
Abstract
A real-time fluorescence resonance energy transfer (FRET) PCR combined with melting curve analysis was developed for the detection of Opisthorchis viverrini in experimentally infected bithynid snails, its first intermediate hosts. The test is based on the fluorescence melting curve analysis of a hybrid between an amplicon from the pOV-A6-specific probe sequence, a 162-bp repeated sequence specific to O. viverrini and specific fluorophore-labeled probes. The real-time FRET PCR could detect as little as a single cercaria artificially introduced in a pool of 30 non-infected snails. The O. viverrini-infected snails were discriminated from non-infected snails and from genomic DNA of other parasite DNAs by their melting temperatures. Sensitivity and specificity of this method were both 100%. Melting curve analysis is a sensitive alternative for the specific detection of O. viverrini-infected snails; it is rapid, allows a high throughput, and can be done on small samples. The assay not only has a high potential for epidemiological surveys of O. viverrini-infected bithynid snails, but also for the detection of cercariae infestations of natural waterways when monitoring transmission sites.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Opisthorchiasis is a disease caused by the liver fluke Opisthorchis viverrini which is endemic throughout Southeast Asian countries including Laos, Cambodia, Vietnam, and Thailand. Approximately nine million people are infected (World Health Organization 1995; Yossepowitch et al. 2004). The disease has become more prevalent in developed countries with the influx of Asian immigrants (Woolf et al. 1984; Schwartz 1986; Peng et al. 1993). A recent survey in Thailand reported that 9.4% of the population were infected (Jongsuksuntigul and Imsomboon 2003). Typical symptoms include upper quadrant abdominal pain, dyspepsia, and fatigue. Gall bladder enlargement can be shown by ultrasonography (Mairiang et al. 1993). In heavily infected cases, cholangitis, biliary calculi, fibrosis of the periportal system, obstructive jaundice, and even bile duct cancer can develop (Schwartz 1980; Harinasuta et al. 1984; Pungpak et al. 1994; Sripa et al. 2007).
The aquatic life cycle of O. viverrini is complex involving two intermediate hosts (bithynid snails and cyprinoid fishes) and one definitive host (fish-eating mammals including humans). Definitive host infection occurs by eating raw or semi-cooked fish infested with the infective stage called metacercariae. After the worms mature in the bile ducts, adults produce eggs which pass through the bile ducts and exit through the feces. If the eggs fall in a body of freshwater, they are ingested by bithynid snails. In the snail, asexual reproduction occurs and results in the daily release of many cercariae. The free-swimming cercariae penetrate the tissue of cyprinoid fishes and transform into metacercariae (Wykoff et al. 1965).
From parasitological surveys, 0.11% of snails were estimated to be infected in any freshwater body in Thailand (Brockelman et al. 1986). However, the parasitological technique is limited by a low parasite burden and has to be done by trained personnel. The cercarial shedding and microscopic examinations for the presence of cercariae in snails are hard work and time consuming. Even trained personnel report false positives if the intermediate hosts are co-infected with other trematodes. False prevalence estimations typically occur because some snails harbor prepatent infections that are not detected by the cercarial shedding method. Because of these reasons, it is at present difficult to indicate real infection intensities in snails.
Thus, the development of a molecular approach for O. viverrini detection in infected snails would be useful, preferably one that is based exclusively on one methodology, such as PCR. Various conventional polymerase chain reaction (c-PCR) assays have been developed to detect O. viverrini DNA in definitive host feces and intermediate hosts (Wongratanacheewin et al. 2001, 2002; Maleewong et al. 2003; Stensvold et al. 2006; Duenngai et al. 2008), but these procedures demand analysis by agarose gel electrophoresis. A gel electrophoresis requires a long time, has a limited throughput, and a tendency to carry-over contamination as well as illusory results. Lately, effective real-time PCR has increasingly replaced the c-PCR due to its greatly improved molecular detection and differential diagnosis of microorganisms belonging to the same genus. Effectual real-time PCR is not only accurate, fast, and can quantify specific DNA in biological specimens (Zarlenga and Higgins 2001), but it also differentiates species or strains of several pathogenic organisms by melting curve analysis (Menard et al. 2005; Hakhverdyan et al. 2006; Abdelbaqi et al. 2007). Moreover, this method provides a high throughput, is performed in a closed system, since it does not need agarose gel electrophoresis for interpretation of the amplicons, and has a wide dynamic range (Walker 2001). The procedure has potential not only for the detection of infectious agents in suspicious biological samples but also for epidemiological surveys and for monitoring elimination strategies of infectious parasites.
However, this procedure has not been applied for the detection of O. viverrini in infected bithynid snails yet. Therefore, this study was designed to use a real-time fluorescence resonance energy transfer (FRET) PCR, another assay format of the real-time PCR, combined with melting curve analysis for the detection of this parasite in infected first intermediate hosts.
Materials and methods
Maintenance of the O. viverrini life cycle and experimental infection in snails
The O. viverrini life cycle was maintained in the first intermediate host, bithynid snails (Bithynia siamensis siamensis), the second intermediate host, cyprinoid fishes (Puntius altus), and a definitive host, Golden Syrian hamsters (Mesocricetus auratus) as previously described (Maleewong et al. 2003). The pure bithynid snails, B. siamensis siamensis, which were cultured and maintained in a laboratory aquarium, were allowed to eat Opisthorchis ova from infected hamsters. After 2 months, the snails were checked for the presence of cercariae by shedding. The free cercariae were taken, kept, and artificially inoculated in tissue aliquots of non-infected snails to determine the sensitivity of the real-time FRET PCR. The whole bodies of the infected snails were separately crushed and kept at −20°C for real-time FRET PCR determination. Non-infected snails were crushed and used to determine the control specificity of the test.
Preparation of specimens for the real-time FRET PCR
DNA samples were extracted from all non-infected and experimentally infected snails including their shells, as well as of the artificially cercaria-inoculated tissue samples. Each specimen was homogenized with disposable polypropylene pestles (Bellco Glass INC. Vineland, NJ, USA) and extracted using the Nucleospin Tissue kit (Macherey-Nagel GmbH & Co., Duren, Germany). The DNA was eluted in 100 μL of distilled water of which 5 μL was used in the real-time PCR reaction. Other parasite DNAs for standardization and specificity evaluations were acquired from O. viverrini, Haplorchis taichui, Centrocestus spp., Echinostoma malayanum, Fasciola gigantica, animal schistosomes, Paragonimus heterotremus, Stellantchasmus spp., or Haplorchoides spp. All fresh or frozen flukes were separately extracted and purified as described above.
Capability of detection
To determine the capability of real-time FRET PCR, non-infected ground snail tissues were separately prepared. Then, individual aliquots of 1, 10, and 30 non-infected snail samples were separately inoculated with 1, 5, 10 and 30 O. viverrini cercariae, respectively. The experiment samples were further used for genomic DNA extraction. The resultant DNAs were used in the real-time FRET PCR.
For a specificity evaluation of the method, 1 ng of genomic DNA from each of Centrocestus spp., H. taichui, F. gigantica, E. malayanum, P. heterotremus, Haplorchoides spp., Stellantchasmus spp., and animal schistosomes was used in the real-time FRET PCR.
Real-time FRET PCR assay
A LightCycler PCR and detection system (Roche Applied Science, Mannheim, Germany) was used for amplification and quantification. The PCR was performed in glass capillaries. A specific primer pair, OV-F (5′ CAG TGA GTG TCT ATT GGC TAA 3′) and OV-R (5′ GTA CTA CTC ATA AGG TTG CGT 3′; Proligo, Singapore), was designed to bind to the pOV-A6-specific DNA probe sequence, as described previously (Sermswan et al. 1991; Genbank Accession No. S80278). This target sequence was used because (1) the sequence was arranged as a tandem repeat and (2) several c-PCR experiments for the detection of O. viverrini DNA have used this sequence as target (Wongratanacheewin et al. 2001, 2002; Maleewong et al. 2003; Stensvold et al. 2006).
For amplification detection, the LightCycler FastStart DNA Master HybProbe Kit (Roche Applied Science) was used as recommended by the manufacturer. Briefly, a pair of adjacent oligoprobes was hybridized to an internal genus-specific repetitive sequence of O. viverrini. One probe was labeled at the 5′ end with the LightCycler Red 640 fluorophore (5′ Red 640-AGA AGG GCG AAA CCG GTC GTG G-Phosphate 3′; OVLC640 probe), and the other was labeled at the 3′ end with 530 fluorescein (5′ GGG ACT GCG CCT ACC TGA TAG CCC-Flou 530 3′; OVFL530 probe; Tib Molbiol, Berlin, Germany). Probes and primers were designed by using the LC probe design software (Roche Applied Science).
The PCR mixture contained LightCycler FastStart DNA Master HybProbe (Roche Applied Science), 2 mM MgCl2, 0.3 μM OV-F primer, 0.3 μM OV-R primer, 0.2 μM OVLC640 probe, and 0.2 μM OVFL530 probe, respectively. The total reaction volume was 20 μL. Samples were run by performing 45 cycles of repeated denaturation (10 s at 95°C), annealing (30 s at 50°C), and extension (10 s at 72°C). The temperature transition rate was 20°C/s. After amplification, a melting curve was produced by heating the product at 20°C/s to 95°C, cooling it to 40°C, keeping it there for 30 s, and then heating it slowly at 0.1°C/s to 75°C. The fluorescence intensity change was measured throughout the slow heating phase. In order to determine the specificity of the oligonucleotide hybridization based on the FRET technique, DNA extracted from parasites other than O. viverrini and non-infected bithynid snails were separately analyzed. Each run contained at least one negative control consisting of 5 μL distilled water.
For improved visualization of the melting temperatures (Tm), melting peaks were derived as previously described (Thanchomnang et al. 2008). Melting curves were used to determine the hybridization of probes and specific PCR products, which were confirmed by conventional agarose gel electrophoresis.
The O. viverrini-positive control plasmid
A positive control plasmid was constructed by cloning a PCR product of the pOV-A6-specific DNA probe sequence into the pCR4-TOPO vector (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. The PCR products were obtained by c-PCR using the primers OV-F and OV-R. The plasmid was produced in Escherichia coli and the nucleotide sequence of the inserted gene was sequenced in both directions. The nucleotide sequence of the cloned repeat revealed an identical structure to the O. viverrini sequence (Sermswan et al. 1991; Genbank Accession No. S80278). The size of the plasmid was 4,118 bp (including the 162-bp pOV-A6 sequence).
Results
Standardization of the real-time FRET PCR
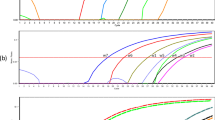
The detection limit of the hybridization real-time PCR was tested with 5 μL of serial dilutions of O. viverrini-positive control plasmid (102 to 1011 copies) and of O. viverrini genomic DNA (3 × 10−8 to 3 ng) in water. The trustworthy limit was approximately at 104 copies of positive control plasmid (Fig. 1) equivalent to 3 × 10−4 ng O. viverrini genomic DNA (figure not shown). Considering the specificity, no fluorescence indicator appeared when purified DNA from Centrocestus spp., H. taichui, F. gigantica, E. malayanum, P. heterotremus, Haplorchoides spp., Stellantchasmus spp., or animal schistosomes was tested.

Amplification plot of fluorescence (y-axis) vs cycle numbers (x-axis) show the analytical sensitivity of the real-time PCR for detecting Opisthorchis viverrini plasmid DNA. a O. viverrini plasmid 1011 copies/reaction; b O. viverrini plasmid 1010 copies/reaction; c O. viverrini plasmid 109 copies/reaction; d O. viverrini plasmid 108 copies/reaction; e O. viverrini plasmid 107 copies/reaction; f O. viverrini plasmid 106 copies/reaction; g O. viverrini plasmid 105 copies/reaction; h O. viverrini plasmid 104 copies/reaction; i O. viverrini plasmid 103 copies/reaction; j O. viverrini plasmid 102 copies/reaction; k distilled water
To estimate the capability of O. viverrini detection, DNA samples from aliquots of 1, 10, and 30 non-infected bithynid snails, artificially inoculated with 1, 5, 10, and 30 O. viverrini cercariae respectively, were amplified (Fig. 2). As little as a single metacercaria could be clearly detected in 30 bithynid snails as revealed by the 162-bp band in the ethidium bromide-stained gel (Fig. 2c, lane 4).

Capability of the real-time FRET PCR for the detection of Opisthorchis viverrini cercariae artificially inoculated into bithynid snails. Ethidium bromide staining patterns of the amplicon products on a 2% agarose gel. Lane m, DNA size markers (50 bp DNA ladder from Invitrogen); lanes 1 and 2, negative and positive controls containing distilled water and O. viverrini plasmid 108 copies/reaction, respectively. Lane 3, amplicon products from non-infected bithynid snails. Amplicon products from 1 (lane 4), 5 (lane 5), 10 (lane 6), and 30 (lane 7) O. viverrini cercariae artificially inoculated into each aliquot of 1 (a), 10 (b), and 30 (c) non-infected bithynid snails. The specific 162-bp products are marked on the right
Real-time FRET PCR detection in experimentally infected and non-infected bithynid snails
Infected and non-infected bithynid snails were separately examined using the real-time FRET PCR assay merged with a melting curve analysis of the specific hybridization probes and PCR products. All of the 30 infected snails were positive for O. viverrini, while all 30 non-infected snails were negative. The melting curve analyses for the O. viverrini DNA are shown in Fig. 3. The sensitivity and specificity of the method were 100% and 100%, respectively. The mean Tm value ± SD of the O. viverrini DNA in the infected snails group was 66.38 ± 0.12.

Representative melting curve analysis of two fluorophore-labeled probes hybridized to the amplification products of the tandem repetitive sequence pOV-A6 DNA from Opisthorchis viverrini. The melting temperature (Tm) of the double-stranded fragment is visualized by plotting the negative derivative of the change in fluorescence divided by the change in temperature in relation to the temperature [−(d/dT) Fluorescence (640/530)]. The turning point of this converted melting curve results in a peak and permits easy identification of the fragment specific Tm. Melting peaks of the positive control plasmid (a), O. viverrini-infected bithynid snails (b–d) and non-infected bithynid snails, Haplorchis taichui, Centrocestus spp., Fasciola gigantica, Echinostoma malayanum, Stellantchasmus spp., Paragonimus heterotremus, Haplorchoides spp., and animal schistosomes as well as the negative control containing no DNA (e)
Discussion
The detection of specific sequences by c-PCR has proved to be valuable for the diagnosis of a variety of parasitic infections in snail intermediate hosts, i.e., Schistosoma spp. (Abath et al. 2006; Abbasi et al. 2007), Fasciola hepatica (Cucher et al. 2006), Echinostoma caproni (Hertel et al. 2003), O. viverrini (Maleewong et al. 2003), and Clonorchis sinensis (Müller et al. 2007). Recently, the real-time FRET PCR-based method has been successfully used for the detection of parasites, e.g., detection of Wuchereria bancrofti (Lulitanond et al. 2004), Brugia malayi (Thanchomnang et al. 2008), Toxoplasma gondii (Brenier-Pinchart et al. 2007), and Plasmodium falciparum (Ojurongbe et al. 2007). In the present study, two individually labeled hybridization probes based on real-time FRET PCR combined with a melting curve analysis for the detection of O. viverrini infection in bithynid snails, the intermediate hosts, were developed for the first time. Two primers were employed to amplify the genus-specific amplicon, which was further exposed by its combined melting peak outline with two hybridizations of fluorophore-labeled probes. This procedure can detect as little as one cercaria implanted in a pool of 30 non-infected bithynid snails. The results showed 100% sensitivity which is possibly useful for application on field samples, such as for determining the prevalence of O. viverrini in areas with a low intensity of infection.
Regarding the specificity of the procedure, no fluorescence appeared and primers did not amplify the 162-bp band when DNA from other digenean flukes belonging to different families than O. viverrini was tested, indicating 100% specificity. Such our real-time FRET PCR could be useful for the differentiation of O. viverrini cercariae from those of other flukes, i.e., Centrocestus spp., H. taichui, F. gigantica, E. malayanum, Stellantchasmus spp., P. heterotremus, Haplorchoides spp., and animal schistosomes.
The real-time FRET PCR protocol described here provides another choice to the available classic and more modern molecular or immunologic methods for the detection of O. viverrini in snails, the first intermediate host. It can also provide information on O. viverrini epidemiology. By virtue of this methodology, the entire protocol starting from the DNA extraction of specimens to conclude the real-time FRET PCR can be completed within 5 h and thus provides a high throughput means since it does not require agarose gel electrophoresis for the analysis of the PCR products. The method also abolishes the need for the laborious and time-consuming microscopic examinations by experienced personnel. A large number of samples can be processed at the same time and only very small sample volumes are necessary. The method is independent of the subjective bias sometimes present in microscopic examinations and avoids confusion with other digenean flukes.
In conclusion, a specific and sensitive real-time FRET PCR for the detection of O. viverrini in snail intermediate hosts is presented here. The test is suitable not only for epidemiological studies and eradication programs for the whole range of intermediate hosts but also for the diagnosis of human infection. An essential and immediate application of this method should be cercarial surveys of natural water bodies in order to monitor transmission sites. This may be a way for the long-term control of the disease.
References
Abath FG, Gomes AL, Melo FL, Barbosa CS, Werkhauser RP (2006) Molecular approaches for the detection of Schistosoma mansoni: possible applications in the detection of snail infection, monitoring of transmission sites, and diagnosis of human infection. Mem Inst Oswaldo Cruz 101(Suppl 1):145–148
Abbasi I, King CH, Sturrock RF, Kariuki C, Muchiri E, Hamburger J (2007) Differentiation of Schistosoma haematobium from related schistosomes by PCR amplifying an inter-repeat sequence. Am J Trop Med Hyg 76:950–955
Abdelbaqi K, Buissonniere A, Prouzet-Mauleon V, Gresser J, Wesley I, Megraud F, Menard A (2007) Development of a real-time fluorescence resonance energy transfer PCR to detect Arcobacter species. J Clin Microbiol 45:3015–3021
Brenier-Pinchart MP, Morand-Bui V, Fricker-Hidalgo H, Equy V, Marlu R, Pelloux H (2007) Adapting a conventional PCR assay for Toxoplasma gondii detection to real-time quantitative PCR including a competitive internal control. Parasite 14:149–154
Brockelman WY, Upatham ES, Viyanant V, Ardsungnoen S, Chantanawat R (1986) Field studies on the transmission of the human liver fluke, Opisthorchis viverrini, in northeast Thailand: population changes of the snail intermediate host. Int J Parasitol 16:545–552
Cucher MA, Carnevale S, Prepelitchi L, Labbé JH, Wisnivesky-Colli C (2006) PCR diagnosis of Fasciola hepatica in field-collected Lymnaea columella and Lymnaea viatrix snails. Vet Parasitol 137:74–82
Duenngai K, Sithithaworn P, Rudrappa UK, Iddya K, Laha T, Stensvold CR, Strandgaard H, Johansen MV (2008) Improvement of PCR for detection of Opisthorchis viverrini DNA in human stool samples. J Clin Microbiol 46:366–368
Hakhverdyan M, Rasmussen TB, Thoren P, Uttenthal A, Belak S (2006) Development of a real-time PCR assay based on primer-probe energy transfer for the detection of swine vesicular disease virus. Arch Virol 151:2365–2376
Harinasuta T, Riganti M, Bunnang D (1984) Opisthorchis viverrini infection: pathogenesis and clinical features. Arzneimittelforschung 34:1167–1169
Hertel J, Haberl B, Hamburger J, Haas W (2003) Description of a tandem repeated DNA sequence of Echinostoma caproni and methods for its detection in snail and plankton samples. Parasitology 126:443–449
Jongsuksuntigul P, Imsomboon T (2003) Opisthorchiasis control in Thailand. Acta Trop 88:229–232
Lulitanond V, Intapan PM, Pipitgool V, Choochote W, Maleewong W (2004) Rapid detection of Wuchereria bancrofti in mosquitoes by LightCycler polymerase chain reaction and melting curve analysis. Parasitol Res 94:337–341
Mairiang E, Haswell-Elkins MR, Mairiang P, Sithithaworn P, Elkins DB (1993) Reversal of biliary tract abnormalities associated with Opisthorchis viverrini infection following praziquantel treatment. Trans R Soc Trop Med Hyg 87:194–197
Maleewong W, Intapan PM, Wongkham C, Wongsaroj T, Kowsuwan T, Pumidonming W, Pongsaskulchoti P, Kitikoon V (2003) Detection of Opisthorchis viverrini in experimentally infected bithynid snails and cyprinoid fishes by a PCR-based method. Parasitology 126:63–67
Menard A, Dachet F, Prouzet-Mauleon V, Oleastro M, Megraud F (2005) Development of a real-time fluorescence resonance energy transfer PCR to identify the main pathogenic Campylobacter spp. Clin Microbiol Infect 11:281–287
Müller B, Schmidt J, Mehlhorn H (2007) Sensitive and species-specific detection of Clonorchis sinensis by PCR in infected snails and fishes. Parasitol Res 100:911–914
Ojurongbe O, Ogungbamigbe TO, Fagbenro-Beyioku AF, Fendel R, Kremsner PG, Kun JF (2007) Rapid detection of Pfcrt and Pfmdr1 mutations in Plasmodium falciparum isolates by FRET and in vivo response to chloroquine among children from Osogbo, Nigeria. Malar J 6:41
Peng HW, Chao HL, Fan PC (1993) Imported Opisthorchis viverrini and parasite infections from Thai labourers in Taiwan. J Helminthol 67:102–106
Pungpak S, Chalermrut K, Harinasuta T, Viravan C, Schelp PF, Hempfling A, Schlattmann P, Bunnag D (1994) Opisthorchis viverrini infection in Thailand: symptoms and signs of infection—a population-based study. Trans R Soc Trop Med Hyg 88:561–564
Schwartz DA (1980) Helminths in the induction of cancer: Opisthorchis viverrini, Clonorchis sinensis and cholangiocarcinoma. Trop Geog Med 32:95–100
Schwartz DA (1986) Cholangiocarcinoma associated with liver fluke infection: a preventable source of morbidity in Asian immigrants. Am J Gastroenterol 81:76–79
Sermswan R, Mongkolsuk S, Panyim S, Sirisinha S (1991) Isolation and characterization of Opisthorchis viverrini specific DNA probe. Mol Cell Probes 5:399–407
Sripa B, Kaewkes S, Sithithaworn P, Mairiang E, Laha T, Smout M, Pairojkul C, Bhudhisawasdi V, Tesana S, Thinkamrop B, Bethony JM, Loukas A, Brindley PJ (2007) Liver fluke induces cholangiocarcinoma. PLoS Med 4:1148–1154
Stensvold CR, Saijuntha W, Sithithaworn P, Wongratanacheewin S, Strandgaard H, Ornbjerg N, Johansen MV (2006) Evaluation of PCR based coprodiagnosis of human opisthorchiasis. Acta Trop 97:26–30
Thanchomnang T, Intapan PM, Lulitanond V, Choochote W, Manjai A, Prasongdee TK, Maleewong W (2008) Rapid detection of Brugia malayi in mosquito vectors using a real-time fluorescence resonance energy transfer PCR and melting curve analysis. Am J Trop Med Hyg 78:509–513
Walker NJ (2001) Real-time and quantitative PCR: applications to mechanism-based toxicology. J Biochem Mol Toxicol 15:121–127
Wongratanacheewin S, Pumidonming W, Sermswan RW, Maleewong W (2001) Development of a PCR-based method for the detection of Opisthorchis viverrini in experimentally infected hamsters. Parasitology 122:175–180
Wongratanacheewin S, Pumidonming W, Sermswan RW, Pipitgool V, Maleewong W (2002) Detection of Opisthorchis viverrini in human stool specimens by PCR. J Clin Microbiol 40:3879–3880
Woolf A, Green J, Levine JA, Estevez EG, Weatherly N, Rosenberg E, Frothingham T (1984) A clinical study of Laotian refugees infected with Clonorchis sinensis or Opisthorchis viverrini. Am J Trop Med Hyg 33:1279–1280
World Health Organization (1995) Control of foodborne trematode infections. Report of a WHO Study Group. World Health Organ Tech Rep Ser 849:1–157
Wykoff DE, Harinasuta C, Juttijudata P, Winn MM (1965) Opisthorchis viverrini in Thailand. The life cycle and comparison with O. felineus. J Parasitol 51:207–214
Yossepowitch O, Gotesman T, Assous M, Marva E, Zimlichman R, Dan M (2004) Opisthorchiasis from imported raw fish. Emerg Infect Dis 10:2122–2126
Zarlenga DS, Higgins J (2001) PCR as a diagnostic and quantitative technique in veterinary parasitology. Vet Parasitol 101:215–230
Acknowledgments
This study received financial support from the Thailand Research Fund, Grant no. BRG50800005. The authors wish to thank Dr. Mark Roselieb for his assistance in the manuscript preparation. The maintenance and care of all experimental animals in this study were in compliance with the Thai law.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Intapan, P.M., Thanchomnang, T., Lulitanond, V. et al. Detection of Opisthorchis viverrini in infected bithynid snails by real-time fluorescence resonance energy transfer PCR-based method and melting curve analysis. Parasitol Res 103, 649–655 (2008). https://doi.org/10.1007/s00436-008-1026-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-008-1026-0