
Abstract
Objective
Clear cell renal cell carcinoma (ccRCC) is the most common type of kidney tumor characterized by the highest mortality rate of the genitourinary cancers, and, therefore, new diagnostic and/or prognostic biomarkers are urgently needed.
Methods
Based on genome-wide DNA methylation profiling in 11 pairs of ccRCC and non-cancerous renal tissues (NRT), the methylation at regulatory regions of ZNF677, FBN2, PCDH8, TFAP2B, TAC1, and FLRT2 was analyzed in 168 renal tissues and 307 urine samples using qualitative and quantitative methylation-specific PCR (MSP).
Results
Significantly higher methylation frequencies for all genes were found in ccRCC tissues compared to NRT (33–60% vs. 0–11%). The best diagnostic performance demonstrated a panel of ZNF677, FBN2, PCDH8, TFAP2B & TAC1 with 82% sensitivity and 96% specificity. Hypermethylation of ZNF677 and PCDH8 in the tissue samples was significantly related to numerous adverse clinicopathologic parameters. For the urine-based ccRCC detection, the highest diagnostic power (AUC = 0.78) was observed for a panel of ZNF677 & PCDH8 (with or without FBN2 or FLRT2) with 69–78% sensitivity and 69–80% specificity, albeit with lower values in the validation cohort. Besides, methylation of PCDH8 was significantly related to higher tumor stage and fat invasion in the study and validation cohorts. Moreover, PCDH8 was strongly predictive for OS (HR, 5.7; 95% CI 1.16–28.12), and its prognostic power considerably increased in combination with ZNF677 (HR, 12.5; 95% CI 1.47–105.58).
Conclusion
In summary, our study revealed novel, potentially promising DNA methylation biomarkers of ccRCC with the possibility to be applied for non-invasive urine-based ccRCC detection and follow-up.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Renal cell carcinoma (RCC) is the most frequent type of renal neoplasm, accounting for 85–90% of all renal malignancies, and is the most lethal cancer of the urinary system (Kabaria et al. 2016; Siegel et al. 2015). RCC encompasses a heterogeneous group of cancers derived from renal tubular epithelial cells with the major subtypes of clear cell RCC (ccRCC), which arises from the proximal convoluted tubule, and comprise 70–80% of all RCC cases (Chow et al. 2010). ccRCC is characterized by the predominance of metastatic disease (~ 90%) as well as accounts for the majority of deaths from kidney cancer (Chow et al. 2010).
Clinically most of the RCC cases are asymptomatic and nowadays discovered incidentally due to extensive use of abdominal computed tomography (CT), ultrasound (US), and magnetic resonance tomography (MRT) for other medical reasons (Jayson and Sanders 1998; Rossi et al. 2018). The critical point for the successful treatment of cancer is an early diagnosis. Meanwhile, 30% of RCC patients have metastases at the time of diagnosis, while an additional 30–50% will develop metastases during follow-up, even if radical surgery has been initially performed (Capitanio et al. 2019). If metastases are present at diagnosis, the probability of 5-year survival may be as low as 10–15%, while among patients with local disease, the 5-years survival rate reaches 95% (Hsieh et al. 2017a). Therefore, it is evident that there is an urgent need to develop new molecular biomarkers for the early diagnosis of ccRCC and to identify patients at high risk of progression.
Normal epigenetic processes, including genome-wide changes in the DNA methylation pattern, are disrupted during the initiation and progression of cancer (McMahon et al. 2017). Hypermethylation of the CpG islands is a common event in various cancer types, including kidney cancer, and is often associated with the transcriptional silencing of tumor-suppressor genes and downstream signaling pathways (Lasseigne and Brooks 2018). During renal cell carcinogenesis, mutations in genes responsible for epigenetic regulation are observed and lead to abundant changes in DNA methylation (The Cancer Genome Atlas Research Network, 2013). Because aberrant DNA methylation is one of the earliest observable molecular changes in cancer, these alterations could be valuable clinical biomarkers for disease diagnosis and/or prognosis. However, despite their potential, no accurate diagnostic or prognostic RCC DNA methylation biomarker has reached the clinic so far.
In urological tumors, especially in the case of RCC, tumor-derived methylated DNA can be easily detectable in the urine samples, which allows the development of non-invasive molecular tests (Larsen et al. 2019). Furthermore, ccRCC is considered to be a heterogeneous malignancy with high intra-tumor and inter-tumor heterogeneity (Gerlinger et al. 2014; Larsen et al. 2019; Hsieh et al. 2017b), that complicates diagnosis and prediction of the course of the disease. DNA methylation in urine bypasses this situation, providing a better reflection of tumor heterogeneity than the tissue sample. In addition, due to the easily available repeatability of the sample acquisition, urine-based biomarkers can be checked periodically in patients at risk, allowing the detection of small tumors at an early stage or following the real-time state of the cancer progression.
In this study, a set of DNA methylation biomarkers, specifically ZNF677, FBN2, PCDH8, TFAP2B, TAC1, and FLRT2, were investigated in the renal tissues as well as urine samples of patients diagnosed with ccRCC and showed valuable clinical utility as well as the promising potential for non-invasive ccRCC detection and prognosis.
Results
Genome-wide DNA methylation analysis
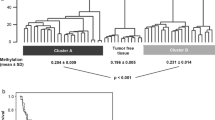
To identify potential DNA methylation biomarkers of ccRCC and to determine the amount of DNA methylation changes in cancerous renal tissues compared to NRT, the genome-wide DNA methylation profile was analyzed in 11 pairs of ccRCC and NRT samples. Among ccRCC, four samples were an early pT1a, while the remaining were of advanced pT3-4 stages. The comparison of cancerous and non-cancerous renal tissue samples revealed significant methylation differences (fold change (FC) ≥ 1.5; P ≤ 0.050) in 766 probes, reflecting 367 genes in total. About a half of differently methylated genes, particularly 175 (48%), were hypermethylated (Supplementary Table S1), and 192 (52%) were hypomethylated. Hierarchical clustering analysis discriminates NRT samples from the ccRCC one (Fig. 1). Comparison of the cases of different tumor stages with NRT revealed the most abundant DNA methylation differences in pT1 tumors. DNA methylation changes in pT1 tumors occurred in 1940 genes (FC ≥ 1.5; P ≤ 0.050) in total, of which 406 (21%) were hypermethylated, and 1526 (79%) were hypomethylated, including eight genes with concurrent changes observed according to different microarray probes. In the pT3-4 tumor stage, DNA methylation differences were less common and observed in 323 genes (FC ≥ 1.5; P ≤ 0.050), of which 210 (65%) were hypermethylated and 112 (35%) hypomethylated, including one overlapped gene. It is worth noting that these diversities between stages can be explained by a small number of samples being compared. However, the comparison of pT3-4 to pT1 revealed methylation differences (FC ≥ 1.5; P ≤ 0.050) in only eight genes. A considerable part of deregulated genes in the separate tumor stages was observed in the ccRCC vs. NRT comparison group as well (Supplementary Table S1; Fig. S1). Relatively scarce methylation differences were found when comparisons according to other clinical-pathological parameters were made. On the contrary, abundant methylation differences were observed among males and females as well as different age groups; however, only one of the genes overlapped with methylation changes determined in the ccRCC vs. NRT comparison group (Supplementary Table S2).

Genome-wide DNA methylation analysis in cancerous and non-cancerous renal tissues. Heat map for hierarchical clustering analysis of 11 pairs of clear cell renal cell carcinoma (ccRCC) and non-cancerous renal tissue (NRT) samples from 11 patients. Only probes indicating significantly differential methylation (FC ≥ 1.5; P ≤ 0.050) between cancerous and non-cancerous tissues are included in the heatmap. The color scale indicates relative methylation level normalized by reference sample (genomic DNA), where − 1 indicates hypomethylated while 1 hypermethylated status of the probe. pT pathological stage, WHO/ISUP World health organization/International Society of Urological Pathology, G grade, yr years, NA not applicable
Based on methylation differences according to the renal tissue histology and/or tumor stage, as well as concerning the number of particular gene-associated probes showing significant methylation differences, six genes ZNF677, FBN2, PCDH8, TFAP2B, TAC1, and FLRT2, were selected for further detailed analysis (Table 1). These genes were selected due to their putative contribution to the cancer hallmarks (according to literature data) and technical feasibility for targeted methylation-specific PCR analysis as well.
Targeted DNA methylation analysis in renal tissue samples and clinical-pathological features
DNA methylation status of ZNF677, FBN2, PCDH8, TFAP2B, TAC1, and FLRT2 was analyzed qualitatively at regulatory regions of the genes in 123 ccRCC and 45 NRT samples using MSP. DNA methylation of all genes was detected at least in one-third of ccRCC samples or even more frequently (from 33.3 to 60.2%) and was significantly more common in cancerous renal tissues as compared to NRT (all P < 0.010; Fig. 2A). Furthermore, quantitative analysis showed that methylation levels of all biomarkers were significantly higher in randomly selected 20 ccRCC than 10 NRT samples (all P < 0.050; Supplementary Fig. S2).

DNA Methylation frequencies of the selected genes according to the renal histology and clinical-pathological parameters. A Methylation frequencies in ccRCC and NRT samples; methylation frequencies according to B patients' gender, C tumor stage, D tumor size, E tumor WHO/ISUP grade, and F tumor necrosis. The box extends from the 25th to 75th percentiles; the line in the box is plotted at median; the plus sign depicts the mean; the whiskers represent the 10–90% range; data values outside the range are marked as dots. ccRCC clear cell renal cell carcinoma, NRT non-cancerous renal tissues, WHO/ISUP World Health Organisation/International Society of Urological Pathology, pT pathological tumor stage. Significant P values are in bold
Diagnostic test selectivity parameters were calculated to evaluate the ability of the biomarkers to distinguish ccRCC and NRT samples (Supplementary Table S3). The separate biomarkers had high specificity (≥ 88.9%) and positive predictive values (≥ 90.4%) for diagnosing ccRCC. The biomarkers were also analyzed in all possible combinations of 2–6 genes, and the panel of five biomarkers, particularly ZNF677, FBN2, PCDH8, TFAP2B & TAC1, was characterized with the best diagnostic potential and reached 82.1% of sensitivity and 95.6% of specificity (Supplementary Table S3).
Aberrant methylation of the genes was further analyzed according to the demographic and clinical-pathological patient's characteristics. Higher methylation frequencies of all investigated genes, except TFAP2B, were observed in males compared to females, and for ZNF677, FBN2, and PCDH8, this occurrence was statistically significant (P < 0.050; Fig. 2B). DNA methylation frequencies in all genes showed an increasing tendency according to the tumor stage, and in the case of ZNF677 and PCDH8, the observed association was statistically significant (P = 0.023 and P = 0.043, respectively; Fig. 2C). Furthermore, the methylated status of ZNF677, PCDH8, TAC1, and FLRT2 was statistically significantly associated with larger tumors (P < 0.050; Fig. 2D). Moreover, the methylation frequencies of all genes were also elevated in the higher-grade tumors, and for PCDH8, this tendency was significant (P = 0.004, Fig. 2E). In addition, frequent methylation of ZNF677 was related to the presence of tumor necrosis (P = 0.007; Fig. 2F).
DNA methylation and overall survival
To investigate the performance of the biomarkers in predicting the progression of ccRCC, overall survival analysis by the Kaplan–Meier curves test was initially performed. The analysis showed significantly lower overall survival rates in ccRCC cases with a methylated status of ZNF677 and FBN2 (P = 0.023 and P = 0.019, respectively; Fig. 3A, B). Although no associations were observed for other single biomarkers (P > 0.050; Supplementary Fig. S3), various combinations of the biomarkers were significantly associated with the poorer overall survival of ccRCC patients (P < 0.050; Fig. 3C–I).

The relationship between methylation status of selected genes and overall survival. Kaplan–Meier survival curves according to the gene methylation status of A ZNF677, B FBN2, and C–I various combinations of two-four biomarkers. For the gene combinations, only panels showing the significant association with the patient's overall survival are depicted. M/U methylated/unmethylated gene status, HR hazard ratio (when gene or panel is methylated). Significant P values are in bold
Univariate Cox proportional hazard analysis confirmed the association of the methylated status of single biomarkers ZNF677, FBN2, and panels of two-four biomarkers with overall survival (model's P < 0.050; Supplementary Table S4) that even outperform the prognostic value of some demographic and clinical-pathological variables. In the multivariate analysis, forward entering of covariates revealed that the methylation status of ZNF677 alone or in a panel with FLRT2 (with or without PCDH8) together with tumor size better predicted overall survival than the clinicopathologic variable alone (model's P < 0.050; Supplementary Table S4). Besides, ZNF677 & FLRT2 also had the same effect in combination with tumor stage and fat invasion (both model's P < 0.001).
Gene expression analysis in renal tissue samples
Expression levels of ZNF677, TFAP2B, TAC1, and FLRT2 were significantly lower in ccRCC tissues as compared to NRT samples (all P < 0.050). No statistically significant differences in mRNA level were observed for FBN2; meanwhile, significantly higher expression of PCDH8 was found in ccRCC as compared to NRT (Fig. 4A–F). Lower expression levels of ZNF677 in ccRCC tissues were significantly associated with methylated promoter status (P < 0.001), while no such correlation was observed for the other selected genes (Supplementary Fig. S4).

Transcriptional expression analysis of the selected genes. A–F Relative expression levels of ZNF677, FBN2, PCDH8, FLRT2, TFAP2B, and TAC1. G A relative expression of ZNF677 according to the clinical-pathological parameters of ccRCC patients. The box extends from the 25th to 75th percentiles; the line in the box is plotted at median; the plus sign depicts the mean; the whiskers represent the 10–90% range. FC fold change, ccRCC clear cell renal cell carcinoma, NRT non-cancerous renal tissues, WHO/ISUP World Health Organisation/International Society of Urological Pathology, pT pathological tumor stage. Significant P values are in bold
In comparison with clinical-pathological parameters, down-regulated ZNF677 was significantly correlated with the higher tumor stage, Fuhrman and WHO/ISUP grade, larger (> 45 mm) tumor size, presence of tumor vascular and fat invasions as well as necrosis (all P < 0.050; Fig. 4G). The lower expression level of FLRT2 and TAC1 was also related to the presence of tumor necrosis and larger (> 45 mm) tumor size, respectively (P = 0.006 and P = 0.035; Supplementary Fig. S4). On the contrary, a higher mRNA level of FBN2 was significantly associated with larger tumors, higher WHO/ISUP grade, and tumor necrosis (P = 0.003, P = 0.020, and P = 0.001 respectively; Supplementary Fig. S4).
In addition, Kaplan–Meier survival analysis and log-rank test results suggested that patients with low expression of ZNF677 had significantly shorter overall survival than patients with high expression of ZNF677 (P = 0.021; Supplementary Fig. S5).
DNA methylation analysis in the urine samples
In urine samples, quantitative DNA methylation analysis of ZNF677, FBN2, PCDH8, TFAP2B, TAC1, and FLRT2 was performed by the qMSP method. The average methylation levels of all genes were significantly higher in the urine of ccRCC cases as compared to the ASC group in both study and validation cohorts (P < 0.050; Fig. 5A, B). As expected, DNA methylation intensity was significantly lower in randomly selected 20 ccRCC patients' urine samples than in paired ccRCC tissues (all P < 0.050; Supplementary Fig. S6).

DNA methylation analysis in urine samples of study and validation cohorts. A, B The gens ZNF677, FBN2, PCDH8, TFAP2B, TAC1, and FLRT2 methylation levels in the urine of patients diagnosed with ccRCC and asymptomatic controls. C, D ROC curve analysis for single biomarkers and E, F the combination of biomarkers in discriminating patients with ccRCC and ASC. ccRCC clear cell renal cell carcinoma, ASC asymptomatic controls, ROC receiver operating characteristic, AUC area under the curve, DSe diagnostic sensitivity, DSp diagnostic specificity. Significant P values are in bold
The power of selected urine biomarkers methylation intensities to discriminate ccRCC cases from asymptomatic controls was analyzed by the ROC curve method. In the study cohort, the area under the curve (AUC) value for all genes was 0.60 or higher, with the highest value for PCDH8, which was 0.71 (all P < 0.050; Fig. 5C). In the validation cohort, AUC values ranged from 0.59 to 0.65 with the highest value identified for PCDH8 as well (all P < 0.050; Fig. 5D). Moreover, panels of two-six biomarkers had a better ability to discriminate between ccRCC and ASC (all P < 0.050; Fig. 5E–F; Supplementary Table S5). In the study cohort, the highest AUC value (0.78) and diagnostic power were observed for the panels, consisting of two—ZNF677 & PCDH8 and three—ZNF677, PCDH8 & FBN2 or ZNF677, PCDH8 & FLRT2 biomarkers (all P < 0.001; Fig. 5E). Although the diagnostic parameters of multi-marker panels were lower in the validation cohort (Fig. 5F; Supplementary Table S5), significantly smaller tumors in this patients group were detected.
Prognostic value of the investigated biomarkers in the urine samples
Aberrant methylation of all genes in the urine samples was further analyzed according to the clinical-pathological characteristics of the disease. A statistically significant higher methylation frequency of PCDH8, TFAP2B, and TAC1 was detected in the urine samples from patients diagnosed with locally advanced ccRCC, particularly characterized by the fat invasion (P = 0.018, 0.050 and 0.042 respectively; Supplementary Table S6) in the study cohort. Moreover, the methylated status of PCDH8 was more common in the case of a higher tumor stage (P = 0.002; Supplementary Table S6). In the validation cohort, PCDH8 sustained significant association with tumor stage and fat invasion (P = 0.042, and P = 0.042, respectively) and additionally correlated to the larger tumor size (P = 0.007; Supplementary Table S6).
Aberrant methylation of all genes was more frequent in the urine samples of the deceased cases, but only for PCDH8 was this tendency significant (P = 0.005; Supplementary Fig. S7). Kaplan–Meier curves and univariate Cox proportional hazard analysis revealed significant associations between the methylated status of PCDH8 and shorter patient's overall survival (P < 0.050; Fig. 6A, Table 2). The prognostic value of PCDH8 even increased in combination with ZNF677 (P < 0.050; Fig. 6B). Moreover, multivariate analysis revealed the methylation status of PCDH8 alone or panel of ZNF677 & PCDH8 is an independent predictor for ccRCC patients OS with HR, 5.7 (95% CI 1.16–28.12) and HR, 12.5 (95% CI 1.47–105.58) respectively, while adjusting according to the most important prognostic factors of ccRCC, including patients' age, gender, tumors stage, size, grade and necrosis (Table 2). Besides, the prognostic value of other combinations of biomarkers was also rather promising (Supplementary Fig. S8, Table S7).

The relationship between methylation status of investigated genes in urine samples and patient's overall survival (study cohort). A, B Kaplan–Meier survival curves according to the methylation status of PCDH8 alone or panel of ZNF677 & PCDH8. M/U methylated/unmethylated gene status, HR hazard ratio (when gene or gene panel is methylated). Significant P values are in bold
Discussion
Malignant kidney tumors account for 2% of the global cancer burden, and their incidence is on the rise (Turajlic et al. 2018a). Renal cell carcinoma (RCC), particularly ccRCC, is the most common type of kidney tumor and is characterized by the highest mortality rate of genitourinary cancers (Kabaria et al. 2016; Siegel et al. 2015). While currently, due to the widespread use of cross-sectional imaging, most RCC cases are detected incidentally at an early stage; more than a quarter of the cases will develop distant metastatic disease responsible for the major health burden of RCC (Capitanio et al. 2019). Another significant problem is the increase of overtreatment of patients with non-life-threatening benign renal masses, especially in the case of small renal tumors (Sohlberg et al. 2019). Thus, it is a vital need for new non-invasive biomarkers, which in combination with imagining data, could provide valuable information about the presence of the disease, its aggressiveness, and possible prognosis.
Using a microarray-based methylation detection platform, our study for the first time identified frequent (33–60%) methylation of ZNF677, TFAP2B, TAC1, and FLRT2 in ccRCC tissues with 89–100% of specificity that was also supported by significantly decreased expression of these genes in ccRCC tissues. Hypermethylation of PCDH8 and FBN2, known from the previous studies (Lin et al. 2014; Morris et al. 2011), was also identified in the current study and showed 100% specificity for ccRCC. The best performing multi-marker combination for ccRCC tissue samples with 82% of sensitivity and 96% of specificity was demonstrated for the five biomarker panel ZNF677, FBN2, PCDH8, TFAP2B & TAC1, that significantly exceeded the diagnostic power of the previously reported methylated gene panel (Costa et al. 2011). Although some studies provided gene panels with stronger diagnostic power (Battagli et al. 2003; Pires-Luís et al. 2017), considerably lower numbers and heterogeneous samples were included in these analyses.
The investigated genes were recognized as potential prognostic ccRCC biomarkers as well. The methylated status and decreased expression of ZNF677 were related to the vast majority of adverse parameters related to the disease prognosis (Warren and Harrison 2018), including tumor stage, grade, necrosis, intravascular and fat invasion, thus presented as an extremely valuable biomarker for ccRCC. While paralleling to the previous report (Lin et al. 2014), frequent methylation of PCDH8 was related to the higher tumor stage and WHO/ISUP grade. Moreover, methylation of ZNF677, PCDH8, FLRT2, and TAC1 was significantly associated with the larger tumor size, a relevant prognostic parameter of renal cancer (Williamson et al. 2019; Li et al. 2021). Survival analysis revealed methylation of ZNF677 and FBN2 as an independent predictor of ccRCC patient's OS in the univariate model; meanwhile, no association between the FBN2 methylation and patient survival was previously found (Ricketts et al. 2014). Besides, multivariate models revealed that ZNF677 alone or in combination with FLRT2 (with or without PCDH8) augmented the prognostic value of tumor size. Moreover, ZNF677 & FLRT2 enhanced the prognostic value of tumor stage and fat invasion—a strong prognostic factor in ccRCC (Bedke et al. 2009). Although van Vlodrop et al. (2017) previously proposed four-gene methylation as a valuable prognostic biomarker for ccRCC, their performance in urine samples has not been assessed yet.
Urine samples are the preferred source of biomarkers for non-invasive detection and/or prognosis of urological cancers. The current study revealed for the first time significantly higher methylation levels of ZNF677, FBN2, PCDH8, TFAP2B, TAC1, and FLRT2 in the urine samples of ccRCC patients in both study and validation cohorts with sensitivities higher than most of the previously reported urine-based epigenetic biomarkers (reviewed in Kubiliute and Jarmalaite 2021). The highest diagnostic power (AUC = 0.78) was observed for a panel of ZNF677 & PCDH8, either with or without FBN2 or FLRT2, with 69–78% of sensitivity, which considerably outperformed the value of the previously reported two-biomarkers panel (Costa et al. 2011). At the same time, the specificity of our biomarker panel was also high and ranged between 69 and 80%, depending on the biomarker combination. Though some previous studies established urine-based biomarker panels with better diagnostic characteristics (Battagli et al. 2003; Hoque et al. 2004), they used a considerably lower number of quite heterogeneous samples and lacked validation. Although the performance of our biomarkers in the validation cohort was of lower significance, it should be highlighted that this group consisted of patients with significantly smaller tumors of potentially lower aggressiveness. On the other hand, the combination of ZNF677, PCDH8 & FLRT2 still were able to detect six out of ten ccRCC in urine samples and correctly classified eight out of each ten suspects. Thus, this inexpensive and simple test method has the potential to detect ccRCC even in the situation of small renal tumors, which can be missed by routine diagnostic methods (Rossi et al. 2018).
The rising amount of incidentally detected ccRCC introduced an important clinical challenge owing to the risk of patients overdiagnosis and overtreatment (Sohlberg et al. 2019). Thus, the ability to predict the possible course of the disease before the radical treatment might reduce this risk. Our study demonstrated that methylation of PCDH8 in urine samples significantly relates to tumor stage and fat invasion in both study and validation cohorts, indicating its prognostic potential. This finding was confirmed in multivariate Cox analysis, where PCDH8, together with the patient's age, gender, and tumor necrosis, retained an independent prognostic value for OS (HR: 5.7), which was considerably enhanced by the combination with ZNF677 (HR: 12.5). To the best of our knowledge, there is only one report presenting urine-based prognostic DNA methylation biomarker of ccRCC (Outeiro-Pinho et al. 2020), and the current study nicely supplements this field of investigations. To sum up, the present study suggests the possibility of predicting ccRCC progression based on only one-two urinary biomarkers that may reduce the risk of patients mistreatment.
The biological function of selected genes may improve the understanding of ccRCC development and progression. ZNF677 (Zinc finger protein 677) encodes the transcription factor and possibly functions as a tumor suppressor, whereas its overexpression inhibits cell proliferation, migration, invasion, induction of cell cycle arrest, and apoptosis (Heller et al. 2015; Li et al. 2018). Another set of three genes, specifically PCDH8, FLRT2, and FBN2, encodes the proteins related to cell migration. Protocadherin-8 PCDH8 (Protocadherin-8) belongs to the protocadherin family and plays multiple roles in cell adhesion, proliferation, differentiation, and migration by both inhibiting (Yu et al. 2008) or promoting (Lin et al. 2018) such processes depending on specific tissue. FLRT2 (Fibronectin leucine-rich transmembrane protein 2) encodes glycosylated membrane protein interacting with fibronectin in either a repulsive or adhesive manner (Flintoff et al. 2014), and its down-regulation due to promoter methylation increases cell proliferation and migration (Bae et al. 2017). FBN2 (fibrillin-2) is an extracellular matrix glycoprotein that regulates TGF-β bioavailability, which can both suppress and promote cancer development (Neuzillet et al. 2015; van Loon et al. 2020). In the case of renal cancer, loss of fibrillin-2 may contribute to a malignant phenotype by giving angiogenic and metastatic advantages (Morris et al. 2011). The most down-regulated gene, TFAP2B (transcription factor AP-2 beta), stimulates cell proliferation, suppresses terminal differentiation, and is indispensable for kidney development (Moser et al. 1997; Eckert et al. 2005). It is worth mentioning that in contrast to other genes, the analyzed TFAP2B sequence encompasses the enhancer region instead of the promoter, of which methylation in the case of RCC has important clinical significance as well (Hu et al. 2014). However, neither expression nor methylation of TFAP2B had any associations with clinical-pathological parameters. Another gene with the markedly reduced expression – TAC1 encodes four products of the neuropeptides family, influencing cell secretion, motility, and inflammatory reactions as well as inhibiting cell proliferation (Rameshwar and Gascon 1996); therefore, it is considered a tumor-suppressor gene that was the most frequently methylated in ccRCC in the current study.
While the main tendency of ZNF677, PCDH8, and FLRT2 as the most promising ccRCC biomarkers in both tissue and urine samples has been confirmed, still existing inconsistencies among sample types are quite confusing. In the present study, urine sediments were used for biomarker analysis, and contaminating urinary bladder cells may have had an impact on the results (Lu and Li 2017). On the other hand, as renal tumors are highly heterogeneous, regional tissue samples of the tumor may not reflect that heterogeneity (Turajlic et al. 2018b), while the analysis of body fluids may reflect a wider spectrum of (epi)genetic alterations. Besides, DNA methylation may significantly vary between the regions in the same CpG island (van Vlodrop et al. 2010). As the PCR primers for tissue and urine analysis are not fully overlapped, this can cause differences in methylation results between these two sources of samples as well. Finally, the differences in the sensitivity and specificity of MSP and QMSP methods (Daniūnaitė et al. 2019) might also cause these variations. However, considering the ability of investigated biomarkers to discriminate patients with ccRCC from asymptomatic controls in both patient's cohorts, we feel confident about the ccRCC-specificity of these urinary biomarkers. Nevertheless, further biomarkers cross-validations, at least in the case of other urological cancers, are highly desirable to prove their applicability for non-invasive precise ccRCC detection and prognosis.
Along with the significant impact of the study in the search for novel ccRCC biomarkers, the investigation has important shortcomings as well. First of all, further biomarkers validation in the independent larger cohort with thorough follow-up data is mandatory to prove their clinical significance, especially considering the relatively small number of samples tested and even smaller in the time-to-event analysis in the current study. In addition, death as an endpoint was used for the survival analysis that has a lower prognostic power as compared to disease-free survival (Sauerbrei et al. 2018), which was not available in the present study. Nonetheless, the novelty of using DNA methylation biomarkers with presumable diagnostic and prognostic value, susceptible for non-invasive urine-based detection, doubtless will stimulate the design of validation studies in larger and independent series.
Conclusions
To sum up, our study demonstrated the ccRCC-specific aberrant methylation of ZNF677, PCDH8, FLRT2, FBN2, TAC1, and TFAP2B amenable for urine-based detection. DNA methylation status of particular genes as an independent variable or in combination with other variables supposedly can predict the overall survival of ccRCC patients. We suggest this novel DNA methylation biomarkers have a significant potential to serve for non-invasive urine-based ccRCC diagnostics and follow-up.
Materials and methods
Tissue and urine samples
Retrospectively recruited patients as the main study cohort were available for this research. Human kidney tissue samples from 123 patients primarily diagnosed with ccRCC who underwent partial or radical nephrectomy without any neoadjuvant therapy at the Urology Centre of Vilnius University Hospital "Santaros Klinikos" (Lithuania) were collected between 2013 and 2016. Non-cancerous renal tissue (NRT) samples were available from 51 ccRCC patients as a control group and collected as previously described (Maleckaite et al. 2019). A small amount of samples were immediately snap-frozen and stored at – 80 °C. From that sample collection, 11 pairs of ccRCC and morphologically normal tissue were used for microarray analysis, while 123 ccRCC and 45 NRT were for validation (Supplementary Table S8). All tissues were sampled and evaluated by an expert pathologist, and the presence of cancerous cells within the particular sample was determined by a histopathologist. Positive surgical margins were obtained in 11 patients, while the remaining cases were negative for surgical margin status. Tumors were categorized based on pathological stage and histological subtype, as described previously (Sobin et al. 2009; Lopez-Beltran et al. 2006), while nuclear differentiation was graded according to the Fuhrman (Fuhrman et al. 1982) and World Health Organization/International Society of Urological Pathology (Delahunt et al. 2013) grading systems. To reduce confounding factors affecting the analyses, patients had to fulfill the following criteria: (1) patients older than 18 years old; (2) clinically and pathologically approved T1–T4 stage without previous treatment; and (3) histologic pure clear cell RCC without sarcomatoid differentiation.
According to the pathological stage (pT) category, approximately half of the tumors, 46% (57/123), were an organ‐confined pT1-2 stage, whereas 50% (61/123) of tumors had signs of invasion, with 57 tumors extending into the renal vein or perirenal and/or renal sinus fat (pT3a) and 4 grossly extending into the vena cava below the diaphragm (pT3b). In addition, 4% (5/123) of the tumors invade beyond Gerota fascia (pT4). For none of the cases were distant metastases diagnosed. The overall survival data were available for 107 of 123 patients with a median follow-up time of 59 months (range 1–79 months). All collected demographic and clinical-pathological data of the investigated cohort are provided in Supplementary Table S8.
Preoperatively voided urine samples were available for all ccRCC patients (study cohort; N = 123). A prospectively collected validation cohort composed of 92 ccRCC patients diagnosed with ccRCC from 2019 July to 2021 April at the Oncourology department of the National Cancer Institute (Vilnius, Lithuania) was included in the analysis as well. Collected demographic and clinical-pathological data of that validation cohort are provided in Supplementary Table S8. An additional voided urine sample from 92 age- and sex-matched asymptomatic volunteers was used as asymptomatic control (ASC) group. All urine samples were processed according to the standard protocol as previously reported (Daniunaite et al. 2014).
The study was conducted following the Declaration of Helsinki. Approval from the Lithuanian Bioethics Committee was obtained before initiating the study and later for the microarray analysis, and all patients gave informed consent for participation.
Genome-wide DNA methylation profiling
For the initial screening, the genome-wide DNA methylation profiling (GEO accession identifier GSE166734) of 11 paired ccRCC and NRT samples with thorough clinical-pathological data was performed to identify potential ccRCC biomarkers. The samples were processed using the two-color Human DNA Methylation 1 × 244 K Microarrays according to the manufacturer's protocol (Agilent Technologies, Santa Clara, CA, USA), as described previously (Daniunaite et al. 2015). Saturated, non-uniform, and outlier probe signals were treated as compromised, and probes undetected in at least one sample were removed from the analysis. The signals were filtered on expression (20.0–100.0th) percentile in the raw data, and all samples had values within the range. Probe annotations were uploaded from the SureDesign platform (https://earray.chem.agilent.com/suredesign). For group comparison, fold change (FC) values were estimated, and a paired (if applicable) or unpaired t-test was applied. The resulting P values were corrected for multiple testing using false discovery rate (FDR). Analysis was performed with GeneSpring GX v14.9 software (Agilent Technologies).
DNA purification and bisulfite conversion
Renal tissue samples were homogenized using liquid nitrogen and cryoPREP™ CP02 Impactor with tissue TUBE TT1 (Covaris, Woburn, MA, USA). Homogenized tissue powder and/or urine sediments were treated for up to 18 h at 55 °C with 10–25 μl of proteinase K (Thermo Scientific™, Thermo Fisher Scientific, Wilmington, DE, USA) and 500 μl of lysis buffer, consisting of 50 mM Tris–HCl pH 8.5, 1 mM EDTA, 0.5% Tween-20 (all from Carl Roth, Karlsruhe, Germany) for tissue samples, and 10 mM Tris–HCl pH 8.0, 1% SDS, 75 mM NaCl (all from Carl Roth) for urine samples. DNA was extracted following the standard phenol–chloroform purification and ethanol precipitation protocol.
For DNA methylation analysis using qualitative or quantitative methylation-specific PCR (MSP or qMSP, respectively), up to 400 ng of purified DNA were modified with bisulfite, using EZ DNA Methylation™ Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer's protocol, except that the initial incubation of samples was performed at 42 °C for 15 min as better results compared to 37 °C were observed.
Qualitative methylation-specific PCR
The MSP primers for unmethylated and methylated DNA for genes ZNF677, FBN2, PCDH8, TFAP2B, TAC1, and FLRT2, were designed to overlap with the location of the microarray probes (if available) or at least next to that probe using Methyl Primer Express® Software v1.0 (Applied Biosystems™, Thermo Fisher Scientific, Carlsbad, CA, USA) and ordered from Metabion (Martinsried, Germany) (Supplementary Table S9). The reaction mix of MSP (25 μl in total) consisted of 1 × PCR Gold Buffer, 2.5 mM MgCl2, 0.4 mM of each dNTP, 1.25 U AmpliTaq Gold® 360 DNA Polymerase (Applied Biosystems™, Thermo Scientific™), 1 μL of 360 GC Enhancer, 1 μM of each primer, and ~ 10 ng of the bisulfite-treated DNA. Thermocycling conditions consisted of 10 min at 95 °C, 35–38 cycles of 45 s at 95 °C, primer annealing for 45 s at 58–65 °C (Supplementary Table S9) and elongation for 45 s at 72 °C, followed by 5–10 min at 72 °C. For each primer pair, methylated control (in vitro fully methylated human leukocyte DNA, MC), unmethylated control (human leukocyte DNA, UC), and no-template control (NTC) was included in all MSP assays. Amplification products were analyzed in 3% agarose gels with 1X TAE buffer and visualized under UV light after ethidium bromide staining (Carl Roth GmbH, Co., KG). An example of MSP analysis results is provided in Supplementary Fig. S9. The individual biomarker was considered as methylated if the amplification product in the sample with primers specific for methylated DNA was detected. Meanwhile, the panel of biomarkers was considered methylated if at least one gene in the particular panel was methylated.
Quantitative methylation-specific PCR
The qMSP primers and probes specific for methylated DNA for genes ZNF677, FBN2, PCDH8, TRAP2B, TAC1, and FLRT2, were designed using Methyl Primer Express® Software v1.0 (Applied Biosystems™, Thermo Fisher Scientific, Carlsbad, CA, USA) and ordered from Metabion (Martinsried, Germany) (Supplementary Table S10). All primers and/or probes overlap at least a fragment of the MSP primers' sequence (Supplementary Table S11). The primers for ACTB, which are not overlapping with CpG dinucleotides, were selected from the previous study (Lehmann et al. 2002) and were used in each run for normalization of the DNA input. The qMSP was performed in triplicates for each set of primers in separate wells. The reaction mix (20 μl in total) consisted of 1 × TaqMan® Universal Master Mix II, no UNG (Applied Biosystems™), 300 nM of each primer, 50 nM of the probe, and ~ 10 ng of 20 bisulfite-converted DNA. All assays were carried out under the following conditions: 95 °C for 10 min followed by 50 cycles of 95 °C for 15 s and 60 °C for 1 min, using the ViiA7 qPCR System (Applied Biosystems™). Only runs wherein MCs provided a positive signal, and the NTC gave no amplification product were considered as valid. The background-based threshold algorithm was applied for the estimation of the cycle of quantification (Cq) value. The methylation level of a particular gene was estimated based on the ΔΔCq algorithm and expressed as a percentage of the MC (Bakavicius et al. 2019).
RNA extraction and cDNA synthesis
Total RNA was extracted from the analyzed tissue samples by MirVana™ miRNA Isolation Kit (Ambion®, Thermo Fisher Scientific, Foster City, CA, USA) following the manufacturer's protocol and used for targeted gene expression analysis by means of quantitative PCR (qPCR). Briefly, homogenized tissue powder was treated with 500 μL Lysis/Binding Buffer for 10 min on ice and 50 μL of miRNA Homogenate Additive for an additional 10 min. The total RNA was extracted with 500 μL of acid-phenol: chloroform and purified with supplied Filter Cartridges. To eluate purified RNA, 100 µL of 95 °C Elution Solution was used. Samples were used immediately or stored at − 40 °C until further use.
Transcriptional gene expression analysis
For qPCR, up to 100 ng of the RNA were reverse transcribed (RT) using Maxima First Strand cDNA Synthesis Kit with ds DNase according to the recommended protocol (Thermo Fisher Scientific). Expression of the genes ZNF677, FBN2, PCDH8, TFAP2B, TAC1, FLRT2 and endogenous control HPRT1 was evaluated using TaqMan® Gene Expression Assays (Hs00737026_m1, Hs00266592_m1, Hs00159910_m1, Hs01560931_m1, Hs00243225_m1, Hs00544171_s1 and Hs02800695_m1, respectively; Applied Biosystems™) in duplicates per gene. The reaction mix (20 μL in total) consisted of 1 × TaqMan® Universal Master Mix II, no UNG (Applied 20 Biosystems™), 0.6 μL of TaqMan® assay, and 2 μL of RT reaction product. Amplification was performed using ViiA7 qPCR System (Applied Biosystems™) under the following thermal cycling conditions: 40 cycles of 95 °C for 15 s and 60 °C for 1 min. NTCs (No-template control) were included in each qPCR run. Relative gene expression values (normalized to HPRT1) in a linear scale were used for the analysis of the results, performed with GenEx v6.0.1 software (MultiD Analyses AB, Göteburg, Sweden). For survival analysis, gene expression levels were categorized as "high" or "low" if the log-transformed values were above or below the mean value of all samples for a particular gene, respectively.
Statistical analysis
Design and analysis of this study were performed according to reporting recommendations for tumor marker prognostic studies (ReMARK) and diagnostic accuracy studies (STARD) guidelines (Sauerbrei et al. 2018; Bossuyt et al. 2015). Statistical analyses were performed using STATISTICA™ v8.0 (StatSoft, Tulsa, OK, USA) and MedCalc® v14.0 software (MedCalc Software, Ostend, Belgium). All quantitative variables were tested for normality (Shapiro–Wilk, Kolmogorov–Smirnov, and Lilliefors tests), and because of abnormal distribution, the nonparametric Wilcoxon–Mann–Whitney test was applied to compare variables between two groups. Meanwhile, the 2-sided Fisher's exact test was applied for the comparison of categorical variables.
For the qMSP data analysis in the urine samples, the ability of biomarkers to distinguish ccRCC and ASC was evaluated by performing receiver operating characteristic (ROC) curve analysis and estimating the area under the curve (AUC) values. The diagnostic test's performance parameters—sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV)—were obtained from the ROC curve analysis and based on the Youden index for the selection of optimal cut-off value. This cut-off value ensured perfect categorization of the samples as positive and negative for the methylation test. For various combinations of biomarkers, logistic regression analysis was applied.
For time-event analysis, Kaplan–Meier curves were used to calculate survival estimates. The univariate and multivariate Cox proportional hazards modeling was performed to estimate the hazard ratio (HR) of death with 95% confidence intervals (CI).
Availability of data and materials
All data supporting the results reported in the article is available from the corresponding author upon a reasonable request.
Abbreviations
- AUC:
-
Area under the curve
- ASC:
-
Asymptomatic control
- ccRCC:
-
Clear cell renal cell carcinoma
- CI:
-
Confidence interval
- MC:
-
In vitro methylated control
- MSP:
-
Methylation-specific polymerase chain reaction
- qMSP:
-
Quantitative methylation-specific polymerase chain reaction
- NRT:
-
Non-cancerous renal tissue
- NTC:
-
No-template control
- HR:
-
Hazard ratio
- UC:
-
Unmethylated control
- M:
-
Methylated
- U:
-
Unmethylated
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the curve
- DSe:
-
Diagnostic sensitivity
- DSp:
-
Diagnostic specificity
- WHO/ISUP:
-
World health organization/International Society of Urological Pathology
References
Bae H, Kim B, Lee H, Lee S, Kang HS, Kim SJ (2017) Epigenetically regulated Fibronectin leucine rich transmembrane protein 2 (FLRT2) shows tumor suppressor activity in breast cancer cells. Sci Rep 7(1):272
Bakavicius A, Daniunaite K, Zukauskaite K, Barisiene M, Jarmalaite S, Jankevicius F (2019) Urinary DNA methylation biomarkers for prediction of prostate cancer upgrading and upstaging. Clin Epigenet 11(1):115
Battagli C, Uzzo RG, Dulaimi E, Ibanez de Caceres I, Krassenstein R, Al-Saleem T et al (2003) Promoter hypermethylation of tumor suppressor genes in urine from kidney cancer patients. Cancer Res 63:8695–8699
Bedke J, Buse S, Pritsch M, Macher-Goeppinger S, Schirmacher P, Haferkamp A et al (2009) Perinephric and renal sinus fat infiltration in pT3a renal cell carcinoma: possible prognostic differences. BJU Int 103(10):1349–1354
Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig L et al (2015) STARD Group. STARD 2015: an updated list of essential items for reporting diagnostic accuracy studies. BMJ 351:h5527
Capitanio U, Bensalah K, Bex A et al (2019) Epidemiology of renal cell carcinoma. Eur Urol 75(1):74–84
Chow WH, Dong LM, Devesa SS (2010) Epidemiology and risk factors for kidney cancer. Nat Rev Urol 7:245–257
Costa VL, Henrique R, Danielsen SA, Knaes M, Patrício P, Morais A et al (2011) TCF21 and PCDH17 methylation: an innovative panel of biomarkers for a simultaneous detection of urological cancers. Epigenetics 6:1120–1130
Daniunaite K, Jarmalaite S, Kalinauskaite N, Petroska D, Laurinavicius A, Lazutka JR et al (2014) Prognostic value of RASSF1 promoter methylation in prostate cancer. J Urol 192(6):1849–1855
Daniunaite K, Serenaite I, Misgirdaite R, Gordevicius J, Unguryte A, Fleury-Cappellesso S et al (2015) Epigenetic regulation of human adipose-derived stem cells differentiation. Mol Cell Biochem 410:111–120
Daniūnaitė K, Jarmalaitė S, Kriukienė E (2019) Epigenomic technologies for deciphering circulating tumor DNA. Curr Opin Biotechnol 55:23–29
Delahunt B, Cheville JC, Martignoni G, Humphrey PA, Magi-Galluzzi C, McKenney J et al (2013) The International Society of Urological Pathology (ISUP) grading system for renal cell carcinoma and other prognostic parameters. Am J Surg Pathol 37:1490
Eckert D, Buhl S, Weber S, Jäger R, Schorle H (2005) The AP-2 family of transcription factors. Genome Biol 6(13):246
Flintoff KA, Arudchelvan Y, Gong SG (2014) FLRT2 interacts with fibronectin in the ATDC5 chondroprogenitor cells. J Cell Physiol 229(10):1538–1547
Fuhrman SA, Lasky LC, Limas C (1982) Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol 6:655–663
Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I et al (2014) Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 46(3):225–233
Heller G, Altenberger C, Schmid B, Marhold M, Tomasich E, Ziegler B et al (2015) DNA methylation transcriptionally regulates the putative tumor cell growth suppressor ZNF677 in non-small cell lung cancers. Oncotarget 6:394–408
Hoque MO, Begum S, Topaloglu O, Jeronimo C, Mambo E, Westra WH et al (2004) Quantitative detection of promoter hypermethylation of multiple genes in the tumor, urine, and serum DNA of patients with renal cancer. Cancer Res 64(15):5511–5517
Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M et al (2017a) Renal cell carcinoma. Nat Rev Dis Primers 3:17009
Hsieh JJ, Manley BJ, Khan N, Gao JJ, Carlo MI, Cheng EH (2017b) Overcome tumor heterogeneity-imposed therapeutic barriers through convergent genomic biomarker discovery: a braided cancer river model of kidney cancer. Semin Cell Dev Biol 64:98–106
Hu CY, Mohtat D, Yu Y, Ko YA, Shenoy N, Bhattacharya S et al (2014) Kidney cancer is characterized by aberrant methylation of tissue-specific enhancers that are prognostic for overall survival. Clin Cancer Res 20(16):4349–4360
Jayson M, Sanders H (1998) Increased incidence of serendipitously discovered renal cell carcinoma. Urology 51(2):203–205
Kabaria R, Klaassen Z, Terris MK (2016) Renal cell carcinoma: links and risks. Int J Nephrol Renovasc Dis 9:45–52
Kubiliute R, Jarmalaite S (2021) Epigenetic biomarkers of renal cell carcinoma for liquid biopsy tests. Int J Mol Sci 22(16):8846
Larsen LK, Lind GE, Guldberg P, Dahl Ch (2019) DNA-methylation-based detection of urological cancer in urine: overview of biomarkers and considerations on biomarker design, source of DNA, and detection technologies. Int J Mol Sci 20(11):2657
Lasseigne BN, Brooks JD (2018) The role of DNA methylation in renal cell carcinoma. Mol Diagn Ther 22(4):431–442
Lehmann U, Langer F, Feist H, Glöckner S, Hasemeier B, Kreipe H (2002) Quantitative assessment of promoter hypermethylation during breast cancer development. Am J Pathol 160(2):605–612
Li Y, Yang Q, Guan H, Shi B, Ji M, Hou P (2018) ZNF677 suppresses Akt phosphorylation and tumorigenesis in thyroid cancer. Cancer Res 78(18):5216–5228
Li L, Shi L, Zhang J, Fan Y, Li Q (2021) The critical impact of tumor size in predicting cancer special survival for T3aM0M0 renal cell carcinoma: A proposal of an alternative T3aN0M0 stage. Cancer Med 10(2):605–614
Lin YL, Wang YL, Fu XL, Ma JG (2014) Aberrant methylation of PCDH8 is a potential prognostic biomarker for patients with clear cell renal cell carcinoma. Med Sci Monit 20:2380–2385
Lin Y, Ge X, Zhang X, Wu Z, Liu K, Lin F et al (2018) Protocadherin-8 promotes invasion and metastasis via laminin subunit γ2 in gastric cancer. Cancer Sci 109(3):732–740
Lopez-Beltran A, Scarpelli M, Montironi R, Kirkali Z (2006) 2004 WHO classification of the renal tumors of the adults. Eur Urol 49:798–805
Lu T, Li J (2017) Clinical applications of urinary cell-free DNA in cancer: current insights and promising future. Am J Cancer Res 7(11):2318–2332
Maleckaite R, Zalimas A, Bakavicius A, Jankevicius F, Jarmalaite S, Daniunaite K (2019) DNA methylation of metallothionein genes is associated with the clinical features of renal cell carcinoma. Oncol Rep 41(6):3535–3544
McMahon KW, Karunasena E, Ahuja N (2017) The roles of DNA methylation in the stages of cancer. Cancer J 23(5):257–261
Morris MR, Ricketts CJ, Gentle D, McRonald F, Carli N, Khalili H et al (2011) Genome-wide methylation analysis identifies epigenetically inactivated candidate tumor suppressor genes in renal cell carcinoma. Oncogene 30(12):1390–1401
Moser M, Pscherer A, Roth C, Becker J, Mücher G, Zerres K et al (1997) Enhanced apoptotic cell death of renal epithelial cells in mice lacking transcription factor AP-2beta. Genes Dev 11(15):1938–1948
Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E et al (2015) Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther 147:22–31
Outeiro-Pinho G, Barros-Silva D, Aznar E, Sousa AI, Vieira-Coimbra M, Oliveira J et al (2020) MicroRNA-30a-5pme: a novel diagnostic and prognostic biomarker for clear cell renal cell carcinoma in tissue and urine samples. J Exp Clin Cancer Res 39(1):98
Pires-Luís AS, Costa-Pinheiro P, Ferreira MJ, Antunes L, Lobo F, Oliveira J et al (2017) Identification of clear cell renal cell carcinoma and oncocytoma using a three-gene promoter methylation panel. J Transl Med 15:149
Rameshwar P, Gascón P (1996) Induction of negative hematopoietic regulators by neurokinin-A in bone marrow stroma. Blood 88(1):98–106
Ricketts CJ, Hill VK, Linehan WM (2014) Tumor-specific hypermethylation of epigenetic biomarkers, including SFRP1, predicts for poorer survival in patients from the TCGA Kidney Renal Clear Cell Carcinoma (KIRC) project. PLoS ONE 9(1):e85621
Rossi SH, Prezzi D, Kelly-Morland C, Goh V (2018) Imaging for the diagnosis and response assessment of renal tumours. World J Urol 36(12):1927–1942
Sauerbrei W, Taube SE, McShane LM, Cavenagh MM, Altman DG (2018) Reporting recommendations for tumor marker prognostic studies (REMARK): an abridged explanation and elaboration. J Natl Cancer Inst 110(8):803–811
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65:5–29
Sobin LH, Gospodarowicz MK, Wittekind C (eds) (2009) TNM classification of malignant tumours, 7th Edition. UICC International Union Against Cancer. 7:310
Sohlberg EM, Metzner TJ, Leppert JT (2019) The harms of overdiagnosis and overtreatment in patients with small renal masses: A mini-review. Eur Urol Focus 5(6):943–945
The Cancer Genome Atlas Research Network (2013) Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499(7456):43–49
Turajlic S, Xu H, Litchfield K, Rowan A, Horswell S, Chambers T et al (2018a) Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell 173:595–610
Turajlic S, Swanton Ch, Boshoff Ch (2018b) Kidney cancer: The next decade. J Exp Med 215(10):2477–2479
van Loon K, Yemelyanenko-Lyalenko J, Margadant C, Griffioen AW, Huijbers EJM (2020) Role of fibrillin-2 in the control of TGF-β activation in tumor angiogenesis and connective tissue disorders. Biochim Biophys Acta Rev Cancer 1873(2):188354
van Vlodrop IJ, Baldewijns MM, Smits KM, Schouten LJ, van Neste L, van Criekinge W et al (2010) Prognostic significance of Gremlin1 (GREM1) promoter CpG island hypermethylation in clear cell renal cell carcinoma. Am J Pathol 176(2):575–584
van Vlodrop IJH, Joosten SC, De Meyer T, Smits KM, Van Neste L, Melotte V et al (2017) A four-gene promoter methylation marker panel consisting of GREM1, NEURL, LAD1, and NEFH predicts survival of clear cell renal cell cancer patients. Clin Cancer Res 23(8):2006–2018
Warren AY, Harrison D (2018) WHO/ISUP classification, grading and pathological staging of renal cell carcinoma: standards and controversies. World J Urol 36(12):1913–1926
Williamson SR, Taneja K, Cheng L (2019) Renal cell carcinoma staging: pitfalls, challenges, and updates. Histopathology 74(1):18–30
Yu JS, Koujak S, Nagase S, Li CM, Su T, Wang X et al (2008) PCDH8, the human homolog of PAPC, is a candidate tumor suppressor of breast cancer. Oncogene 27:4657–4665
Acknowledgements
The authors would like to thank Aušra Šumskaitė, Rūta Matuliavičiūtė, and Eugenijus Ganža for their assistance in DNA samples preparation and MSP analysis.
Funding
This work was funded by the 2014–2020 European Union Structural Funds according to the activity "Intelligence. Joint science-business projects" grant No. J05-LVPA-K-04–0029. SJ and KZ were supported by the European Social Fund according to the activity "Development of students' ability to carry out R&D activities" under Measure No.09.03.3-LMT-K-712 "Development of Scientific Competences of Scientists, other Researchers and Students through Practical Research Activities" (grant No. 09.03.3-LMT-K-712–15-0214 to SJ). The study was also partially supported by the Research Council of Lithuania (RCL) grant No. S-MIP-17/54.
Author information
Authors and Affiliations
Contributions
RK: performed the DNA methylation and gene expression analysis in the tissue samples and urine samples of the validation cohort, analyzed the experimental and clinical data and drafted the manuscript; KZ: performed the DNA methylation analysis in urine samples of the study cohort, contributed to gene expression analysis; AZ, AU, RS and AB: collected the clinical data and revised the analysis; RS and AZ: critically revised the manuscript; AU and FJ: coordinated the patient selection, supervised the clinical data analysis and was involved in the conception of the study; SJ: designed the research, supervised the analysis of the clinical and experimental data, and revised the manuscript critically for important intellectual content. All authors read and approved the manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work is appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
RK and SJ are inventors of the patent application No. PCT/IB2021/052532. The remaining authors declare that they have no conflict of interest.
Ethics approval and consent to participate
The study was approved by the Lithuanian Bioethics Committee (Nr. 158200˗18/12˗1077˗585 for Study cohort and Nr. 158200-18/12-1077-585 for Validation cohort), and written informed consent was obtained from all patients.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kubiliūtė, R., Žukauskaitė, K., Žalimas, A. et al. Clinical significance of novel DNA methylation biomarkers for renal clear cell carcinoma. J Cancer Res Clin Oncol 148, 361–375 (2022). https://doi.org/10.1007/s00432-021-03837-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-021-03837-7