
Abstract
In the poultry industry, Eimeria spp. is one of the important pathogens which cause significant economic losses. We have previously generated a chicken monoclonal antibody (mAb), 6D-12-G10, with specificity for an antigen located in the apical cytoskeleton of Eimeria acervulina and with cross-reactive among Apicomplexan parasites, including other Eimeria spp., Toxoplasma, Neospora, and Cryptosporidium spp. Furthermore, the protein of Cryptosporidium parvum recognized by the 6D-12-G10 has been identified as elongation factor-1α (EF-1α). In the present study, to identify the target molecule of E. acervulina by the mAb, we performed two-dimensional Western blotting analysis. Finally, we found two positive molecules which are identified as EF-1α and a related protein. Our previous finding using C. parvum and the results in this study suggest that EF-1α could be associated with the invasion facilitated by the cytoskeleton at the apical region of zoites.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Protozoan parasites of the phylum Apicomplexa include a large number of medically or veterinary important species. Among them, Toxoplasma gondii causes encephalitis and birth defects in humans and in a wide range of animals, Plasmodium is the etiological agent of malaria, Cryptosporidium causes watery diarrhea and mortality in immunocompromised individuals, and Eimeria induces lethal diarrhea in livestock. All the apicomplexans are obligate intracellular parasites, and their biological cycle involves the invasion of host cells, growth and division, and release after lysis of the host cells. The invasive zoites possess a collection of unique organelles, which form the apical complex at the apical region (Morrissette and Sibley 2002). These organelles include the rhoptries, micronemes, and dense-granule organelles, as well as apical polar ring and subpellicular microtubules as cytoskeleton-associated structures (Santos et al. 2009). Although the functions of the organelles have not been fully elucidated, they are suggested to be associated with motility, adhesion, invasion, and formation of the parasitophorous vacuole (Dubremetz et al., 1998; Blackman and Bannister 2001).
In the poultry industry, Eimeria has been recognized as the most significant protozoan parasite owing to its worldwide prevalence and potential to cause economic losses (Dalloul and Lillehoj 2006). Rather than using antibody strategies with laboratory animals, we have previously generated monoclonal antibodies (mAb) against Eimeria acervulina (a species that infects chicken) using the chicken immune system to identify target vaccine molecules as an alternative to chemoprophylactic methods (Lillehoj et al. 1994; Sasai et al. 1996; Constantinoiu et al. 2003). In previous studies, a chicken mAb-designated 6D-12-G10 significantly inhibited the invasion of T lymphocytes by sporozoites in vitro and recognized the conoid of the sporozoites (Sasai et al. 1996 and 1998). The 6D-12-G10 is the first and only mAb that recognized the conoid of the apical complex. Moreover, 6D-12-G10 appears to show high cross-reactivity with related parasites, including six other chicken Eimeria spp., Neospora caninum, T. gondii, and Cryptosporidium spp. (Sasai et al. 1998; Matsubayashi et al. 2005 and 2013). Further analyses in Cryptosporidium parvum revealed that the protein recognized by 6D-12-G10 was elongation factor-1α (EF-1α). Although Cryptosporidium spp. lack a conoid, it was suggested that EF-1α was associated with the invasion supported by the cytoskeleton located at the apical region of zoites (Matsubayashi et al. 2008 and 2013). However, the molecule of E. acervulina as the original antigen of the chicken mAb remains unknown. In the present study, we found that this molecule corresponded to the conoid antigen in E. acervulina.
Materials and methods
Chicks and parasites
The USDA strain of E. acervulina was maintained at the Laboratory of Parasitic Diseases of the National Institute of Animal Health (NIAH; Tsukuba, Ibaraki, Japan) and was used throughout this study. The parasites were maintained by passage in 2- to 3-week-old chicks (Nisseiken, Tokyo, Japan). The chicks were housed in wire-floored cages in coccidian-free rooms and with free access to feed and water not supplemented with anticoccidial drugs or antibiotics. The animals were treated in accordance with protocols approved by the Animal Care and Use Committee of NIAH (approval no. 13-090). The study animals were orally inoculated with 2 × 105 oocysts, and feces were collected after 6–8 days. Oocysts were purified from feces using the sugar flotation method. The purified oocysts were sporulated by incubation at 28 °C in 2.5 % potassium dichromate (Wako, Osaka, Japan) for 3–4 days.
Preparation of sporozoites
For purification of sporozoites, the sporulated oocysts (5 × 108) were treated with sodium hypochlorite (Nacalai Tesque, Kyoto, Japan) for 20 min at 4 °C and washed with phosphate-buffered saline (PBS). The sporozoites were ruptured by vortexing with glass beads for 2–3 min to release the sporocysts. The sporocysts were treated in excystation medium, 0.25 % (w/v) trypsin (Merck, Darmstadt, Germany), and 1 % (w/v) taurodeoxycholic acid (Sigma, STL, USA) in Hanks’ balanced salt solution (Sigma), pH 7.4, at 41 °C in a 5 % CO2 incubator for 90–120 min. The sporozoites were purified by two methods. Namely, the sporozoites were obtained by DEAE-cellulose columns (DE52; GE Healthcare, Tokyo, Japan) (Schmatz et al. 1984) or by centrifugation at 106×g for 2 min according to a modification of a previously described method (Stotish et al. 1977).
For one-dimensional polyacrylamide gel electrophoresis (1-DE), the purified sporozoites were diluted in PBS, and then, the antigens were extracted as previously reported (Sasai et al. 1998). After centrifugation at 600×g for 5 min, the aliquoted supernatants were treated with sample buffer (125 mM Tris, 4 % SDS, 20 % glycerol, 10 % β-mercaptoethanol, 0.0025 % bromophenol blue) and were heated at 95 °C for 4 min. For two-dimensional polyacrylamide gel electrophoresis (2-DE), the purified sporozoites were dissolved in 1.0–1.5 ml of rehydration/sample buffer containing 8 M urea, 2.0 % CHAPS, 50 mM dithiothreitol, 0.2 % Bio-lyte 3/10, and 0.001 % bromophenol blue (Bio-Rad, CA, USA). The solution was centrifuged at 600×g for 5 min, and the supernatant was aliquoted and stored at –80 °C until use. Protein concentration was determined using the Pierce™ Coomassie Blue Assay Kit (Thermo Fisher Scientific, MA, USA).
One- and two-dimensional polyacrylamide gel electrophoresis and Western blotting
For 1-DE, 20–40 μg of prepared E. acervulina antigens was resolved on 4 % stacking/12 % resolving sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) at 200 V constant voltages (Laemmli 1970), and the separated proteins were blotted to polyvinylidene difluoride (PVDF) membrane (Immobilon Transfer Membranes, Millipore, Bedford, Massachusetts) using the Mini Trans-Blot® Electrophoretic Transfer Cell (Bio-Rad). 2-DE was performed using the Protean IEF Cell kit (Bio-Rad) according to the manufacturer’s protocol. Briefly, for first-dimension electrophoresis, ReadyStrip™ pH 3–10 IPG strips (7 cm, Bio-Rad) were rehydrated for 12 h with 150 μl of 100 μg of the sporozoite extract. The focusing conditions were 20 min at 250 V, followed by voltage ramping to 4000 V over a 2-h interval and electrophoresis for 2.5 h at 4000 V at 20 °C. For second-dimension electrophoresis, proteins were resolved on 4 % stacking/12 % gradient SDS-PAGE gels at 200 V. Second-dimension gels were blotted to a PVDF membrane (Millipore) and analyzed by Western blotting as described below.
Western blot analysis was performed as previously reported (Matsubayashi et al. 2005). Briefly, a cell culture supernatant containing mAb 6D-12-G10 (Sasai et al. 1996) and a chicken-purified IgG as a control (1:1000, Sigma) were used as the primary antibodies, and the bound antibodies were detected using horseradish peroxidase-labeled rabbit anti-chicken IgG F(ab)′2 fragment (1:1000, Cortex Biochem., CA, USA) and developed using the TMB Membrane Peroxidase Substrate System (Kirkegaard & Perry Laboratories, MD, USA) according to the manufacturer’s instructions.
ELISA
To confirm the reactivity against the antigen prepared for 1-DE, antigens extracted from sporozoite (50 μl/well at 50 μg/ml) was coated onto flat-bottomed 96-well enzyme-linked immunosorbent assay (ELISA) plates (Becton Dickinson). Non-specific binding was blocked by 1 % bovine serum albumin in PBS for 1 h at 37 °C. The mAb 6D-12-G10 or control (purified normal chicken IgG (1:1000, Sigma)) was distributed at 50 μl/well. Following incubation at 37 °C for 1 h, plates were washed with PBS containing 0.05 % Tween 20 (PBST) and incubated at 37 °C for 1 h with 50 μl/well of HRP-labeled rabbit anti-chicken IgG F(ab)′2 fragment (1:1000, Cortex Biochem., California, USA). Plates were washed with PBST and developed using TMB microwell peroxidase substrate (KPL) according to the manufacturer’s instructions. The single wavelength absorbance was measured at 450 nm by iMark™ Microplate Absorbance Reader (Bio-Rad).
Identification of proteins
The E. acervulina antigen recognized by mAb 6D-12-G10 was identified via NH2-terminal amino acid sequencing in combination with liquid chromatography/tandem mass spectrometry (LC-MS/MS), as previously reported (Matsubayashi et al. 2013). Briefly, for LC-MS/MS analysis, the positive spots detected in the 2-DE gel after staining with Coomassie blue (Bio-rad) were excised, digested with trypsin (Promega, WI, USA), and resolved via high-performance liquid chromatography on a C-18 column (0.1 × 50 mm; Michrom BioResources, MA, USA) coupled to a Q-TOF2 mass spectrometer equipped with a nanoelectrospray ionization source (Waters Micromass, McKinley Scientific, NJ, USA). Positive ion tandem mass spectra were acquired, and the MS/MS data were used to search a non-redundant NCBI protein database using the MASCOT software (Matrix Science, MA, USA).
Ethics statement
Any animals were not scarified for the purpose of this study. Any human participants were not involved in this study. All the authors have no conflict of interest.
Results
To identify the molecule of E. acervulina recognized by the mAb 6D-12-G10, the 1-DE and 2-DE Western blotting analyses were conducted. After purifications of sporozoites by DEAE-cellulose columns or the centrifugal elutriation, antigens were extracted in PBS for 1-DE. As results, no positive bands were observed in these antigens by Western blotting. By ELISA, absorbance values of these antigens did not show any significant reactivity compared with those of the control. Thus, it was suggested that the molecules recognized the mAb could not be extracted in PBS.
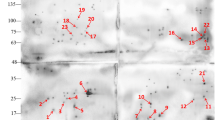
Subsequently, after purification by DEAE-cellulose columns, sporozoites were treated with rehydration/sample buffer for 2-DE, and however, any positive spots against the mAb 6D-12-G10 were not seen by 2-DE Western blotting. While using the antigens prepared for 2-DE after the centrifugal elutriation, some spots were detectable by Western blotting with mAb 6D-12-G10 (Fig. 1a). When using the control antibody, chicken IgG, on the other hand, most of spots other than two spots that were indicated by two arrows in Fig. 1a were immunostained (Fig. 1b). These results suggest that these two spots were the target molecules of the mAb 6D-12-G10 in E. acervulina. These two spots were named provisionally as spot nos. 1 and 2, respectively. Molecular weight and isoelectric point (pI) of these spot nos. 1 and 2 were calculated each 37.8 kDa and pI 7.39 and 39.0 kDa and pI 7.49, respectively. Figure 1c shows Coomassie blue staining profile of the blotting membrane, and these spots were identified as a single distinct protein.

Two-dimensional Western blot analysis of E. acervulina antigens stained with mAb 6D-12-G10 (a), normal chicken IgG (b), and the proteins separated by two-dimensional gel electrophoresis were stained with Coomassie blue (c). Arrows indicate positive spots. The positions of the molecular mass standards are indicated on the right. pH values are indicated at the top
The tryptic peptides derived from the proteins reactive to 6D-12-G10 were analyzed by LC-MS/MS. The significant hits with >10 % sequence coverage after database search of the tandem mass spectra using Mascot are shown in Table 1 and Figs. 2 and 3. The protein in spot no. 1 was identified as E. acervulina EF-1α as a significant hit. LC-MS/MS analysis indicated that the tryptic peptides in spot no. 2 corresponded to two predicted proteins with 23 % sequence coverage of E. acervulina EF-1α protein and 18.6 % coverage of E. acervulina transhydrogenase.

Amino acid sequences of E. acervulina transhydrogenase and EF-1α (NCBI database accession nos. CDI76761 and CDI83049). The tryptic peptides identified by LC-MS/MS analysis using E. acervulina antigen (spot no. 1) and recognized by mAb 6D-12-G10 are underlined

Amino acid sequences of E. acervulina transhydrogenase and EF-1α (NCBI database accession nos. CDI76761 (a) and CDI83049 (b)). The tryptic peptides identified by LC-MS/MS analysis using E. acervulina antigen (spot no. 2) and recognized by mAb 6D-12-G10 are underlined
Discussion
The E. acervulina conoid antigen had previously been recognized by one-dimensional electrophoretic separation followed by Western blotting as a 21-kDa protein (Sasai et al. 1996). Since then, we tried to identify the protein, and however, we could not detect the positive bands so far. In the present study, we concluded that the positive protein could not be detected by extraction in PBS unlike the method as described previously. About 2-DE Western blotting, we could identify the positive spots of two distinct proteins (with 39.0 and 37.8 kDa, respectively) only by the centrifugal purification, but not by DEAE-cellulose columns. Although the differences in molecular weight between our results and previous findings cannot be completely explained, one possible reason might be the methods of purification for sporozoites or by the soluble reagents used for 2-DE, including urea and CHAPS. Additionally, we speculate that the reproducibility of purification for sporozoites including protein or epitope preservations by DEAE-cellulose columns might not be always realized depending on the researchers. Furthermore, the conoid is thought to intermittently protrude beyond the apical end of zoites before the invasion (Hu et al. 2002), and thus, it remains unknown that the antigens could be secreted or released at this moment. Instead of the complicated and troublesome purifications using cellulose columns, we obtained sporozoites using a light centrifugation, as described above, without losing a large number of zoites and in a short period (within 30 min), and successfully identified the positive spots.
With regard to the 39.0-kDa antigen (spot no. 2), it corresponded to two distinct proteins: EF-1α, which was identical to the protein in spot no. 1, and as transhydrogenase. In general, transhydrogenases couple the transfer of reducing equivalents (hydride ion equivalents) between NAD(H) and NADP(H) to the translocation of protons across membranes (Hatefi and Yamaguchi 1996). In Eimeria, transhydrogenases are present in refractile bodies located in the cytoplasm, in the central area of sporozoites (Vermeulen et al. 1993), but not at the apical region, and little information on these enzymes is available for this parasite. To date, the whole genome of E. acervulina has not been fully sequenced, and therefore, genetic information is limited unlike Eimeria tenella. Actually, the number of expressed sequence tag of E. acervulina, which is deposited in GenBank, is 20,354, in contrast with 67,101 of E. tenella. Although details remain unknown, on the basis of this information, we speculate that the EF-1α isoforms reported in other organisms (Choi et al. 2009; Becker et al. 2013) or proteins homologous to EF-1α may also be present in E. acervulina. The slight differences in molecular weight and pI value between the isoforms may be due to the small database available for Eimeria. Although further studies on whole genome sequencing are needed, the proteins that possess the epitope recognized by 6D-12-G10 were found in two spots, which were thought to be EF-1α (spot no. 1, with 37.8 kDa) and EF-1α-like protein (spot no. 2, with 39.0 kDa).
EF-1α is a highly conserved ubiquitous protein involved in translation and functionally carries aminoacyl-tRNA to the A site of the ribosome in a GTP-dependent reaction (Riis et al. 1990). EF-1α is reported to have many other functions, including cell growth, motility, protein turnover, and signal transduction (Ridgley et al. 1996; Ransom-Hodgkins 2009). In addition, EF-1α can bind actin filaments and microtubules and regulate their assembly and cross-linking (Yang et al. 1990; Doyle et al. 2011). The results of our previous study on C. parvum suggest that EF-1α is associated with the cytoskeleton at the apical region and is an essential component of the invasion apparatus of the parasite (Matsubayashi et al. 2013). The distinct localization of EF-1α in zoites, i.e., on the apical membranes of C. parvum and in the conoid of E. acervulina, may be related to differences in the mechanism of invasion, considering that Eimeria sporozoites invade both T lymphocytes and intestinal epithelial cells, whereas C. parvum sporozoites infect only the surface microvilli of epithelial cells. Of note, our present results suggest that EF-1α of Apicomplexan parasites including EF-1α-associated protein can mediate an active process in the cytoskeleton; therefore, the elucidation of the role of this protein may help identify vaccine candidates against parasite infection.
References
Becker M, Kuhse J, Kirsch J (2013) Effects of two elongation factor 1A isoforms on the formation of gephyrin clusters at inhibitory synapses in hippocampal neurons. Histochem Cell Biol 140:603–609
Blackman MJ, Bannister LH (2001) Apical organelles of Apicomplexa: biology and isolation by subcellular fractionation. Mol Biochem Parasitol 117:11–25
Choi WI, Kim Y, Kim Y, Yu MY, Park J, Lee CE, Jeon BN, Koh DI, Hur MW (2009) Eukaryotic translation initiator protein 1A isoform, CCS-3, enhances the transcriptional repression of p21CIP1 by proto-oncogene FBI-1 (Pokemon/ZBTB7A). Cell Physiol Biochem 23:359–370
Constantinoiu CC, Lillehoj HS, Matsubayashi M, Hosoda Y, Tani H, Matsuda H, Sasai K, Baba E (2003) Analysis of cross-reactivity of five new chicken monoclonal antibodies which recognize the apical complex of Eimeria using confocal laser immunofluorescence assay. Vet Parasitol 118:29–35
Dalloul RA, Lillehoj HS (2006) Poultry coccidiosis: recent advancements in control measures and vaccine development. Expert Rev Vaccines 5:143–163
Doyle A, Crosby SR, Burton DR, Lilley F, Murphy MF (2011) Actin bundling and polymerisation properties of eukaryotic elongation factor 1 alpha (eEF1A), histone H2A-H2B and lysozyme in vitro. J Struct Biol 176:370–378
Dubremetz JF, Garcia-Réguet N, Conseil V, Fourmaux MN (1998) Apical organelles and host-cell invasion by Apicomplexa. Int J Parasitol 28:1007–1013
Hatefi Y, Yamaguchi M (1996) Nicotinamide nucleotide transhydrogenase: a model for utilization of substrate binding energy for proton translocation. FASEB J 10:444–452
Hu K, Roos DS, Murray JM (2002) A novel polymer of tubulin forms the conoid of Toxoplasma gondii. J Cell Biol 156:1039–1050
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lillehoj HS, Sasai K, Matsuda H (1994) Development and characterization of chicken-chicken B cell hybridomas secreting monoclonal antibodies that detect sporozoite and merozoite antigens of Eimeria. Poult Sci 73:1685–1693
Matsubayashi M, Kimata I, Iseki M, Lillehoj HS, Matsuda H, Nakanishi T, Tani H, Sasai K, Baba E (2005) Cross-reactivities with Cryptosporidium spp. by chicken monoclonal antibodies that recognize avian Eimeria spp. Vet Parasitol 128:47–57
Matsubayashi M, Takase H, Kimata I, Nakagawa H, Tani H, Sasai K, Baba E (2008) Electron microscopic observation of cytoskeletal frame structures and detection of tubulin on the apical region of Cryptosporidium parvum sporozoites. Parasitology 135:295–301
Matsubayashi M, Teramoto-Kimata I, Uni S, Lillehoj HS, Matsuda H, Furuya M, Tani H, Sasai K (2013) Elongation factor-1α is a novel protein associated with host cell invasion and a potential protective antigen of Cryptosporidium parvum. J Biol Chem 288:34111–34120
Morrissette NS, Sibley LD (2002) Cytoskeleton of apicomplexan parasites. Microbiol Mol Biol Rev 66:21–38
Ransom-Hodgkins WD (2009) The application of expression analysis in elucidating the eukaryotic elongation factor one alpha gene family in Arabidopsis thaliana. Mol Genet Genomics 281:391–405
Ridgley EL, Xiong ZH, Kaur KJ, Ruben L (1996) Genomic organization and expression of elongation factor-1 alpha genes in Trypanosoma brucei. Mol Biochem Parasitol 79:119–123
Riis B, Rattan SI, Clark BF, Merrick WC (1990) Eukaryotic protein elongation factors. Trends Biochem Sci 15:420–424
Sasai K, Lillehoj HS, Hemphill A, Matsuda H, Hanioka Y, Fukata T, Baba E, Arakawa A (1998) A chicken anti-conoid monoclonal antibody identifies a common epitope which is present on motile stages of Eimeria, Neospora, and Toxoplasma. J Parasitol 84:654–656
Sasai K, Lillehoj HS, Matsuda H, Wergin WP (1996) Characterization of a chicken monoclonal antibody that recognizes the apical complex of Eimeria acervulina sporozoites and partially inhibits sporozoite invasion of CD8+ T lymphocytes in vitro. J Parasitol 82:82–87
Santos JM, Lebrun M, Daher W, Soldati D, Dubremetz JF (2009) Apicomplexan cytoskeleton and motors: key regulators in morphogenesis, cell division, transport and motility. Int J Parasitol 39:153–162
Schmatz DM, Crane MS, Murray PK (1984) Purification of Eimeria sporozoites by DE-52 anion exchange chromatography. J Protozool 31:181–183
Stotish RL, Simashkevich PM, Wang CC (1977) Separation of sporozoites, sporocysts, and oocysts of Eimeria tenella by centrifugal elutriation. J Parasitol 63:1124–1126
Vermeulen AN, Kok JJ, van den Boogaart P, Dijkema R, Claessens JA (1993) Eimeria refractile body proteins contain two potentially functional characteristics: transhydrogenase and carbohydrate transport. FEMS Microbiol Lett 110:223–229
Yang F, Demma M, Warren V, Dharmawardhane S, Condeelis J (1990) Identification of an actin-binding protein from Dictyostelium as elongation factor 1a. Nature 347:494–496
Acknowledgments
We are grateful to Dr. Y. Fukuta, from the APRO Life Science Institute, Inc., for the LC-MS/MS analyses. This study was supported in part by the Grants-in-aid for Scientific Research 23580445 (to K. S., H. T., M. F., and M. M.) and 25450436 (to M. M.) from the Ministry of Education, Culture, Sports, and Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics statement
Any animals were not scarified for the purpose of this study. Any human participants were not involved in this study. All the authors have no conflict of interest.
Rights and permissions
About this article

Cite this article
Matsubayashi, M., Minoura, C., Kimura, S. et al. Identification of Eimeria acervulina conoid antigen using chicken monoclonal antibody. Parasitol Res 115, 4123–4128 (2016). https://doi.org/10.1007/s00436-016-5185-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-016-5185-0