
Abstract
Understanding the molecular mechanisms underlying bone development is a fundamental and fascinating problem in developmental biology, with significant medical implications. Here, we have identified the expression patterns for 36 genes that were characteristic or dominant in the consecutive cell differentiation zones (mesenchyme, precartilage, cartilage) of the tip section of the developing velvet antler of red deer Cervus elaphus. Two major functional groups of these genes clearly outlined: six genes linked to high metabolic demand and other five to tumor biology. Our study demonstrates the advantages of the antler as a source of mesenchymal markers, for distinguishing precartilage and cartilage by different gene expression patterns and for identifying genes involved in the robust bone development, a striking feature of the growing antler. Putative roles for “antler” genes that encode α-tropomyosine (tpm1), transgelin (tagln), annexin 2 (anxa2), phosphatidylethanolamine-binding protein (pebp) and apolipoprotein D (apoD) in intense but still controlled tissue proliferation are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Deer stags provide one of the most spectacular phenomena of tissue regeneration in the animal kingdom: the development of the antlers. Antlers are male secondary sexual characteristic and used for display and in combat. Antlers are bony appendages of the deer head, which are cast and re-grow each year. They are the only example of regeneration of a complete organ in mammals (Goss 1983; Bubenik and Bubenik 1990). Antlers develop from pedicles that grow during puberty to become permanent protuberances from the deer frontal bone. The development of antlers is a modified endochondral ossification process (Rucklidge et al. 1997).
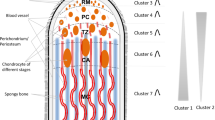
After casting, antler re-growth is initiated at the distal rim of the pedicle from resident stem cell population, as suggested by Li and Suttie (2001). These mesenchymal cells sequentially proliferate and differentiate into chondroblasts and chondrocytes associated with the formation of cartilage. The transition from undifferentiated cells to chondrocytes is gradual, and the direction of differentiation is disto-proximal. Although the matrix of antler cartilage is biochemically similar to that of other hyaline cartilages, it is well vascularized. This feature is explained by the metabolic demands of fast growth. On the cartilage scaffold, ossified bony trabeculae are formed by mineral deposition. Calcification gradually occludes the blood supply to the antler. Antler growth slows and then stops and transformation to bone is completed (Fig. 1) (Banks and Newbrey 1983). At the end, the “velvet” skin is shed; the antler is “polished” and ready for the rutting season.

Histology of developing antler. a Parasagittal section of the velvet antler tip. b Alcian blue/alizarin red stainings show the levels of glycoseaminoglycan (colour blue) and calcium (fuchsia) in the extracellular matrix. c Hematoxylin/eosin stainings. Bar: 0.1 mm
The developing antlers covered with velvet-like skin are the fastest growing mammalian organs. Their maximal elongation rate can be around 2 cm at each tip of the branches per day, corresponding to 100–150 g daily growth in weight in the Danube–Drava National Park of Hungary and Croatia, where a capital antler of red deer Cervus elaphus may weigh 14–17 kg. This extremely rapid growth localizes at the tip of the velvet antlers, and includes multiple tissues: skin, mesenchyme, cartilage, bone, blood vessels and nerves (for a comprehensive overview see a recent review by Price and Allen 2004).
We started systematic investigations with the aim to identify tissue- and organ-specific gene expressions in the tip of the velvet antler. In this way, we expect to get a deeper insight into the genetic background of the robust bone growth that is a natural attribute of red deer antlers.
In this paper, we present an Amplified Restriction Fragment Polymorphism (AFLP)-based selection of 36 “antler” genes. These genes are classified functionally, and their expressions are ordered into patterns by aligning with tissues mesenchyme, precartilage and cartilage. More detailed characterizations are presented and particular attention is given to those five genes that seem to be significant for the very intense, but controlled tissue proliferation, and hence for the tumor biology.
Materials and methods
Collection of tissue samples
Antlers, grown for 80 days, were collected from the Deer Farm of Pannon Equestrian Academy, Bőszénfa, Hungary, during commercial harvest. The distal 5 cm of the tips was removed and dissected (kept in ice) from freshly chopped velvet antlers and prepared for the further investigations within 30 min after chopping. Positions of different tissue layers (reserve mesenchyme, precartilage, cartilage) of the growing antlers were determined as described by Li et al. (2002). Since the potential for cross-region contamination was believed to be considerable, precautions were taken: the transition zones (easily recognized by the naked eyes) were sliced out with some “pure” tissues from both uplayer and downlayer. These slices (∼0.3–0.5 cm thick) were discarded. The slices (∼0.5 cm thick) containing the “pure” tissues were cut in two parts, one of them was fixed, sectioned, stained and used for histological control, the other part was stored in liquid nitrogen and kept for mRNA preparations. Fetuses were collected from hinds shot for selection purposes in the middle of December. The fetal growth plates were dissected, carefully cleaned of periosteum and other tissues remnants and cut in two parts. As in the case of the antler samples, one part was prepared for histological control; the other for mRNA isolations, within 30 min after the hind was shot.
mRNA purification and cDNA synthesis
mRNA samples from three different tissue layers of the velvet antler and from fetus, long bone growth plates were isolated using PolyATtract®System 1000 (Promega) according to the manufacturer’s protocol. Samples were then exposed to 3 U of DNaseI treatment at 37°C for 30 min. First strand cDNA synthesis was performed using SMART™ cDNA synthesis technology according to the SMART™ cDNA Library Contruction Kit User Manual (Clontech). Five hundred nanograms of mRNA was used as template. Reactions were carried out under RNase-free conditions at 42°C for 60 min.
AFLP-based differential display
The AFLP technology is based on the selective PCR-based amplification of a subset of restriction fragments (Vos et al. 1995; Money et al. 1996). We used the AFLP Analysis System I (Gibco) protocol with the exception that the double-stranded cDNAs (250 ng from each sample) were digested simultaneously with two restriction endonucleases, EcoRI and MseI. AFLP reactions with primers having none of the selective nucleotides (called preamplification) were performed for 20 cycles with the following cycle profile: 30 s DNA denaturation step at 94°C, 60 s annealing step at 56°C, and 60 s extension step at 72°C. AFLP reactions with primers having three selective nucleotides (called selective amplification) were performed for 40 cycles with the following cycle profile: 30 s DNA denaturation step at 94°C, 30 s annealing step, and 60 s extension step at 72°C. The annealing temperature in the first cycle was 65°C, than reduced by 0.7°C in each cycle for the next 11 cycles, and was 56°C in the last 28 cycles. All amplification reactions were performed in a GeneAmp® PCR System 2700 thermocycler (Applied Biosystems). The EcoRI-primers for selective amplification reactions were end-labeled using [γ33P]ATP and T4 polinucleotid kinase. PCR products were separated on 6% denaturing polyacrylamide gels and visualized by exposure to Kodak film for ∼16 h. The resultant banding patterns were analyzed.
cDNA fragments cloning and re-testing AFLP pattern
Amplified restriction fragment polymorphism fragments were excised from the polyacrylamide gels (i.e. where the strongest signals were detected) as a band of approximately 1 mm wide. DNA fragments were cloned into pBluescript® II KS (Stratagene) with standard protocol and transformed into Escherichia coli DH5α competent cells. From the resultant colonies, recombinant plasmids were isolated and used to identify fragments giving the AFLP signal. Since quantities of mRNAs isolated from four tissues (i.e. mesenchyme, precartilage, cartilage and fetus growth plate) were limited, to have enough quantity for Northern analyses, we had to amplify the hybridization probes. First the mRNA pools were converted to the complementary ssDNA strands using SMART™ cDNA synthesis technology as seen earlier in “mRNA purification and cDNA synthesis”. In a second step, these ssDNA pools served as templates for the linear amplifications by PCR of 25 cycles. Therefore, the resulting PCR products consisting of ssDNA pools of the sense strands (corresponded to cognate mRNA pools of the start) were subjected to [α32P] labeling and used for hybridization probes in Northern blottings. (see Fig. 2). The hybridization patterns of each fragment were rigorously checked by series of washings (of increasing stringency).

Schematic summary of the pathway that led to identification of genes with enhanced expression in velvet antler
Northern blot analysis
Tissue samples (approximately 300 mg) were homogenized in guanidinium-thiocyanate, then extracted with acidic phenol–chloroform. RNAs were precipitated from the aqueous phase with isopropyl-ethanol and dissolved in 50 μl RNase-free water. RNA concentrations were determined by measuring the absorbance at 260 nm. Ten microgram aliquots of total RNA were separated by electrophoresis in 1.2% formaldehyde–agarose gels. RNA was blotted to Hybond N+ filter (Amersham) according to the manufacturer’s protocol. Fifty nanograms of cDNA fragments were labeled with [α32P]dATP using random hexamers and E. coli DNA Polymerase I Large (Klenow) Fragment. The hybridization was performed at 65°C in the Perfecthyb TM Plus buffer (Sigma). After hybridization, the filters were washed at the same temperature according to the manufacturer’s protocol. Signals were visualized using a phosphorimager.
Construction and screening of lambda cDNA libraries
cDNA libraries were constructed from the three antler tissue zones mentioned earlier in “Collection of tissue samples” section using the SMART cDNA Library Construction Kit (Clontech) according to the manufacturer’s protocol. Briefly, 1 μg mRNA samples were reverse transcribed with CDSIII oligo (dT) primer and SMART IV oligo provided by the kit using SuperScript II RNaseH-minus reverse transcriptase (Invitrogene). Each reaction volume was 20 μl. Long Distance PCRs were performed in 50 μl reaction volume using 1 μl from the first strand cDNA reaction, 1 μl dNTP mix (10 mM each), 5 μl 10× BD Advantage2 PCR Buffer, 10 μM primers, 1 μl 50× Advantage Polymerase Mix and 1 μl (5 U) KlenTaq LA DNA Polymerase Mix (Sigma). Cycling parameters were 94°C for 5 min (initial denaturation), 25 cycles at 94°C for 1 min and 68°C for 7 min. Five microliter double-stranded cDNA samples were checked by electrophoresis in 1% agarose gel and after phenol–chloroform purification and ethanol precipitation 3 μg digested with SfiI. Size fractionation was done by using SizeSep 400 (Pharmacia) chromatography column and the resulting product was ligated into the SfiI digested, dephosphorilated λTriplEx2 vector. For in vitro packaging 1 μl ligation reactions were used and 1–2 × 106 recombinant pfu were further propagated in XL1 Blue strain to get amplified libraries with 109–1010 pfu/ml. Library screening was performed according to the standard protocol (Sambrook et al. 1989). Approximately 2–3 × 104 phages were plated on Petri dishes of 180 mm diameter and plaques were lifted to GeneScreen hybridization transfer membrane (Perkin–Elmer Life Sciences). The AFLP fragments were re-amplified and 50 ng were labeled with [α32P]dATP using the Pre-MC, PreT primers by random priming protocol. PreT: CGTAGACTGCGTAGGAATTCT, Pre-MC: GCACGATGAGTCCTGAGTAAC. Plaque hybridizations were carried out in two rounds; “positive” phages were isolated and converted to plasmids in E. coli BM25.8 strain. Plasmid DNA was purified with QIAprep Spin Miniprep Kit (Qiagen) and sequenced.
Quantitative real-time PCR
Quantitative real-time RT-PCR (qRT-PCR) analysis was performed using a LightCycler instrument and Relative Quantification Software Ver.4 (Roche) as follows: initial denaturation at 95°C for 10 min, amplification for 50 cycles of denaturation (95°C, 15 s; ramp rate 2°C/s) annealing (60°C, 10 s; ramp rate 2°C/s) and extension (72°C, 20 s; ramp rate 2°C/s). Each PCR reaction consisted of 1× BSA, 20 pmol TaqMan fluorescence probe (for amplicon detection, Bio-Science Ltd.), 2× TaqMan® Universal PCR Master Mix (Applied Biosystems, Manufactured by Roche), 20 pmol forward primer, 20 pmol reverse primer, cDNA template (5 ng of mRNA converted to cDNA). Sequences of the primers and probes are shown in Table 1. The mRNA level was normalized to β-actin mRNA level. The results of three independent reactions were averaged. A single online fluorescence reading for each sample was taken at the end of extension step. Quantitative results were expressed by the identification of the second derivative maximum points, which marked the cycles, where the second derivatives of the fluorescence signal curve are at the maximum.
In situ hybridization
The velvet antler tip was dissected and fixed in 4% (w/v) paraformaldehyde in PBS. Blocks were embedded in low-melting-point paraffin (Paraplast, Sigma) and sectioned vertically. Sections were mounted on slides coated with (3-aminopropyl)triethoxy-silane. Sections were then dewaxed, rehydrated and pretreated with Proteinase K and acetic anhydride, and hybridized overnight. RNA probes were labeled with digoxigenin-UTP (Roche) by in vitro transcription of the T7 RNA polymerase (cDNA template was cloned in λTriplEx2 vector in the antisense orientation behind T7 promoter). The hybridization temperatures chosen were specific for the probes. After hybridization, the sections were washed in 2×SSC, 50% formamide, 2×SSC, for 30 min each at the hybridization temperature. Then the sections were treated with RNase A for 30 min at 37°C and then washed twice in 0.2×SSC. Digoxigenin was detected using sheep anti-digoxigenin alkaline phosphatase Fab fragments (Roche), followed by an alkaline phosphatase reaction using nitrotetrazolium blue chloride and 5-bromo-4-chloro-3-indolyl phosphate as substrate. Sections were mounted using Kaiser’s glycerin gelatine.
Western blot analysis
Reserve mesenchyme, precartilage, cartilage and fetus tissue powder (ground in a mortar in liquid nitrogen) were treated with lysis buffer (100 mg tissue powder in 0.4 ml buffer: 0.0625 M Tris–HCl, 2% SDS, protease inhibitor) and sonicated. SDS-PAGE separation of 1 μl of the non-reduced supernatant was performed on 10% NuPAGE Novex Bis–Tris gel in MOPS-SDS running buffer (50 mM MOPS, 50 mM Tris base, 0.1% SDS, 1 mM EDTA pH 7.7) (Invitrogen). Proteins resolved by SDS-PAGE were electroblotted onto a Hybond-C Extra nitrocellulose membrane (Amersham Biosciences) for 1 h at 30 V in NuPAGE Transfer buffer (25 mM Bis–Tris, 2 mM Bicine, 1 mM EDTA, pH 7.2) containing 10 % metanol.
The blot was blocked in TBS containing 5% powdered nonfat dry milk. Antigens were identified with a polyclonal antibody raised against the N-terminus of red deer Annexin 2 protein (manufactured by Sigma) diluted 1:2,000, bound antibodies were detected by alkaline phosphatase conjugated goat anti rabbit polyclonal antibody (Sigma) diluted 1:30,000 in the same buffer as used by Sambrook et al. (1989). Detection was by an alkaline phosphatase reaction using nitrotetrazolium blue chloride and 5-bromo-4-chloro-3-indolyl phosphate as substrate.
Immunohistochemistry
Tissue distribution of Annexin 2 (Anxa2) protein in paraffin-embedded sections of velvet antler tip was visualized by using polyclonal antibody from rabbit raised against red deer Annexin 2 protein (ordered from Sigma). Standard immunohistochemistry was carried out using peroxidase-conjugated goat anti-rabbit IgG antiserum and 3-amino-9-ethylcarbazole (AEC) as substrate. Sections were dewaxed, rehydrated and digested with Proteinase K. To avoid nonspecific binding of the antibodies, the sections were incubated for 2 h with 20% (w/v) normal goat serum in TBS. The primary antibody was diluted 1:200 and incubated overnight at 4°C with the sections, the secondary antibody was diluted 1:100 and incubated for 1 h at room temperature.
Results
Differentially expressed genes in the chondrogenic tissue of velvet antler, screened by AFLP
In this section, we describe the steps from screening to confirmation of genes expressed differently in three antler tissue regions. We have chosen three successive sections from the tip of the velvet antler, including: mesenchyme, precartilage and cartilage (Fig. 1). The sections were taken as proposed by Li et al. (2002) with modifications described in experimental procedures, and were compared to growth plate cartilage isolated from long bones of red deer fetuses. The screening steps and the rationale of why fetal growth plate was chosen for normalization control are summarized in Fig. 2 and detailed below.
Step 1
Amplified Restriction Fragment Polymorphism Differential Display was applied in two parallels in order to compare the expressions in the four tissues by combining 32 primer pairs. Forty AFLP fragments were detected and isolated showing differences in their expressions in pair wise comparisons: antler tissue(s) versus fetal growth plate, i.e. no or very slight expression seen in fetal growth plate versus stronger expression was observed in at least one of the three antler tissues. The purity of all tissue samples was checked histologically: (1) the mesenchymal samples were “pure”, i.e. contained characteristic proliferating small spindle-shaped cells; (2) the “purity” of the precartilageous samples were more than 90% for (pre)chondroblasts, and few percent chondrocytes were generally also detected; (3) samples from the antler cartilage consisted of only chondrocytes (i.e. no (pre)chondroblasts or osteoblasts were recorded) with mineralization in the matrix and blood vessels seen (Fig. 1b, c); (4) the fetal growth plates consisted of ∼90% chondrocytes and ∼10% osteoblasts, with few blood vessels. No (pre)chondroblasts and mineralization were recorded in this cartilage. The above histological patterns led to the rationale that by subtracting the expressions of fetal growth plate from that of antler mesenchyme and precartilage samples would result in the detection of AFLP fragments specific for the expressions both in the antler mesenchymal cells and in the (pre)chondroblasts. In the case of the antler cartilage samples, the “subtraction” of the fetal growth plate expression reveals the elevated expressions arising predominantly from the antler chondrocytes and in a lesser extent from the perivascular cells.
Step 2
Refining the AFLP selections. The 40 AFLP cDNA bands were cloned into pBluescript KS plasmid vector and were retested by four parallel Southern hybridizations using radioactively labeled total cDNA mixtures prepared from the cognate four tissues (the same as used before for the “AFLP” step). Thirty-six out of these 40 fragments gave identical expression pattern as before in the “AFLP” screening (i.e. step 1) (see more details in Materials and methods).
Step 3
Classification of gene expression classes. The expressions of the 36 AFLP isolates were evaluated by visual comparisons and classified as strong, medium, weak and not detectable (denoted with s, m, w, and n, respectively, in Table 2). With these variables expression patterns were aligned to each AFLP. Six classes of expression pattern were found with 4–10 specimens in each: dominant expression in mesenchyme (I), in mesenchyme and precartilage (II), in all three antler tissues (III), in precartilage (IV), in precartilage and cartilage (V), in cartilage (VI) (Table 2). The 36 fragments were sequenced and sequence similarity searches were carried out in NCBI databases (http://www.ncbi.nlm.nih.gov)(Table 2., last column). Signals for two groups of genes emerged from the 36 fragments. One group with six genes can be linked to transcriptional and translational machinery: AF 11.24.21 (class I), AF 09.05.10 (class II), AF 08.09.01, AF 11.24.04 and AF 11.24.22 (class III), AF 11.24.28 (class VI), the other group with five genes to tumor biology. The latter group was selected for further investigations: in order to identify and characterize novel genes that are involved in intense bone growth, AF 08.09.09 (class I), AF 09.05.07 (class II), AF 08.09.00 and AF 07.08.05 (class III), AF 01.28.10 (class V).
Cloning genes cognate for AFLP-s
From the three antler cDNA pools (prepared from the three corresponding tissues) three cDNA libaries were made and used for targeting and isolating the cognate genes for AF 08.09.09; AF 09.05.07; AF 08.09.00; AF 07.08.05; AF 01.28.10; (i.e. “tumor biology” group, see previous section and Table 2.). The clones in the libraries which hybridized with the corresponding AFLP probe were isolated and those carried the longest inserts were sequenced, and tested against NCBI databases. The previous BLAST searches (Table 2.) were confirmed and sequences were submitted to GenBank as new red deer annotated genes: i.e. AF 08.09.09 for α-tropomyosin (tpm1, GenBank accession number: DQ239919), AF 09.05.07 for transgelin (tagln GenBank accession number: DQ412697), AF 08.09.00 corresponded to the gene for red deer annexin 2 (anxa2, GenBank accession number: DQ239920), AF 07.08.05 for phosphatidylethanolamine binding protein (pebp, previously called prostatic binding protein, GenBank accession number: DQ412698) and AF 01.28.10 for apolipoprotein D (apoD, GenBank accession number: DQ2399221). In all cases high identity scores (89–98%) were calculated for the human, bovine and mouse orthologs (Table 3).
mRNA expression pattern confirmed by Northern hybridization
The presence of mRNA for genes cloned (see previous section) was demonstrated in tissue samples by Northern hybridization using total length cDNA as hybridization probes. Total RNA for blotting was extracted from mesenchyme, precartilage and cartilage of the velvet antler as well as from the fetal growth plate. All labeled cDNA probes gave unique patterns for hybridization signal, consequently unique expressions patterns for the three tissues, in perfect agreement with AFLP data (Fig. 3).

Northern blot analysis. FC foetal cartilage, RM reserve mesenchyme, PC precartilage, C cartilage
Quantification of gene expressions
Quantitative real-time RT-PCR analyses for α-tropomyosin (tpm1), annexin 2(anxa2), apolipoprotein D (apoD) were performed to confirm the expression differences. Values were computed as: the ratio of the mRNA levels of our genes to the mRNA levels of β-actin (the reference gene) in an antler tissue was normalized to the ratio of the same two mRNAs in the fetal growth plate (the reference sample). As seen in Fig. 4, the expression level of the gene for annexin2 (anxa2) decreased from mesenchymal region to cartilaginous regions in conformity with Northern hybridization (described above). The α-tropomyosin (tpm1) mRNA level was found 2.5-fold increased in the mesenchyme and lessened toward the cartilaginous tissues. In contrast, apolipoprotein D (apoD) expression was not observed in mesenchyme, but increased dramatically in precartilage (∼26-fold) and cartilage (∼67-fold).

Relative mRNA levels of tpm1, anxa2 and apoD from different tissue. Black columns: relative mRNA levels in reserve mesenchyme zone of velvet antler, grey in precartilage zone, white in cartilage. β-actin mRNA levels were used as a controls
Spatial localization of the gene expression revealed by in situ hybridization
In situ hybridizations were performed to determine cell types that express the genes investigated. Parasagittal sections of velvet antler were hybridized with α-tropomyosin (tpm1), annexin 2(anxa2), and apolipoprotein D (apoD) antisense RNA probes. As seen in Fig. 5g–i, expression of annexin 2 (anxa2) mRNA was detected in all antler tissues. However, the hybridization signal in the mesenchymal region was the strongest. The endothelial cells of blood vessels gave also a positive signal. Expression of α-tropomyosin (tpm1) gene concentrated to the apical mesenchyme cells, and became weaker as differentiation proceeded downstream. Prechondroblasts still gave a positive signal. In cartilage, α-tropomyosin (tpm1) gene was expressed only around vascular channels in fibroblast-like cells (Fig. 5a–c). Expression of the apolipoprotein D (apoD) gene was restricted to chondrocytes. Hybridization signal was detectable neither in mesenchyme, nor in precartilage (Fig. 5d–f).

In situ localization of gene expression in velvet antler tissues. Analysis of 8 μm thick longitudinal section from mesenchyme region (A/a, D/d, G/g, J), from precartilage (B/b, E/e, H/h, K) and cartilage (C/c, F/f, I/i, L, M). A/a–C/c Expression of α-tropomyosin (tpm1) is most striking in mesenchyme cells and fibroblast-like cells at the periphery of vascular channels (arrow). D/d–F/f apolipoprotein D (apoD) mRNA detection. Chondrocytes express mRNA of apolipoprotein D. Hybridization signal cannot be seen in the less differentiated regions. G/g–I/i Detection of annexin 2 (anxa2) mRNA. The mesenchyme region gave the greatest number of positive cells. In the zones of precartilage and cartilage expression of annexin 2 (anxa2) is also high. J–L Immunolocalization of Annexin 2 protein. Annexin 2 is detectable in the mesenchyme cells (J), chondroblasts (K), chondrocytes and at periphery of blood vessel (arrowhead) (L). Immunohistochemistry signal can be seen at periphery of chondrocytes (M). Bar: 0.25 mm in A–L, 0.1 mm in c, 0.006 mm in a, b, d, e, f, g, h, i and M
Immunolocalization of red deer Annexin 2
Annexin2 (anxa2) is a member of the annexin multigene family. Multiple alignment of family members revealed that they share high sequence similarity and differ only in their relatively short N-terminal domains, which give them identity. The repetitions of annexin domains make it difficult to detect expression of annexin 2 alone by means of in situ hybridization. This fact prompted us to immunolocalise Annexin 2 (Anxa2) by producing an antibody against the N-terminal tail of the protein, specific to red deer Annexin 2 (Anxa2) protein.
The Western blot verified that the antiserum reacted specifically with the red deer Annexin 2 (Anxa2) (data not shown). In mesenchyme, precartilage and cartilage regions of velvet antler, the Annexin 2 (Anxa2) protein was observed on the periphery of the cells, where associated with the cell membrane (Fig. 5m). Deposition of protein in the matrix was only seen in the perivascular tissue although it should have been in the matrix in other tissues (Kirsch et al. 2000). One explanation may be that while Annexin 2 (Anxa2) proteins form macromolecular complexes in the matrix, they use the N-terminal domains as the reacting surfaces (i.e. its reacting epitop was masked from the antiserum used in the investigation) (Fig. 5k–l).
Discussion
The velvet antler of red deer may provide a unique model for studies on mammalian bone development. This could be particularly true for the well-separated mesenchyme mass located at the tip, which could be used in pursuit of the earliest gene expression events of the developmental pathway from (multipotent) mesenchyme toward hard bone. Furthermore, the longitudinal segregation of precartilage from the cartilage makes possible to separate gene expressions in these two tissues. Other characteristics of the “antler pathway” add further values to this biological model: (1) the whole cycle of antler regeneration is repeated every year, (2) this complex development involves the most rapid (“robust”) growth of animal tissues, with intense vascularization and nerve regeneration, (3) and this robust proliferative growth proceeds within a controlled path and apparently avoids malignancy, an aspect that may be significant for cancer research.
We took these advantages of the antler model and combined them with recombinant DNA technology in order to unearth genes whose expression points either to the early (i.e. mesenchymal toward bone) or to the robust way (i.e. stronger in antler cartilage than in fetal-skeletal cartilage) of bone development. In the first step, we used AFLP screen and achieved approximately 90% fidelity in the score of expression differences as shown by the confirmatory Southern hybridizations with the cognate pKS clones (of the AFLP-s): 36 out of 40 of the pKS clones displayed a hybridization pattern concordant with the cognate AFLP expression pattern. The Northern, qRT-PCR, and in situ identifications of the tissue specific expressions of the further investigated five “tumor-linked” genes validates the “gene screening”. Although our investigations here have been carried on with genes at the cDNA levels, we believe that using these cDNAs and applying the method of single oligonucleotide nested (SON)-PCR (Antal et al. 2004) the genomial copies of the “antler” genes would also be available in many cases.
Blast analyses (Table 2) pointed clearly to two groups of genes enriched among the 36 genes taken randomly after the AFLP based selections. The first group contained six genes (for ribosomal components and transcriptional factors) whose elevated expression can be explained by the high metabolic demands when vigorous cell proliferation takes place. This phenomenon was also documented for yeast (Kressler et al. 1999). The second group consisted of five genes (α-tropomyosine (tpm1), transgelin (tagln), annexin 2 (anxa2), phosphatidylethanolamine-binding protein (pebp) and apolipoprotein D (apoD)) whose elevated expression could be associated with tumor biology, and were known to be expressed at a strikingly decreased level in malignant tumors (see discussion below). Furthermore, three of these genes annexin 2 (anxa2), phosphatidylethanolamine-binding protein (pebp) and apolipoprotein D (apoD), can be linked to the robust development of the antler; their expressions are upregulated in the cartilage of the growing antlers, compared to the fetal-skeletal cartilage.
Appearance of the genes α-tropomyosine (tpm1), transgelin (tagln) and annexin 2 (anxa2) in the AFLP selection was not surprising. α-tropomyosine (tpm1) and transgelin (tagln) have been known for long for their mesenchyme-specific role (Goodwin et al. 1991; Lowson et al. 1997). The expression of annexin 2 (anxa2) has been reported to be involved in the formation of calcium channels and in mineralization around hypertrophic chondrocytes and osteoblasts (Kirsch et al. 2000; Gillette and Nielsen-Preiss 2004).
The strong expression of apolipoprotein D (apoD) in chondrocytes was a new finding. So far, apoD mRNA has been detected during embryogenesis in the precartilageous condensation of Meckel’s cartilage, in the basio-occipital bone primordium and in sclerotomal condensations of vertebral bodies (Sanchez et al. 2002). Apolipoprotein D (ApoD), which was identified in the plasma HDL fraction (McConathy and Alaupovic 1973), belongs to the lipocalin superfamily and shows significant sequence homology to other proteins like human retinol binding protein (RBP) or insect bilin binding protein (BBP) (Rassart et al. 2000). Generally, it is regarded as a multifunctional and multi ligand binding protein with special emphases on small hydrophobic molecules. Apolipoprotein D (apoD) mRNA is expressed in wide variety of tissues, and the sites of its expression vary from species to species (Seguin et al. 1995). In humans and rabbit, expression is predominant in the cerebellum, adrenal and spleen, while in mouse it is restricted mainly to the central nervous system (CNS) (Rassart et al. 2000). The apolipoprotein D (apoD) mRNA expression was mostly linked to fibroblasts, mainly around the blood vessels, and in the nervous system to astrocytes and oligodendrocytes. Contrary to this expression pattern, apolipoprotein D (apoD) was expressed primarily in chondrocytes in the antler tip and only faint traces were detected in the perivascular tissue.
On antlerogenesis and tumors: although the α-tropomyosine (tpm1), transgelin (tagln), annexin 2 (anxa2), phosphatidylethanolamine-binding protein (pebp) and apolipoprotein D (apoD) genes were expressed in apparently different patterns in the developing antlers, as shown in Table 2 and demonstrated by Fig. 3, they share a common feature with regard to tumorgenesis, namely their expression decreases in malignant tumors, and in contrast, at least for three of them their overexpression lessens the aggressiveness of malignant phenotypes. This phenomenon has been shown for anxa2 in osteosarcomas (Gillette et al. 2004), for α-tropomyosine (tpm1) in breast and lung malignant transformations (Mahadev et al. 2002), and phosphatidylethanolamine-binding protein (pebp) in prostate cancer (Fu et al. 2003; Keller et al. 2005). For transgelin (tagln), abolishment of its expression has also been observed in lung and colon cancers, and regarded as an early signal for the developing tumor (Shields et al. 2002). The apolipoprotein D (apoD) expression decreases in a wide variety of malignant tumors (Sanchez et al. 1992a, b). The reciprocal expression patterns of apolipoprotein D (apoD) in the antler versus tumors may be indicative of the controlled way the intense tissue proliferation proceeds in the growing antler. Piecing together the expression pattern of α-tropomyosine (tpm1), transgelin (tagln), annexin 2 (anxa2) and phosphatidylethanolamine-binding protein (pebp), in the antler with their suppressive effects on malignancy can envision a functional network. In the antler mesenchyme, α-tropomyosin (tpm1) and less profoundly, transgelin, (tagln) slow down the vigorous cell proliferation and make conditions favorable toward differentiation. Downstream of this pathway, annexin 2 (anxa2) acts by channeling the cells toward chondrogenesis. Behavior of apolipoprotein D (apoD) fitted well into this line-up. Its expression is also inversely related to proliferation, and hence it may be linked to a more differentiated tissue state (Rassart et al. 2000). The developing antler provides supporting evidence for this: from the tip toward the pedicle proliferation decreases continuously along the path of differentiation, while apolipoprotein D (apoD) expression increases from mesenchyme to cartilage.
The very high DNA sequence homologies between red deer and human orthologous genes (Table 3) encourage us to find a room for the cDNA microarray technology in further studies of bone development. Our extended experiments (not shown) indicate that the antler mRNA and cDNA samples hybridize with high fidelity to human and mouse cDNA microarrays, and provide a good handle for developing an antler cDNA microarray with manifold impacts on future directions. One of them could point toward the better understanding of the genetic background of specifically upregulated gene expression in the developing antlers, but applications in biomedical, agricultural and conservation biological researches and developments also deserve consideration.
References
Antal Zs, Rascle C, Fèvre M, Bruel C (2004) Single oligonucleotide nested PCR: a rapid method for isolation of genes and their flanking regions from expressed sequence tags. Curr Genet 46:240–246
Banks WJ, Newbrey JW (1983) Light microscopic studies of the ossification process in developing antlers. In: Brown RD (ed) Antler development in Cervidae. Caesar Kleburg Wildlife Research Institute, Kingsville, pp 231–260
Bubenik AB, Bubenik GA (1990) Horns, pronghorns and antlers: evolution, morphology, physiology, and social significance. Springer, Berlin Heidelberg New York
Fu Z, Smith PC, Zhang L, Rubin MA, Dunn RL, Yao Z, Keller ET (2003) Effects of raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J Natl Cancer Inst 95:878–889
Gillette JM, Nielsen-Preiss SM (2004) The role of annexin 2 in osteoblastic mineralization. J Cell Sci 117:441–449
Gillette JM, Chan DC, Nielsen-Preiss SM (2004) Annexin 2 expression is reduced in human osteosarcoma metastases. J Cell Biochem 92:820–832
Goodwin LO, Lees-Miller JP, Leonard MA, Cheley SB, Helfman DM (1991) Four fibroblast tropomyosin isoforms are expressed from the rat α-tropomyosin gene via alternative mRNA splicing and the use of two promoters. J Bio Chem 266:8408–8415
Goss RJ (1983) Deer antler: regeneration, evolution and function. Academic, New York
Keller ET, Fu Z, Brennen M (2005) The biology of prostate cancer metastasis suppressor protein: Raf kinase inhibitor protein. J Cell Biochem 94:273–278
Kirsch T, Harrison G, Golub EE, Nah HD (2000) The roles of Annexins and types II and X collagen in matrix vesicle-mediated mineralization of growth plate cartilage. J Biol Chem 275:35577–35583
Kressler D, Linder P, Cruz J (1999) Protein trans-acting factors involved in ribosome biogenesis in Saccharomyces cerevisiae. Mol Cell Biol 19:7897–7912
Li C, Clark DE, Lord EA, Stanton JL, Suttie JM (2002) Sampling technique to discriminate the different tissue layers of growing antler tips for gene discovery. Anat Rec 268:125–130
Li C, Suttie JM (2001) Deer antlerogenic periosteum: a piece of postnatally retained embryonic tissue? Anat Embryol 204:375–388
Lowson D, Harrison M, Shapland C (1997) Fibroblast transgelin and smooth muscle SM22-alpha are the same protein, the expression of which is down-regulated in many cell lines. Cell Motil Cytoskeleton 38:250–257
Mahadev K, Ravel G, Bharadwaj S, Willingham MC, Lange EM, Vonderhaar B, Salomon D, Prasad GL (2002) Suppression of the transformed phenotype of breast cancer by tropomyosin-1. Exp Cell Res 279:40–51
McConathy WJ, Alaupovic P (1973) Isolation and partial characterization of apolipoprotein D: a new protein moiety of the human plasma lipoprotein system. FEBS Lett 37:178–182
Money T, Reader S, Qu LJ, Dunford RP, Moore G (1996) AFLP-based mRNA fingerprinting. Nucleic Acid Res 24:2616–2617
Price J, Allen S (2004) Exploring the mechanisms regulating regeneration of deer antlers. Philos Trans R Soc Lond 359:809–822
Rassart E, Bedirian A, Carmo SD, Guinard O, Sirois J, Terrisse L, Milne R (2000) Apolipoprotein D. Biochim Biophys Acta 1482:185–198
Rucklidge GJ, Milne G, Bos KJ, Farquharson C, Robins SP (1997) Deer antler does not represent a typical endochondral growth system: immunoidentification of collagen type X but little collagen type II in growing antler tissue. Comp Biochem Physiol B Biochem Mol Biol 118:303–308
Sambrook J, Fritsch EF, Maniatis T (1989) Methods of screening. In: Nolan C (ed) Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Sanchez D, Ganforinina MD, Martinez S (2002) Expression pattern of the lipocalin apolipoprotein D during mouse embryogenesis. Mech Dev 110:225–229
Sanchez LM, Diez-Itza I, Vizoso F, Lopez-Otin C (1992a) Cholesterol and apolipoprotein D in gross cystic disease of the breast. Clin Chem 38:695–698
Sanchez LM, Vizoso F, Diez-Itza I, Lopez-Otin C (1992b) Identification of the major protein components in breast secretions from women with benign and malignant breast cancer diseases. Cancer Res 52:95–100
Seguin D, Desforges M, Rassart E (1995) Molecular characterization and differential mRNA tissue distribution of mouse apolipoprotein D. Brain Res Mol Brain Res 30:242–250
Shields JM, Rogers-Graham K, Der CJ (2002) Loss of transgelin in breast and colon tumors and in RIE-1 cells by Ras deregulation of gene expression through Raf-independent pathways. J Biol Chem 277:9790–9799
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acid Res 23:4407–4417
Acknowledgment
The authors are indebted to Dr. János Nagy for providing access to deer fetus and developing antler samples, to Professor Péter Lakatos for access to quantitative real-time qRT-PCR, to Professor Péter Péczely for kind help in histology, to Magdolna Tóth Péli, Csilla Sánta Török and Kornélia Szóráth Gál for excellent technical assistance. They are also thankful to the constant interest of Dr. Tibor Vellai, Professors Sankar Adhya, Péter Lakatos and László Sugár, to the critical reading of the manuscript for Kriszta Takács Vellai. This work was supported by grants OTKA T032205 to L.O. and T032255 to P.P., T034729 and T60659 to F.D., T049608 to I.K., T43272 to Zs.B. from the Hungarian Scientific Research Foundation; by grants OM 0028/2001, OM 0278/2001 to L.O. and P.P., OM 0320/2004 to L.O., OM 255/2002 to F.D. from the National Research and Development Program NKFP; and by grants MTA/TKI/AKT-F 2003–2006 to L.O. from the Hungarian Academy of Sciences, and from the Ministry of Health, Social and Family Affairs (454/2003) to L.O and Ministry of Agriculture and Regional Development (76-a/2000, 31/a/2001) to L.O., by grant FKFP 0021/2002 from the Ministry of Education to the Ph.D. school for Biology of Eötvös Loránd University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Ruiz.
Rights and permissions
About this article
Cite this article
Molnár, A., Gyurján, I., Korpos, É. et al. Identification of differentially expressed genes in the developing antler of red deer Cervus elaphus . Mol Genet Genomics 277, 237–248 (2007). https://doi.org/10.1007/s00438-006-0193-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-006-0193-x