiss P, Toth G, Endre G, Kiss GB (2005) Significant microsynteny with new evolutionary highlights is detected between Arabidopsis and legume model plants despite the lack of macrosynteny. Mol Gen Genomics 274:644–657
Klaer R, Kuhn S, Tillmann E, Fritz HJ, Starlinger P (1981) The sequence of IS4. Mol Gen Genet 181:169–175
Ku H-M, Vision T, Liu J, Tanksley SD (2000) Comparing sequenced segments of the tomato and Arabidopsis genomes: large-scale duplication followed by selective gene loss creates a network of synteny. Proc Natl Acad Sci USA 97:9121–9126
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinformatics 5:150–163
Letunic I, Copley RR, Pils B, Pinkert S, Schultz J, Bork P (2006) SMART 5: domains in the context of genomes and networks. Nucleic Acids Res 34:D257–D260
Liu J, Van Eck J, Cong B, Tanksley SD (2002) A new class of regulatory genes underlying the cause of pear-shaped tomato fruit. Proc Natl Acad Sci USA 99:13302–13306
Mahé L, Combes M-C, Lashermes P (2007) Comparison between a coffee single copy chromosomal region and Arabidopsis duplicated counterparts evidenced high level synteny between the coffee genome and the ancestral Arabidopsis genome. Plant Mol Biol 64:699–711
Martin GB, Brommonschenkel SH, Chunwongse J, Frary A, Ganal MW, Spivey R, Wu T, Earle ED, Tanksley SD (1993) Map-based cloning of a protein kinase gene conferring disease resistance in tomato. Science 262:1432–1436
Morales M, Orjeda G, Nieto C, van Leeuwen H, Monfort A, Charpentier M, Caboche M, Arús P, Puigdomènech P, Aranda MA, Dogimont C, Bendahmane A, Garcia-Mas J (2005) A physical map covering the nsv locus that confers resistance to melon necrotic spot virus in melon (Cucumis melo L). Theor Appl Genet 111:914–922
Mudge J, Cannon SB, Kalo P, Oldroyd GE, Roe BA, Town CD, Young ND (2005) Highly syntenic regions in the genomes of soybean, Medicago truncatula, and Arabidopsis thaliana. BMC Plant Biol 5:15
Nieto C, Morales M, Orjeda G, Clepet C, Monfort A, Sturbois B, Puigdomenech P, Pitrat M, Caboche M, Dogimont C, Garcia-Mas J, Aranda M, Bendahmane A (2006) An eIF4E allele confers resistance to an uncapped and non-polyadenylated RNA virus in melon. Plant J 48:452–462
Parra G, Blanco E, Guigó R (2000) Geneid in drosophila. Genome Res 10:511–515
Périn C, Hagen LS, De Conto V, Katzir N, Danin-Poleg Y, Portnoy V, Baudracco-Arnas S, Chadoeuf J, Dogimont C, Pitrat M (2002) A reference map of Cucumis melo based on two recombinant inbred line populations. Theor Appl Genet 104:1017–1034
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33:W116–W120
Ramakrishna W, Bennetzen JL (2003) Genomic colinearity as a tool for plant gene isolation. Methods Mol Biol 236:109–122
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277
Rossberg M, Theres K, Acarkan A, Herrero R, Schmitt T, Schumacher K, Schmitz G, Schmidt R (2001) Comparative sequence analysis reveals extensive microcolinearity in the lateral suppressor regions of the tomato, Arabidopsis and Capsella genomes. Plant Cell 13:979–988
Schmidt R (2002) Plant genome evolution: lessons from comparative genomics at the DNA level. Plant Mol Biol 48:21–37
Schwarz G, Michalek W, Mohler V, Wenzel G, Jahoor A (1999) Chromosome landing at the Mla locus in barley (Hordeum vulgare L) by means of high-resolution mapping with AFLP markers. Theor Appl Genet 98:521–530
Staden R (1996) The Staden sequence analysis package. Mol Biotechnol 5:233–241
Stanke M, Schöffmann O, Morgenstern B, Waack S (2006) Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7:62
The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815
Tuskan GA, et al (2006) The genome of black cottonwood, Populus trichocarpa (Torr. and Gray). Science 313:1596–1604
van Leeuwen H, Monfort A, Zhang H-B, Puigdomenech P (2003) Identification and characterisation of a melon genomic region containing a resistance gene cluster from a constructed BAC library, microcolinearity between Cucumis melo and Arabidopsis thaliana. Plant Mol Biol 51:703–718
van Leeuwen H, Garcia-Mas J, Coca M, Puigdomenech P, Monfort A (2005) Analysis of the melon genome in regions encompassing TIR-NBS-LRR resistance genes. Mol Genet Genomics 273:240–251
Yan HH, Mudge J, Kim DJ, Larsen D, Shoemaker RC, Cook DR, Young ND (2003) Estimates of conserved microsynteny among the genomes of Glycine max, Medicago truncatula and Arabidopsis thaliana. Theor Appl Genet 106:1256–1265
Yeh R-F, Lim LP, Burge CB (2001) Computational inference of homologous gene structures in the human genome. Genome Res 11:803–816
Acknowledgments
The authors thank Mercè Miquel from the Sequencing Service of IBMB-CSIC (Barcelona, Spain) for the sequencing of shotgun clones. W. D. is recipient of a postdoctoral contract from the Centre de Recerca en Agrigenòmica (CRAG).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Y. Van de Peer.
Wim Deleu and Víctor González contributed equally to this work. The nucleotide sequences of BACs 1-21-10 and 13J4 are available in the DDBJ/EMBL/GenBank databases under the accession numbers EF188258 and EF657230, respectively.
Rights and permissions
About this article
Cite this article
Deleu, W., González, V., Monfort, A. et al. Structure of two melon regions reveals high microsynteny with sequenced plant species. Mol Genet Genomics 278, 611–622 (2007). https://doi.org/10.1007/s00438-007-0277-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-007-0277-2