
Abstract
Small mammals play an essential role as disseminators of pathogens because they reach high population densities and have ubiquitous distributions. In the Northern Hemisphere rodents are well recognized as reservoirs for tick-borne bacteria of the Anaplasmataceae family and also apicomplexan protozoans. In contrast, South American rodents hosting these microorganisms have been rarely identified. In this study, we collected blood from rodents and marsupials in northern Chile and screened for Anaplasmataceae bacteria and apicomplexan protozoa. Overall, 14.7% of the samples were positive for Babesia, Hepatozoon, and Sarcocystidae using conventional PCR assays targeting the structural 18S rRNA locus (18S). Phylogenetic analyses performed with amplicons derived from 18S and cytochrome c oxidase (COI) gene provided evidence of a Babesia sp. belonging to the Babesia microti group in Phyllotis darwini, and a novel Babesia genotype in P. darwini and Abrothrix jelskii. Furthermore, four novel genotypes of Hepatozoon retrieved from Abrothrix olivacea, P. darwini, and Oligoryzomys longicaudatus, formed independent lineages within a clade that includes additional Hepatozoon spp. detected in South American rodents. Moreover, an incidental finding of a previously detected apicomplexan, herein designated as Sarcocystidae sp., was recorded in Thylamys opossums with a high prevalence, indicating a possible specific association with these mammals. Phylogenetic analysis of Sarcoystidae sp. clearly demonstrated its relatedness to apicomplexans detected in Australian marsupials. Our results expand the range of mammals hosting tick-borne apicomplexans in South America, highlight a novel clade consisting of South American babesias, and report for the first time the B. microti group infecting rodents in the region.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Small mammals are recognized hosts of bacteria of the Anaplasmataceae family and of apicomplexans with importance in veterinary and public health (Goodman et al. 2005; Perles et al. 2019). Due to their ubiquity, high population densities, and implications in food webs, small mammals play an important role in the dissemination of pathogens (Han et al. 2015). However, knowledge in relation to whether these vertebrates naturally carry pathogenic tick-borne bacteria or protozoans in South America is still emerging and requires further study.
The Anaplasmataceae family of bacteria includes intracellular obligate gram-negative alphaproteobacteria that infect ticks, and some strains cause disease in humans (Rar and Golovljova 2011). While pathogenic species of Anaplasma, Ehrlichia, and “Candidatus Neoehrlichia” are known to infect wild rodents in the Northern Hemisphere (Rar and Golovljova 2011), significantly less is known regarding vertebrate hosts of these bacteria in Southern latitudes of the globe. In particular, research on tick-borne bacterial pathogens is still incipient in Chile.
The Apicomplexa are obligate parasites with more than 6,000 described species, some of which may cause severe disease to their hosts and provoke significant economic losses worldwide (Votýpka et al. 2017). Decades ago, the genus Babesia attracted the attention of scientists because of a clinical disease that affected humans and domestic animals (Homer et al. 2000; Schreeg et al. 2016). In this context, it is currently known that species in the Babesia microti complex are zoonotic agents in Asia, Europe, and North America, with rodents carrying pathogenic strains in some cases (Goethert 2021). Although B. microti has never been detected in wild animals in South America, a B. microti-like agent was identified using molecular methods in humans from Bolivia (Gabrielli et al. 2016), suggesting that these parasites may represent neglected pathogens in the region.
Animals can also develop illness when infected with apicomplexan parasites such as species in the genus Hepatozoon (Merino et al. 2009; de Sousa et al. 2017; Perles et al. 2019). Hepatozoon spp. are intraerythrocytic and intraleukocytic parasites with a heteroxenous life cycle that involves vertebrates and invertebrates as intermediate and definitive hosts, respectively (Smith 1996). In South America, studies based on the detection of the Hepatozoon 18S rRNA structural locus (18S hereafter) suggested that in nature, these parasites use rodents and other small mammals as intermediate and paratenic hosts (Smith 1996; Merino et al. 2009; Wolf et al. 2016; Muñoz-Leal et al. 2019; Perles et al. 2019; Alabí et al. 2021). Hepatozoon spp. are transmitted to vertebrates through the ingestion of infected ectoparasites (Smith 1996). Interestingly, ticks of the genus Ixodes associated with rodents have be suggested as potential vectors in Chile (Muñoz-Leal et al. 2019).
Chilean small mammals are represented by 72 species: 67 belong to the order Rodentia and five to Marsupialia (D’elía et al. 2020; Mejías et al. 2021). Although the role of Chilean rodents and marsupials as reservoirs of pathogenic microorganisms is still obscure, DNA of Anaplasmataceae, Bartonella, Borrelia, Hepatozoon, and Mycoplasma has been detected previously (Merino et al. 2009; Müller et al. 2018, 2020; Muñoz-Leal et al. 2019; Thomas et al. 2020; Alabí et al. 2020, 2021). Therefore, it seems likely that widening the repertoire of species submitted to molecular analyses searching for those agents would contribute to the understanding of eco-epidemiological cycles. To address this objective, we targeted DNA of bacterial and apicomplexan agents in a range of small mammals captured in northern Chile.
Material and methods
Study area and collection of samples
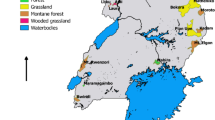
Collections were performed during July of 2018 (Austral winter) in seven localities of northern Chile roughly between latitudes 19 and 31°S as follows: Parinacota and Socoroma (Arica and Parinacota Region), Enquelga, Chusmiza, and Pampa del Tamarugal National Reserve (PTNR) (Tarapacá Region), Llanos de Challe National Park (LCNP), and Bosque Fray Jorge National Park (BFJNP) (Atacama Region and Coquimbo regions, respectively) (Fig. 1). This area of northern Chile is characterized by constant dryness along the year, with small peaks of humidity in summer (Luebert and Pliscoff 2017), and includes the core of the Atacama Desert, the driest ecosystem on earth (Clarke 2006).

Map of Northern Chile with the localities where collections were performed, highlighted with black and white symbols. Administrative regions: I, Tarapacá; III, Atacama; IV, Coquimbo; XV, Arica and Parinacota. Abbreviations: NP, National Park; NR, National Reserve
To capture rodents and marsupials, we set an average of 113 Sherman-like traps baited with oats along transects that remained active for two nights per locality (100 h in total), totalizing 1139 traps/night. Animals were manipulated as recommended by Sikes (2016). Briefly, we used a 300 g scale (Pesola) to weight the animals, and an intraperitoneal injection with a ketamine (60 mg/kg)-xylazine (3 mg/kg) solution to restrain them (Carpenter and Marion 2018). Approximately 20 μL of blood was obtained by nicking the caudal vein. Samples were preserved in sterile tubes with 96% ethanol (Sigma-Aldrich). Animals were identified morphologically in situ using a taxonomic guide (Patton et al. 2015), and released at their capture sites after recovering from anaesthesia.
DNA extraction and gene amplification
Total genomic DNA was extracted from blood samples employing the DNeasy Blood & Tissue Kit (QIAGEN, Germany), and eluted in 40 μL of Buffer AE (10 mM Tris–Cl; 0.5 mM EDTA, pH 9.0). Successful DNA extractions were checked through a conventional PCR targeting the mammalian gapdh (glyceraldehyde-3-phosphate dehydrogenase) gene (Birkenheuer et al. 2003). Positive samples were subsequently screened for Anaplasmataceae (16S rRNA), Piroplasmida spp. (18S and COI), and Hepatozoon spp. (18S) DNA using conventional PCR protocols. A touchdown PCR to amplify a fragment of the mammalian cytb gene was used to genetically identify positive animals (Leite and Patton 2002). Primers and PCR thermal conditions used in this study are provided in Table 1.
Each PCR reaction was performed by adding 2 μL genomic DNA into a mix of 2.5 μL DreamTaq Buffer, 0.5 μL dNTPs (0.2 mM), 0.3 μL DreamTaq Polymerase, 1 μL of each primer (10 pmol), and 17.7 μL of ultra-pure water. Amplicons were submitted to electrophoresis into 2% agarose gels, stained with SYBR Safe (Life Technologies/Thermo Fisher Scientific, Carlsbad, CA), and visualized by UV transillumination. Products of the expected size were purified and sequenced in both directions at the AUSTRAL-omics facility, in Universidad Austral de Chile (Valdivia, Chile). Positive controls included DNA of Ehrlichia canis, Hepatozoon canis, and Babesia canis previously obtained from infected dog blood.
Phylogenetic analyses
Sequences were quality-checked and edited with Geneious Prime® version (v) 2021.2.2 (www.geneious.com). Pairwise comparisons were performed with BLASTn (https://blast.ncbi.nlm.nih.gov), and similar sequences were downloaded from GenBank (https://www.ncbi.nlm.nih.gov) to construct alignments with MAFFT (Katoh and Standley 2013).
Phylogenies were built using Bayesian Inference (BI) and Maximum Likelihood (ML) methods with MrBayes 3.2.2. (Ronquist et al. 2012) and IQ-TREE v 1.6.12 (Nguyen et al. 2015), respectively. BI best evolutionary models were selected using the MrBayes command “lset nst = mixed rates = gamma” (Huelsenbeck 2004; Ronquist et al. 2012). Two independent tests of 107 generations and four MCMC chains were run, with sampling of trees every 1,000 generations removing the first 25% as burn-in. Tracer v 1.7.1 was used to confirm the effective sample size values (ESS) and the correlation of Markov chains (Rambaut et al. 2018). Bayesian posterior probabilities (BPP) with values ≥ 0.70 were considered to represent strong statistical support (Huelsenbeck and Rannala 2004). ModelFinder was used to select best nucleotide substitution model for ML analyses (Kalyaanamoorthy et al. 2017). We used rapid hill-climbing and stochastic disturbance methods to evaluate the robustness of the inferred tree with 1,000 ultrafast bootstrapping pseudo-replicates. Ultrafast bootstrap values ≥ 70%, between 70 and 94%, and > 95% were interpreted as low, medium, and strong statistical support values respectively (Minh et al. 2013).
Results
A total of 102 small mammals belonging to 10 species and three families were captured (Table 2). The production of amplicons of the expected size for gapdh corroborated successful DNA extractions in all samples. While PCR screenings for Anaplasmataceae yielded negative results, 15/102 (14.7%), samples were positive for apicomplexan DNA (Table 2). Sequences of mammalian cytb were obtained for 13/15 positive animals, confirming the identities of five species of rodents and one marsupial (Table S1). GenBank accession numbers for nucleotide sequences generated in this study are available in Table S2.
Babesia
Two genotypes of Babesia 18S were obtained (Babesia sp. LC87, and Babesia sp. LC77, hereafter). The Babesia sp. LC87 genotype (537 bp) was obtained from two specimens of Phyllotis darwini (Rodentia: Cricetidae) and two Abrothrix jelskii (Rodentia: Cricetidae) in LCNP and Parinacota, respectively. BLASTn comparisons for this genotype yielded an identity of 94.8% (439/463 bp, 83% query cover, 7 gaps, 0 E value) with Theileria sp. ex Damaliscus lunatus clone TS23-6 (HQ179765), amplified from the common tsessebe (Damaliscus lunatus, Artiodactyla: Bovidae) in South Africa (Brothers et al. 2011). The Babesia sp. LC77 genotype (546 bp) was retrieved from a single specimen of P. darwini captured at LCNP. BALSTn comparisons for Babesia sp. LC77 revealed an identity of 98.6% (489/496 bp, 90% query cover, 0 gaps, 0 E value) with B. microti isolate Kv21 (MG062780), detected in Ixodes persulcatus from Russia (Livanova et al. 2018).
ML and BI phylogenetic reconstructions for 18S showed 11 discrete phylogenetic clades (Fig. 2). With strong support for Babesia sp. LC87 clustering into a monophyletic clade with a Babesia sp. detected in Thrichomys pachyurus (Rodentia: Echymidae) and a Babesia sp. detected in Phyllostomus discolor (Chiroptera: Phyllostomidae) from Brazil (clade XI, Fig. 2). In contrast, the Babesia sp. LC77 genotype is grouped within the B. microti complex, branching basally to several strains of this group detected in the Northern Hemisphere (clade I, Fig. 2).

Bayesian and Maximum Likelihood phylogenies for a subset of Piroplasmida spp. inferred using an alignment (1,721 bp) of the gene encoding 18S rDNA. Calculated substitution models for BI and ML were M125, M191, M134, M200, M136, M40, and M189, and TIM3 + F + R6, respectively. Best models were chosen using the Bayesian Information Criterion (BIC) (Schwarz 1978). Values of Bayesian Posterior probability/ML Bootstrap are indicated above or below main branches. The position of Babesia spp. characterized in the present study is highlighted in bold. Roman numbers indicate phylogenetic lineages of the Piroplasmida order according to Jalovecka et al. (2019) and Ikeda et al. (2021); I: B. microti group, II: Monotremata group, III: Western group, IV: Marsupialia group, V: Percei group, VI: Rhinocerotidae group, VII: Cytauxzoon, VII: Equus group, IX: Theileria sensu stricto, X: Babesia sensu stricto, XI: Babesia spp. detected in South American mammals
Amplicons of COI target (922 bp) were obtained only in the sample from which Babesia sp. LC77 was retrieved, showed an identity of 86.7% (794/916 bp, 98% query cover, 6 gaps, 0 E value) with B. microti strain RI (LN871600) isolated from humans (Cornillot et al. 2012). In accordance with this genetic similarity, ML and BI phylogenies for COI confirmed that Babesia sp. LC77 belongs to the B. microti group (clade I; Fig. 3).

Bayesian Inference and Maximum Likelihood phylogenies for a subset of Piroplasmida spp. inferred using an alignment (1,401 bp) of the gene encoding cytochrome c oxidase I (COI). Calculate substitution models for BI and ML were M136, M40, M125, M191, and M198 (part1), M93, M184, and M155 (part2), M201, M162, M200; M189, M203, M134, M138, and M198 (part3), and TN + F + I + G4 (part1), TVM + F + R3 (part2), GTR + F + I + G4 (part3), respectively. Best models were chosen using the Bayesian Information Criterion (BIC) (Schwarz 1978). Values of Bayesian Posterior probability/ML Bootstrap are indicated above each branch. The position of Babesia spp. characterized in the present study is highlighted in bold
Hepatozoon
Four genotypes of Hepatozoon 18S were identified. Three identical sequences were recovered from three different specimens of P. darwini captured in BFJNP. A consensus of 544 bp designated as Hepatozoon sp. BFJ69 represents those sequences. Hepatozoon sp. BFJ69 was identical to Hepatozoon sp. isolate HepIxo-281 (MH174345), detected in ticks of the Ixodes sigelos group collected at the same locality (Muñoz-Leal et al. 2019). A second genotype of 544 bp (Hepatozoon sp. LC82 hereafter) was obtained from two specimens of P. darwini collected in BFJNP and LCNP. BLASTn comparisons of this second haplotype yielded 99.8% of identity with Hepatozoon sp. isolate HepIxo-284 (MH174344), obtained from the same Ixodes ticks at BFJNP (Muñoz-Leal et al. 2019).
Two additional Hepatozoon genotypes of 566 and 577 bp were recovered from Abrothrix olivacea (Rodentia: Cricetidae) collected in BFJNP (Hepatozoon sp. BFJ7 hereafter) and Oligoryzomys longicaudatus (Rodentia: Cricetidae) from Socoroma (Hepatozoon sp. Soc48 hereafter), respectively. Hepatozoon sp. BFJ7 showed an identity of 99.8% (565/566 bp, 100% query cover, 0 gaps, 0 E value) with Hepatozoon sp. AS7 (FJ719819) detected in Abrothrix sanborni (Rodentia: Cricetidae) from Chiloé Island, in southern Chile (Merino et al. 2009). On the other hand, Hepatozoon sp. Soc48 showed an identity of 98.6% (569/577 bp, 100% query cover, 1 gap, 0 E value) with Hepatozoon sp. HepIxo-281 (MH174345) recovered from ticks of the I. sigelos group in BFJNP (Muñoz-Leal et al. 2019).
Phylogenetic analyses performed for Hepatozoon 18S yielded a logic topology, separating the genus into two large clades: one composed by Hepatozoon spp. detected in amphibians, small mammals, reptiles, and ticks, and a second one conformed by species detected in canids. The genotypes characterized herein formed four independent lineages within a large clade composed by Hepatozoon spp. detected in South American rodents and their ticks (Fig. 4).

Bayesian and Maximum Likelihood phylogenies for a subset of Hepatozoon spp. inferred using an alignment (1,662 bp) of the gene encoding 18S rRNA gene. Calculate substitution models for BI and ML were M85, M15, M177, M147, M134, and M179, and HKY + F + G4, respectively. Best models were chosen using the Bayesian Information Criterion (BIC) (Schwarz 1978). Values of Bayesian Posterior probability/ML Bootstrap are indicated above, below, or arrowing major branches. The position of Hepatozoon spp. characterized in the present study is highlighted in bold
Sarcocystidae
Interestingly, the primers targeting DNA of Piroplasmida order amplified a 577-bp fragment of 18S that upon BLASTn analyses did not match any piroplasmids species. The amplicons were produced form two specimens of Thylamys elegans (Didelphimorphia: Didelphidae) and a single specimen of Thylamys pallidior (Didelphimorphia: Didelphidae), captured at BFJNP and Socoroma, respectively. Sequences obtained from the two species of Thylamys were identical and showed 100% sequence similarity with a undetermined apicomplexan denominated as Sarcocystidae sp. TE1 (577/577, 100% query cover, 0 gaps, 0 E value, EU443095), detected in the blood of T. elegans captured in Chile (Merino et al. 2010). Considering the high similarity with the sequence reported by Merino et al. (2010), and that the identity of these apicomplexans remains unsolved, we opted to designate it as Sarcocystidae sp. BFJ35 until additional genes can be analysed.
From a phylogenetic point of view, Sarcocystidae sp. BFJ35 formed a monophyletic clade with sequences of sarcocystids detected in the Australian marsupials Petaurus australis (Diprotodontia: Petauridae) and Acrobates pygmaeus (Diprotodontia: Acrabatidae) (Fig. 5) (Zhu et al. 2009; Holz et al. 2020). Collectively, the undetermined apicomplexan parasites detected in Acrobates, Petaurus, and Thylamys form an independent lineage within the Sarcocystidae family.

Bayesian and Maximum Likelihood phylogenies for a subset of apicomplexan protozoans. inferred using an alignment (1,992 bp) of the gene encoding 18S rRNA. Calculate substitution models for BI and ML were M40, M134, M136, M162, M138, and M125, and TIM2 + F + R3, respectively. Best models were chosen using the Bayesian Information Criterion (BIC) (Schwarz 1978). Values of Bayesian Posterior probability/ML Bootstrap are indicated above, below, or arrowing major branches. The position of Sarcocystidae sp. BFJ35 characterized in the present study is highlighted in bold
Discussion
To elucidate the role of wild mammals in the tick-host-microorganism reservoir system is important for recognizing areas where outbreaks of tick-borne pathogens might occur in nature (Mills 1998). Although previous research in Chile looking for tick-borne Anaplasmataceae identified “Candidatus Neoehrlichia chilensis” in Abrothrix sp., Mus musculus (Rodentia: Murinae), and Ixodes ticks collected upon P. darwini and Octodon degus (Rodentia: Octodontidae) (Müller et al. 2018; Muñoz-Leal et al. 2019), in our study, none of the tested animals was positive. Importantly, we used blood samples to perform the screenings whereas previous studies extracted DNA from the spleen to detect these agents (Müller et al. 2018), likely increasing the sensitivity of the assays. While our samples were negative for Anaplasmataceae, we detected novel genotypes of Babesia and Hepatozoon in rodents and expanded the distributional and host range of an apicomplexan of the Sarcocystidae family that infects marsupials.
Babesia
Babesia species are transmitted by ticks and merge their life cycles with wild mammals that maintain the infection in nature (Karshima et al. 2021). The B. microti group is of medical importance in the Northern Hemisphere, and five clades currently define its diversity (Goethert and Telford 2003). Human pathogenic strains belong to B. microti sensu stricto (Goethert and Telford 2003), and are represented mostly by Eurasian and North American isolates (Goethert 2021). Remarkably, a Babesia sp. 99% identical with B. microti sensu stricto detected in blood from inhabitants of the Bolivian Chaco (Gabrielli et al. 2016) represents the sole genetic evidence of this blood parasite in South America. Despite being a human pathogen, the identity of the vector or vertebrate reservoir of this agent is yet to be elucidated. Herein, we characterized amplicons derived from the 18S and COI of a Babesia sp. (Babesia sp. LC77) belonging to the B. microti group in Phyllotis rodents from Chile, suggesting that these mammals could act as a reservoir. Our results, combined to the record of Gabrielli et al. (2016) from Bolivia, indicate that the B. microti group is likely underrepresented in South America, and that neglected human-pathogenic strains circulate in the region.
The role of rodents as hosts for Babesia spp. has been barely assessed in South American ecosystems. However, Babesia spp. have been detected in T. pachyurus (Wolf et al. 2016), Thrichomys fosteri (de Sousa et al. 2018), Rattus norvegicus, and Oligoryzomys nigripes from Brazil (Gazeta et al. 2004). Herein, we characterized an additional genotype, Babesia sp. LC87, detected in P. darwini and A. jeslkii, therefore adding novel hosts for these tick-borne apicomplexans in the continent. Interestingly, Babesia sp. LC87 clusters phylogenetically within a recently described array of babesias associated with bats and rodents from Brazil (Wolf et al. 2016; Ikeda et al. 2021) (Fig. 2). It is important to note that we are aware that our sequences are relatively short, and that analyses employing longer sequences of 18S might yield a different tree topology. However, the phylogenies derived from previous studies do mirror our analyses (de Sousa et al. 2018; Ikeda et al. 2021), and support the occurrence of a South American lineage of Babesia spp. (Fig. 2). Although the vectors of these agents in the region remain unidentified, it is well known that Ixodes ticks transmit Babesia in the Northern Hemisphere (Karshima et al. 2021). In Chile, the Ixodes sigelos group of ticks are common parasites of Abrothrix and Phyllotis rodents (Muñoz-Leal et al. 2019; Landaeta-Aqueveque et al. 2021), so attempts to understand the ecoepidemiology of Babesia sp. LC77 and LC87 should consider those ticks as potential vectors.
Hepatozoon
Ten years ago, Merino et al. (2009) reported an Hepatozoon sp. in A. olivacea and A. sanborni at Chiloé island. Recently, Ixodes ticks collected upon Abrothrix longipilis (Rodentia; Cricetidae) and P. darwini were positive to this apicomplexan as well (Muñoz-Leal et al. 2019). Herein, we report P. darwini as new host for Hepatozoon in Chile, therefore expanding the distribution of agents of this genus within the country. Collectively, these results point that rodent of genera Abrothrix, Oligoryzomys, and Phyllotis are common intermediate hosts for Hepatozoon along Chilean ecosystems. Importantly, the four genotypes of Hepatozoon characterized in this study were retrieved from cricetid rodents, and clustered into a large monophyletic group including sequences retrieved from other rodents of this family and their ticks (Fig. 4a). This fact supports the hypothesis that cricetid rodents and their ectoparasites could maintain enzootic cycles of a natural group of Hepatozoon spp. in the region (Muñoz-Leal et al. 2019). Although the detection of Hepatozoon in ticks feeding on small mammals does not demonstrate any transmission capacity (Giannelli et al. 2013), these ectoparasites should not be ruled out as potential vectors (Muñoz-Leal et al. 2019).
Recently, Alabí et al. (2021) reported DNA of Hepatozoon in synanthropic rodents (M. musculus and R. rattus) and O. longicaudatus from Valdivia, in southern Chile. Given that the 18S sequences generated by Alabí et al. (2021) corresponded to a different region of the locus, we were unable to include them in our phylogenies. However, according to our phylogenetic inferences (Fig. 4), and those of Alabí et al. (2021), discrete lineages of Hepatozoon would be associated with specific genera of native rodents, likely denoting events of coevolution (Poulin and Keeney 2008). A similar pattern of association between Hepatozoon spp. and their rodent hosts was also reported by Merino et al. (2009), yet this trend seems to be an exception, since Hepatozoon species are considered by some authors to be rather generalist parasites with low specificity for their vertebrate hosts (Smith 1996; Telford et al. 2001). The fact that in Chile, Hepatozoon species appear to be associated with specific genera of rodents is intriguing and suggests that the evolutionary history of these mammals may be shaping the diversity of the parasites (Hoberg and Brooks 2010). To test this hypothesis, future research should aim to retrieve complete Hepatozoon 18S sequences together with data for fast-evolving loci such as COI, and target previously unstudied rodent species.
Sarcocystidae
The detection of apicomplexans in mouse opossums of genus Thylamys is an incidental finding that reflects the low specificity exhibited by the primers used in this study to amplify 18S of piroplasmid species. Moreover, our findings are not a novelty in Chile. In fact, Merino et al. (2010) reported an undescribed apicomplexan species related to the Sarcocystidae family in T. elegans. Although the number of Thylamys analysed in our study was low (n = 3), it is noteworthy that all the specimens were positive. In this context, the previous detection of a Sarcocystidae sp. in Chilean opossums also showed a high prevalence of infection (Merino et al. 2010). In contrast, all rodents (n = 99) sampled in our study were negative to this apicomplexan, indicating a degree of specificity for Thylamys opossums. Indeed, specific associations between Sarcocystidae parasites and their hosts have been reported to occur in other ecosystems (Šlapeta et al. 2003).
The relatedness of Sarcocystidae sp. BFJ35 genotype detected in Chilean opossums with apicomplexans from Australian marsupials is particularly interesting (Fig. 5), and suggests that a common ancestor that infected marsupials diverged with them 45–50 million years ago during the Eocene era, after the split of Antarctica and Australia (Nilsson et al. 2004; Merino et al. 2010). It is pertinent to note that there is evidence that this novel apicomplexan taxon can disseminate and cause severe liver, spleen, and lung infection, and as such, it would represent a threat to marsupial populations with conservation issues (Holz et al. 2020). In the meantime, the Sarcocystidae sp. associated with Australian and Chilean marsupials remains as an understudied taxon that clearly deserves more attention (Duszynski 2016).
Parasites of the Sarcocystidae family have heteroxenous life cycles involving carnivores as definitive hosts, and other vertebrates such as marsupials, as intermediate hosts (Votýpka et al. 2017). Oocysts of these parasites are released into the environment through faeces and reach novel hosts after ingestion of contaminated food or water (Votýpka et al. 2017; Holz et al. 2020). While the ecology of Sarcocistidae sp. BFJ35 is still unclear, the study of owls and foxes that usually prey on Thylamys spp. (Jaksic Andrade 1993; Carevic et al. 2013; Valladares Faúndez et al. 2018) should shed light on the definitive hosts of this novel apicomplexan species in Chile.
References
Alabí AS, Monti G, Otth C et al (2021) Genetic diversity of Hepatozoon spp. in rodents from Chile. Rev Bras Parasitol Veterinária 30:e012721. https://doi.org/10.1590/s1984-29612021082
Alabí AS, Monti G, Otth C et al (2020) Molecular survey and genetic diversity of Hemoplasmas in rodents from Chile. Microorganisms 8:1493. https://doi.org/10.3390/microorganisms8101493
Almeida AP, Marcili A, Leite RC et al (2012) Coxiella symbiont in the tick Ornithodoros rostratus (Acari: Argasidae). Ticks Tick Borne Dis 3:203–206. https://doi.org/10.1016/j.ttbdis.2012.02.003
Birkenheuer AJ, Levy MG, Breitschwerdt EB (2003) Development and evaluation of a seminested PCR for detection and differentiation of Babesia gibsoni (Asian genotype) and B. canis DNA in canine blood samples. J Clin Microbiol 41:4172–4177. https://doi.org/10.1128/JCM.41.9.4172-4177.2003
Brothers PS, Collins NE, Oosthuizen MC et al (2011) Occurrence of blood-borne tick-transmitted parasites in common tsessebe (Damaliscus lunatus) antelope in Northern Cape Province, South Africa. Vet Parasitol 183:160–165. https://doi.org/10.1016/j.vetpar.2011.06.015
Carevic FS, Carmona ER, Muñoz-Pedreros A (2013) Seasonal diet of the burrowing owl Athene cunicularia Molina, 1782 (Strigidae) in a hyperarid ecosystem of the Atacama desert in northern Chile. J Arid Environ 97:237–241. https://doi.org/10.1016/j.jaridenv.2013.07.008
Carpenter JW, Marion CJ (2018) Exotic Animal Formulary. Elsevier
Clarke JDA (2006) Antiquity of aridity in the Chilean Atacama Desert. Geomorphology 73:101–114. https://doi.org/10.1016/j.geomorph.2005.06.008
Cornillot E, Hadj-Kaddour K, Dassouli A et al (2012) Sequencing of the smallest Apicomplexan genome from the human pathogen Babesia microti†. Nucleic Acids Res 40:9102–9114. https://doi.org/10.1093/nar/gks700
D’elía G, Jhoann CH, Ossa G et al (2020) Lista actualizada de los mamíferos vivientes de Chile. Boletín Del Mus Nac Hist Nat 69:67–98
de Sousa KCM, Fernandes MP, Herrera HM et al (2017) Molecular detection of Hepatozoon spp. in domestic dogs and wild mammals in southern Pantanal, Brazil with implications in the transmission route. Vet Parasitol 237:37–46. https://doi.org/10.1016/j.vetpar.2017.02.023
de Sousa KCM, Fernandes MP, Herrera HM et al (2018) Diversity of piroplasmids among wild and domestic mammals and ectoparasites in Pantanal wetland, Brazil. Ticks Tick Borne Dis 9:245–253. https://doi.org/10.1016/j.ttbdis.2017.09.010
Duszynski DW (2016) Species Inquirendae in Marsupials. In: The biology and identification of the Coccidia (Apicomplexa) of Marsupials of the World. Elsevier, Academic Press, pp 155–173. https://doi.org/10.1016/C2014-0-02739-6
Gabrielli S, Totino V, Macchioni F et al (2016) Human babesiosis, Bolivia, 2013. Emerg Infect Dis 22:1445–1447. https://doi.org/10.3201/eid2208.150195
Gazeta GS, Carvalho RW, Avelar RF et al (2004) Ocorrência de Babesia sp em pequenos roedores no Brasil. Arq Bras Med Veterinária e Zootec 56:741–744. https://doi.org/10.1590/S0102-09352004000600007
Giannelli A, Ramos RAN, Dantas-Torres F et al (2013) Experimental evidence against transmission of Hepatozoon canis by Ixodes ricinus. Ticks Tick Borne Dis 4:391–394. https://doi.org/10.1016/j.ttbdis.2013.03.001
Goethert HK (2021) What Babesia Microti is now. Pathogens 10:1168. https://doi.org/10.3390/pathogens10091168
Goethert HK, Telford SR (2003) What is Babesia microti? Parasitology 127:301–309. https://doi.org/10.1017/S0031182003003822
Goodman J, Dennis D, Sonenshine D (2005) Tick-borne diseases of humans. ASM Press, Washington
Han BA, Schmidt JP, Bowden SE, Drake JM (2015) Rodent reservoirs of future zoonotic diseases. Proc Natl Acad Sci 112:7039–7044. https://doi.org/10.1073/pnas.1501598112
Hoberg EP, Brooks DR (2010) Beyond vicariance: integrating taxon pulses, ecological fitting, and oscillation in evolution and historical biogeography. In: Morand S, Krasnov BR (eds) The Biogeography of Host-Parasite Interactions. University Press, Oxford, pp 7–20
Holz PH, Koehler AV, Gasser RB, Dobson E (2020) Disseminated protozoal infection in a wild feathertail glider (Acrobates pygmaeus) in Australia. Int J Parasitol Parasites Wildl 13:46–50. https://doi.org/10.1016/j.ijppaw.2020.07.012
Homer MJ, Aguilar-Delfin I, Telford SR et al (2000) Babesiosis. Clin Microbiol Rev 13:451–469. https://doi.org/10.1128/CMR.13.3.451-469.2000
Huelsenbeck JP (2004) Bayesian phylogenetic model selection using reversible jump Markov chain Monte Carlo. Mol Biol Evol 21:1123–1133. https://doi.org/10.1093/molbev/msh123
Huelsenbeck JP, Rannala B (2004) Frequentist properties of Bayesian posterior probabilities of phylogenetic trees under simple and complex substitution models. Syst Biol 53:904–913. https://doi.org/10.1080/10635150490522629
Ikeda P, Menezes TR, Torres JM et al (2021) First molecular detection of piroplasmids in non-hematophagous bats from Brazil, with evidence of putative novel species. Parasitol Res 120:301–310. https://doi.org/10.1007/s00436-020-06985-w
Jaksic Andrade F (1993) The components of predation on small mammals in semiarid Chile: preliminary results. Rev Chil Hist Nat 66:305–321
Kalyaanamoorthy S, Minh BQ, Wong TKF et al (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589. https://doi.org/10.1038/nmeth.4285
Karshima SN, Karshima MN, Ahmed MI (2021) Animal reservoirs of zoonotic Babesia species: a global systematic review and meta-analysis of their prevalence, distribution and species diversity. Vet Parasitol 298:109539. https://doi.org/10.1016/j.vetpar.2021.109539
Katoh K, Standley DM (2013) MAFFT Multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Landaeta-Aqueveque C, Moreno Salas L, Henríquez A et al (2021) Parasites of native and invasive rodents in Chile: ecological and human health needs. Front Vet Sci 8:e643742. https://doi.org/10.3389/fvets.2021.643742
Leite Y, Patton J (2002) Evolution of South American spiny rats (Rodentia, Echimyidae): the star-phylogeny hypothesis revisited. Mol Phylogenet Evol 25:455–464. https://doi.org/10.1016/S1055-7903(02)00279-8
Livanova NN, Fomenko NV, Akimov IA et al (2018) Dog survey in Russian veterinary hospitals: tick identification and molecular detection of tick-borne pathogens. Parasit Vectors 11:591. https://doi.org/10.1186/s13071-018-3161-5
Luebert F, Pliscoff P (2017) Sinopsis bioclimática y vegetacional de Chile, 2nd edn. Editorial Unviersitaria, Santiago, Chile, p 384
Mejías C, Castro‐Pastene CA, Carrasco H, et al (2021) Natural history of the relict marsupial Monito del Monte at the most extreme altitudinal and latitudinal location. Ecosphere 12:e03577. https://doi.org/10.1002/ecs2.3577
Merino S, Martínez J, Vásquez RA, Šlapeta J (2010) Monophyly of marsupial intraerythrocytic apicomplexan parasites from South America and Australia. Parasitology 137:37–43. https://doi.org/10.1017/S0031182009990710
Merino S, Vásquez RA, Martínez J et al (2009) Molecular characterization of an ancient Hepatozoon species parasitizing the ‘living fossil’ marsupial ‘Monito del Monte’ Dromiciops gliroides from Chile. Biol J Linn Soc 98:568–576. https://doi.org/10.1111/j.1095-8312.2009.01302.x
Mills J (1998) Ecologic studies of rodent reservoirs: their relevance for human health. Emerg Infect Dis 4:529–537. https://doi.org/10.3201/eid0404.980403
Minh BQ, Nguyen MAT, von Haeseler A (2013) Ultrafast approximation for phylogenetic Bootstrap. Mol Biol Evol 30:1188–1195. https://doi.org/10.1093/molbev/mst024
Müller A, Gutiérrez R, Seguel M et al (2020) Molecular survey of Bartonella spp. in rodents and fleas from Chile. Acta Trop 212:105672. https://doi.org/10.1016/j.actatropica.2020.105672
Müller A, Monti G, Otth C et al (2018) “Candidatus Neoehrlichia chilensis” sp. nov.: molecular detection and characterization of a novel Anaplasmataceae in wild rodents from Valdivia, southern Chile. Transbound Emerg Dis 65:357–362. https://doi.org/10.1111/tbed.12815
Muñoz-Leal S, Lopes MG, Marcili A et al (2019) Anaplasmataceae, Borrelia and Hepatozoon agents in ticks (Acari: Argasidae, Ixodidae) from Chile. Acta Trop 192:91–103. https://doi.org/10.1016/j.actatropica.2019.02.002
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol Biol Evol 32:268–274. https://doi.org/10.1093/molbev/msu300
Nilsson MA, Arnason U, Spencer PBS, Janke A (2004) Marsupial relationships and a timeline for marsupial radiation in South Gondwana. Gene 340:189–196. https://doi.org/10.1016/j.gene.2004.07.040
Parola P, Roux V, Camicas J-L et al (2000) Detection of ehrlichiae in African ticks by polymerase chain reaction. Trans R Soc Trop Med Hyg 94:707–708. https://doi.org/10.1016/S0035-9203(00)90243-8
Patton JL, Pardiñas UFJ, D’Elía G (2015) Mammals of South America, vol 2. University of Chicago Press
Perles L, Roque ALR, D’Andrea PS et al (2019) Genetic diversity of Hepatozoon spp. in rodents from Brazil. Sci Rep 9:10122. https://doi.org/10.1038/s41598-019-46662-2
Poulin R, Keeney DB (2008) Host specificity under molecular and experimental scrutiny. Trends Parasitol 24:24–28. https://doi.org/10.1016/j.pt.2007.10.002
Rambaut A, Drummond AJ, Xie D et al (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904. https://doi.org/10.1093/sysbio/syy032
Rar V, Golovljova I (2011) Anaplasma, Ehrlichia, and “Candidatus Neoehrlichia” bacteria: pathogenicity, biodiversity, and molecular genetic characteristics, a review. Infect Genet Evol 11:1842–1861. https://doi.org/10.1016/j.meegid.2011.09.019
Ronquist F, Teslenko M, van der Mark P et al (2012) MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. https://doi.org/10.1093/sysbio/sys029
Thomas R, Santodomingo AM, Muñoz-Leal S et al (2020) Rodents as potential reservoirs for Borrelia spp. in northern Chile. Rev Bras Parasitol Veterinária 29:e000120. https://doi.org/10.1590/s1984-29612020029
Schreeg ME, Marr HS, Tarigo JL et al (2016) Mitochondrial genome sequences and structures aid in the resolution of Piroplasmida phylogeny. PLoS ONE 11:e0165702. https://doi.org/10.1371/journal.pone.0165702
Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6(2):461–464. https://doi.org/10.1214/aos/1176344136
Sikes RS (2016) 2016 Guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. J Mammal 97:663–688. https://doi.org/10.1093/jmammal/gyw078
Šlapeta JR, Modrý D, Votýpka J et al (2003) Evolutionary relationships among cyst-forming coccidia Sarcocystis spp. (Alveolata: Apicomplexa: Coccidea) in endemic African tree vipers and perspective for evolution of heteroxenous life cycle. Mol Phylogenet Evol 27:464–475. https://doi.org/10.1016/S1055-7903(03)00018-6
Smith TG (1996) The genus Hepatozoon (Apicomplexa: Adeleina). J Parasitol 82:565. https://doi.org/10.2307/3283781
Telford J, Wozniak EJ, Butler JF (2001) Haemogregarine specificity in two communities of Florida snakes, with descriptions of six new species of Hepatozoon (Apicomplexa: Hepatozoidae) and a possible species of Haemogregarina (Apicomplexa: Haemogregarinidae). J Parasitol 87:890–905. https://doi.org/10.1645/0022-3395(2001)087[0890:hsitco]2.0.co;2
Valladares Faúndez P, Urrutia Osorio N, Álvarez Henríquez N, Alvarado Orellana S (2018) Comparación de la dieta del pequén (Athene cunicularia) a nivel intra e interespecífico en el desierto de Atacama, Chile. Interciencia 43:93–97
Votýpka J, Modrý D, Oborník M et al (2017) Apicomplexa. Handbook of the Protists. Springer International Publishing, Cham, pp 567–624
Wolf RW, Aragona M, Muñoz-Leal S et al (2016) Novel Babesia and Hepatozoon agents infecting non-volant small mammals in the Brazilian Pantanal, with the first record of the tick Ornithodoros guaporensis in Brazil. Ticks Tick Borne Dis 7:449–456. https://doi.org/10.1016/j.ttbdis.2016.01.005
Zhu BY, Hartigan A, Reppas G et al (2009) Looks can deceive: molecular identity of an intraerythrocytic apicomplexan parasite in Australian gliders. Vet Parasitol 159:105–111. https://doi.org/10.1016/j.vetpar.2008.10.031
Acknowledgements
We thank Constanza Aguilera, Angel Oviedo, Ignacio Troncoso, and Juan Uribe for their collaboration in field and laboratory work. To Dr. Jonas Moraes-Filho for providing positive controls to perform genetic screenings. To Dr. Douglas McIntosh for kindly providing advice on English language. This paper is dedicated to the memory of Dr. Daniel González-Acuña, who made significant contributions to the study of parasites in Chile and passed away while this study was being elaborated.
Funding
This study was supported by the “Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT)” N° 1170972 and by the ANID BECAS/Scholarship Program/DOCTORADO NACIONAL/2019–21190078 and 2020–21200182. Funders had no role in the study design, data collection, analysis, preparation of the manuscript, and decision to publish it.
Author information
Authors and Affiliations
Contributions
Adriana M. Santodomingo: conceptualization, data curation, methodology, formal analysis, investigation, writing—original draft, and writing—review and editing. Richard S. Thomas: conceptualization, formal analysis, investigation, methodology, and writing—review and editing. Julian F. Quintero-Galvis: formal analysis, methodology, and review and editing. Diana M. Echeverry-Berrio: methodology, writing—original draft, and writing—review and editing. María C. Silva-de la Fuente: methodology, and review and editing. Lucila Moreno-Salas: formal analysis, investigation, project administration, review and editing. Sebastián Muñoz-Leal: conceptualization, data curation, methodology, resources, formal analysis, investigation, project administration, writing—original draft, and writing—review and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Procedures performed in this study were verified and approved by the Bioethics Committee of School of Veterinary Sciences, Universidad de Concepción (Form CBE-19–2017). Captures of small mammals and field work in national parks and reserves were authorized by the Servicio Agrícola y Ganadero (SAG; Resolution No. 1532/2019 and 9071/2018), and the Corporación Nacional Forestal (CONAF; Permits 39/2018; 67/2019; 05/2018; 76/2018; 66/2018), respectively.
Competing interests
The authors declare no competing interests.
Additional information
Handling Editor: Una Ryan
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Santodomingo, A.M., Thomas, R.S., Quintero-Galvis, J.F. et al. Apicomplexans in small mammals from Chile, with the first report of the Babesia microti group in South American rodents. Parasitol Res 121, 1009–1020 (2022). https://doi.org/10.1007/s00436-022-07452-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-022-07452-4