
Abstract
Introduction
Next-generation sequencing (NGS)-based assays to understand various mutations and co-occurrence of genomic alterations in non-small cell lung cancer (NSCLC) have enabled understanding of treatment impact on clinical outcomes.
Methods
This retrospective study was conducted in 1353 formalin-fixed paraffin-embedded (FFPE) tissues from surgically resected, pre-TKI-treated NSCLC patients with identified gene alterations. Genomic DNA and RNA extraction was followed by NGS library preparation and sequencing using the Ion Ampliseq Colon and Lung Cancer Gene Panel V2 and the AmpliSeq RNA Lung Cancer Research Fusion Panel.
Results
A total of 2328 alterations in 25 genes were detected from the 1293 patients. DNA mutations and RNA fusions co-occurred in 27 patients with TP53 being the most common co-occurring DNA mutation (43.8%) with concurrent ALK fusions. Analysis of the 975 patients with EGFR mutations revealed that the incidence of dual EGFR L858R/T790M mutations was higher compared to EGFR 19del/T790M, and the mean allele fraction (MAF) of T790M was lower compared to 19del in dual EGFR 19del/T790M patients.
Conclusion
NSCLC patients represented genetically heterogeneous subgroup with a high frequency of co-occurring mutations in cancer-associated pathways. This diverse mutational profile may have key clinical and research implications for understanding the variability of treatment outcome in pre-TKI-treated NSCLC population. The differences in the MAF of EGFR T790M may determine different responses to TKI therapy in patients harboring dual mutations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Treatment of advanced non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) mutations includes EGFR-tyrosine kinase inhibitors (TKIs) and has resulted in higher survival rates when compared to the standard chemotherapy regimen (Yang et al. 2015). Despite remarkable efficacy against T790M-related resistance mutation-positive NSCLC (Mok et al. 2017), genetically driven resistance to TKIs has emerged, thereby limiting their prolonged effectiveness (Suda and Mitsudomi 2014). It has been speculated that co-occurring genetic alterations might play an underlying role in resistance mechanisms, thereby potentially explaining the diversity of individual patient outcomes (Hong et al. 2018; Schildgen and Schildgen 2018). Although previous study has hinted at the potential role of TP53 mutations in poor therapeutic response, specific co-occurring genetic alterations affecting the clinical outcomes of EGFR TKI in addition to primary driver mutations remain poorly studied (Yu et al. 2018). Some of the recent studies have suggested the presence of T790M at a low frequency within the tumor cells before the initiation of TKI therapy that eventually becomes dominant clone following TKI therapy (Godin-Heymann et al. 2007; Inukai et al. 2006). However, due to the limited sensitivity of the different detection methods, the reported rates of de novo resistance varied. The study by Su et al. has reported a shorter progression-free survival in NSCLC patients harboring de novo T790M mutation treated with EGFR TKI therapy (Su et al. 2012).
Compared to ARMS, FISH and IHC, NGS has enabled simultaneous screening of a variety of genetic alterations with relatively low input of nucleic acids (Butler et al. 2016; Jurgens et al. 2016; Zheng et al. 2016). It provides a cost- and tissue-efficient alternative to current single-gene assessment methods. NGS-based hybrid capture assays not only allows identification of hotspot mutations but also the assessment of unknown alterations, from a single FFPE specimen or serum sample (Drilon et al. 2015). Amplicon-based NGS has clinical significance at deeper sequencing depths to facilitate increased detection sensitivity for mutations in heterogeneous or low purity tumor samples (Grasso et al. 2015; Youssef et al. 2018). These options have enabled the study of a complex genomic condition of NSCLC easier and accessible.
As per the recent studies, co-occurring genetic alterations are frequently observed and cooperate with the primary genetic driver as co-drivers to promote tumor progression and limit targeted therapy response (Blakely et al. 2017; Hong et al. 2018). Further, there are evidences of de novo mutation associated with worse clinical benefits in NSCLC patients treated with EGFR TKI therapy (Liu et al. 2017), thereby necessitating the detection of this subset in NSCLC patients. In this study, we performed concurrent DNA and RNA NGS assays in 1336 pre-TKI-treated NSCLC tumor samples harboring identified gene alterations using the Ion AmpliSeq Colon and Lung Cancer Research Panel V2 and the Ion AmpliSeq RNA Fusion Lung Cancer Research Panel.
Materials and methods
Patients and samples
This retrospective study assessed FFPE tissues from surgically resected NSCLC with identified gene alterations using PCR, IHC, and FISH. A total of 1353 NSCLC patient samples from five different institutions (Peking University Cancer Hospital & Institute, West China Hospital, Chinese PLA General Hospital, The Affiliated Cancer Hospital of Zhengzhou University, Beijing Chest Hospital, from 2015 to 2017) were used to perform NGS. Tumor tissue samples were reviewed by a pathologist to make certain that cancer cells accounted for ≥ 30%. This study was approved by the ethics committee of Peking University Cancer Hospital. All patients signed informed consents for their tumor samples to be used in future studies. All clinical data and samples were received anonymously.
DNA and RNA extraction from clinical samples
Genomic DNA was extracted from unstained FFPE samples using the TIANamp FFPE DNA Kit (TIANGEN, Beijing, China) following the manufacturer’s instructions. DNA was quantified using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and the Qubit 2.0 Flurometer. DNA quality check was performed by electrophoresis using 5 ng DNA in a 1% agarose gel. DNA samples with bands < 500 bps after agarose gel electrophoresis were excluded. RNA was isolated using the RecoverAll™ Total Nucleic Acid Isolation Kit and RNA integrity was evaluated using the Qubit™ RNA HS Assay Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
NGS library preparation and sequencing
Ten nanograms of DNA was used to prepare libraries using the Ion AmpliSeq™ Library Kit 2.0 and Ion Ampliseq Colon and Lung Cancer Panel V2 (Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer’s instructions. The panel consisted of a primer pool of 92 amplicons covering 504 hotspot mutations in 22 genes, including EGFR, ALK, ERBB2, ERBB4, FGFR1, FGFR2, FGFR3, MET, DDR2, KRAS, PIK3CA, BRAF, AKT1, PTEN, NRAS, MAP2K1, STK11, NOTCH1, CTNNB1, SMAD4, FBXW7, TP53.
RNA libraries were prepared with 30 ng of RNA using the AmpliSeq RNA Lung Cancer Research Fusion Panel. The panel consisted of 83 pairs of unique primers in a single pool that included: (i) primers that allowed the amplification and detection of 70 known ALK, RET, ROS1, and NTRK1 fusion transcripts; (ii) primers located in the 5′ and 3′ regions of ALK, RET, ROS1, and NTRK1 mRNA genes; (iii) primers that targeted five housekeeping genes to serve as internal controls.
The libraries were purified using AMPure XP beads (Beckman coulter Co, Ltd.) and quantified using the Ion Library TaqMan® Quantitation Kit (ThermoFisher Scientific, Inc., Waltham, MA, USA). Multiplex barcoded libraries were enriched by clonal amplification using emulsion PCR on Ion Sphere particles (Ion PI™ Template OT2 200 Kit v3, Life Technology, CA, USA) and loaded onto an Ion PI™ Chip. Sequencing was performed on the Ion Proton platform using the Ion PI™ Sequencing 200 Kit v3 according to the manufacturer’s instructions.
NGS data analysis
Variant calling was performed using the Ion Reporter Software (version 5.6). Sequencing was considered successful if the mean sequencing depth was 2000. If the criterion was not met, the sample was excluded from the analysis. Called variants were only accepted if the allele frequency (AF) was ≥ 1%.
Amplification-refractory mutation system (ARMS) PCR
DNA mutations in FFPE tumor samples, including EGFR, KRAS, NRAS, BRAF, etc., were carried out by ADx-ARMS Test Kits (Xiamen AmoyDx Biomedical Technology Co., Ltd.). After the reaction, the fluorescent signal curves and the threshold line were used to interpret the mutation results.
Fluorescence in situ hybridization (FISH)
FISH results were used to classify each tumor as ERBB2 amplified or nonamplified in accordance with the standard practice. FFPE tissue sections were hybridized with probes to ERBB2 and the centromere region of chromosome 17 (CEP17) in the PathVysion ERBB2 FISH assay (Abbott-Vysis Co., Ltd.). FISH results were reported as average ERBB2 copy number and ERBB2/CEP17 ratio. All FFPE samples were reviewed by two individual pathologists to determine ERBB2 status.
ALK rearrangement status was assessed by FISH using the Apart FISH Probe Kit (Abbott-Vysis Co., Ltd.). A tumor was considered ALK rearrangement positive if more than 15% of 50 (minimum) or 100 analyzed tumor cells showed split probes signals or isolated orange signals in accordance with published IASLC guidelines (IASLC Atlas of ALK Testing in Lung Cancer).
Statistical analysis
The experimental data were presented as percentages and were analyzed with the two-tailed Student’s t test. Pearson’s correlation was used to assess relation between EGFR variant type and T790M. The threshold of P < 0.05 was considered as statistically significant.
Results
Patient characteristics
A total of 1353 NSCLC patients were enrolled in this study, of which 1336 tumor samples were used to perform amplicon-based NGS. The remaining samples were excluded due to insufficient quantity and/or poor sample quality. Median age of our cohort patients was 60 years (21–83 years). There were 541 (40.49%) male and 795 (59.51%) female patients. The pathological features of patient tumor samples showed that 1263 (94.54%) patients diagnosed with adenocarcinoma, 43 (3.22%) with squamous carcinoma, and 27 (2.02%) with adenosquamous carcinoma (Supplementary Table 1).
Overall mutation status detected by NGS
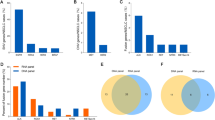
The mutation status in 1336 tumor samples, including DNA mutations and RNA fusions, has been identified previously by ARMS or FISH in the hospital where the patient was first treated. The known tumor mutation status was evaluated using the Ion Ampliseq Colon, and Lung Cancer Panel V2, AmpliSeq RNA Lung Cancer Research Fusion Panel. The DNA mutations (insertion, deletion and point mutation) from 1230 samples, RNA fusions from 90 samples, co-occurring DNA mutations and RNA fusions from 27 samples, and the remaining 43 samples had no detectable mutations using the aforementioned panels (Fig. 1a). Among the detected genetic mutations, the range of DNA mutations and RNA fusions was 1–9 and 1–2 per sample, respectively (Fig. 1b). A total of 2328 alterations were detected from the 1293 patients, where point mutations accounted for 71%, deletion mutations accounted for 21%, fusion mutations accounted for 5%, and insertional mutations accounted for 3% (Fig. 1c).

Overall mutational spectrum. a Venn diagram showing mutation status, including DNA mutations detected in 1230 samples, RNA fusion detected in 90 samples, and 43 samples had no detectable mutation. b Incidence of detected genetic mutations (DNA mutation and RNA fusion) for each sample. The detected DNA mutations ranged from 1 to 9, and RNA fusion ranged from 1 to 2 per sample. c Pie chart of mutation type for the 2328 mutations detected in 1293 tumor samples. The proportion of SNP, deletion mutations, fusion mutations and insertion mutations is shown
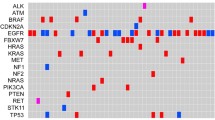
Co-occurring genetic alterations
Lung cancer-associated genetic alterations were listed in the mutation spectrum for analyzing the detailed pattern of these mutations (Fig. 2a). This included fusions, truncations, in-frame and missense mutations. EGFR had the highest rate of variation, accounting for 73% of 1336 samples. As an important tumor suppressor and oncogene, TP53 and KRAS mutations were detected in 38% and 8% of the samples, respectively. TP53, SMAD4, PIK3CA, CTNNB1, and PTEN mutations occurred either alone or alongside with EGFR mutations. KRAS mutations and fusions (including ALK, RET, ROS1) rarely co-occurred with EGFR. Of the 1293 mutation detected patients, 25 genes were identified to have 2338 variants (Supplementary Table 2). Frequency of EGFR mutations was the highest (47%), followed by TP53 (23%) and KRAS (5%) (Fig. 2b).

Co-occurring genetic alterations. a Genetic alterations and frequency identified by NGS in 1336 samples. Each sample occupies a vertical column. The alternations of genes are classified as fusions, truncating mutation, inframe and missense mutations. b Pie chart with frequency of each driver alteration for the 2328 mutations detected in 1293 samples. EGFR was the most common occurring DNA mutation (47%), followed by TP53 (23%) and KRAS (5%)
Co-occurring DNA mutations and RNA fusions in 27 patients
Co-occurring DNA mutations and RNA fusions were identified in 27 patients (Fig. 3). TP53 was the most common co-occurring DNA mutation, present in 43.8% (7/16), 71.4% (5/7) and 50% (2/4) of the samples with concurrent ALK, ROS1 and RET fusions, respectively. There were five patients with concurrent drug-related EGFR/ERBB2 variations and ALK/ROS1 fusions (Table 1), which were confirmed by subsequent Sanger sequencing.

Co-occurring DNA mutations and RNA fusions. Co-occurring DNA mutations and RNA fusions detected in 27 patients were plotted. Each sample occupies a vertical column. The alternations of genes are classified as fusions, truncating mutation, inframe and missense mutations, which were denoted in different colors
Alteration of EGFR T790M with 19del and L858R in pre-TKI-treated NSCLC patients
It was seen that 73% (975/1336) of the patients harbored EGFR mutations (Fig. 2a). Of the 2328 mutations identified from 1293 patients, EGFR mutations accounted for 47% (Fig. 2b). As shown in Table 2, EGFR exon19 deletion and exon21 L858R substitution mutations were the highest two variant types. The frequency of EGFR exon19 deletions was higher compared to exon21 L858R mutations (42.5% vs. 36.8%). In this study, 57 patients (5.2%) had detectable T790M mutations. Exon 20 insertion mutations were detected in 34 patients (3.1%).
Analysis of 975 patients with EGFR mutation revealed high prevalence of co-occurring genetic alterations. The most common co-occurring genetic alteration was TP53, present in 368/975 (37.7%) of all EGFR-mutated samples. Further analysis revealed that 184/464 (39.7%) of the samples harbored TP53 co-occurring mutations with EGFR exon19 deletions, 138/401 (34.4%) samples with EGFR L858R mutations, and 30/57 (52.6%) of samples with EGFR T790M mutations. Additionally, 10/975 (1.0%) samples had co-occurring EGFR and KRAS mutations, and 53/975 (5.4%) samples had co-occurring EGFR and PIK3CA mutations.
Further, analysis of EGFR exon19 deletion, exon21 L858R and exon20 T790M mutation showed that the mean allele fraction (MAF) of EGFR exon21 L858R, exon19 deletion and exon20 T790M was 23.3% (95% CI 25.86–30.02%), 37.1% (95% CI 35.17–39.04%), and 20.9% (95% CI 17.80–28.85%), respectively (Fig. 4a). MAF of EGFR exon19 deletion was higher compared to exon21 L858R, but the difference was not statistically significant. The MAF of T790M mutation was lower when compared to EGFR exon19 deletion and exon21 L858R mutation.

The alteration of EGFR T790M between 19del and L858R in pre-TKI-treated NSCLC patients. a MAF of the three variants of EGFR (exon19 deletion, exon21 L858R, exon20 T790M). The mean allele fraction (MAF) of exon21 L858R, exon19 deletion and exon20 T790M was 23.3%, 37.1% and 20.9%, respectively. **P > 0.05. b Correlation of MAFs in NSCLCs with co-occurring L858R/T790M and 19del/T790M mutations. Among the 57 samples harboring T790M mutations, 31 samples had co-occurring L858R/T790M mutations, and 20 samples had co-occurring 19del/T790M mutations
Among the 57 samples harboring T790M mutations, 31 samples had L858R mutations, and 20 samples had exon19 deletions. For patients with concurrent L858R/T790M mutations, the MAF of these two mutations was very close, whereas the MAF of T790M was lower compared to 19del in samples with co-occurring EGFR 19del/T790M mutations (Fig. 4b).
RNA Fusions
As shown in Fig. 1a and Supplementary Table 3, 106 RNA fusions were detected in 90 patients. There were seven types of ALK fusions, the most frequent type of fusions in this study. Among 76 ALK fusions, the most common ALK-EML4 fusion variant was ALK-E6aA20 (35.5%) and ALK-E13A20 (26.3%), and frequently co-occurred in the same samples. The most common RET and ROS fusion variants were KIF5B-RET.K15R12 and CD74-ROS1.C6R34, respectively.
Discussion
Recommendations from current guidelines suggest testing of molecular variations in terms of genetic mutations for all patients with NSCLC to aid clinicians to first- and second-line targeted therapies with durable clinical responses (Hirsch et al. 2016). Accurate molecular characterization using NGS, involving gene panels, offers a quick and cost-effective multiple gene analysis allowing a more comprehensive tumor mutation profiling (Lim et al. 2016; Lusebrink et al. 2018a, b). Results from the current study represent an extensive and comprehensive analysis of co-occurring genomic alterations in pre-TKI-treated NSCLC patients. The current data suggest that differences in co-occurring molecular events may partly explain the clinical heterogeneity in patients with EGFR mutant NSCLC.
Lung cancer tissues exhibiting EGFR mutations demonstrate an equal distribution of mutations in exon19 and the L858R point mutation in exon21 (Peng et al. 2018). Although patients with EGFR exon19 deletions and L858R mutations have similar benefits to EGFR TKIs, several recent studies have suggested that the benefit of EGFR TKIs is greater for patients with exon19 deletions than exon21 L858R substitutions (Lee et al. 2015; Yang et al. 2015; Zhang et al. 2014). However, patients randomly assigned to chemotherapy harboring exon21 L858R substitutions had significantly longer PFS compared to patients with exon19 deletions (Ke and Wu 2016); these findings have been inconsistent (Rosell et al. 2012; Wu et al. 2014). A recent, single-arm meta-analysis involving 1770 patients revealed that de novo T790M mutations was significantly less frequent with 19del compared to L858R mutations (OR 0.59; 95% CI 0.44–0.80; P < 0.001). However, the acquired resistance of T790M, i.e., after the TKI therapy, was higher with 19del when compared to L858R mutations (53% vs. 36%; OR 1.87; P < 0.001) (Chen et al. 2016; Liang et al. 2018). This observation was consistent with our study wherein the T790M mutation was less with 19del compared to L858R in pre-TKI patients. We also found that the MAF of T790M was lower compared to 19del in samples with co-occurring EGFR 19del/T790M mutations (Fig. 4b). T790M has been identified as a common mutation in pre-TKI-treated NSCLC samples using NGS. Osimertinib is an oral EGFR‐TKI with high affinity for mutated EGFR with T790M mutation (Soejima et al. 2017). The pooled analysis of AZD9291 First Time in Patients Ascending Dose Study (AURA) extension trial and the AURA2 trial has demonstrated clinical benefits of Osimertinib as second- and third-line therapy in terms of higher objective response rate [66% (95% confidence interval [CI] 61–70%)], progression-free survival (PFS) [9.9 months (95% CI, 9.5–12.3 months)] and median overall survival [26.8 months (95% CI 24.0–29.1 months)] (Ahn et al. 2019). Further, the AURA3 trial including the Japanese subgroup analysis study has demonstrated the superior clinical efficacy and safety of Osimertinib in comparison to platinum-pemetrexed chemotherapy (Akamatsu et al. 2018; Mok et al. 2017). Based on the results of AURA3 trial, FDA has approved Osimertinib as second-line therapy for the treatment of EGFR T790M mutation-positive NSCLC whose disease has progressed on or after EGFR TKI therapy. The detection of de novo T790M mutation will aid the clinicians in initiating the therapy with Osimertinib upfront (Gregorc et al. 2018). The AZENT trial (ClinicalTrials. gov, NCT02841579) assessing the efficacy and safety of Osimertinib as first-line therapy in advanced NSCLC patients with de novo T790M mutation will highlight the importance of primary T790M mutation. T790M mutations in 19del patients are more likely to be selected and enriched. Future clinical studies are needed to confirm this hypothesis.
There were 27 patients harboring DNA mutations and RNA fusion variants (Fig. 1a). Analysis of these samples with fusion mutations found that the highest frequency of co-occurring DNA mutations was TP53, followed by EGFR (Fig. 3). We observed that 43.8% of samples were harboring TP53 mutations with ALK fusions, 71.4% of ROS fusions and 50% of RET fusions (Fig. 3). Previous findings have shown that TP53 mutations reduced the responsiveness to crizotinib and worsened prognosis in NSCLC patients with ALK rearrangements (Wang et al. 2018). Larger cohorts are needed to assess the impact of TP53 mutations on response to targeted drugs in NSCLC patients with ALK/ROS1/RET rearrangements. Furthermore, another recent study showed that co-occurrence of TP53 mutation with an EGFR mutation significantly shortened PFS and OS in EGFR TKI 1st/2nd pretreated samples (Canale et al. 2017; Yu et al. 2018). This observation thereby postulates that it is not just important to analyze the major driver mutations to assess therapeutic response and clinical outcomes but also to conduct comprehensive genome sequencing in patients harboring an EGFR mutation.
Five patients had co-occurring EGFR/ERBB2 drug-related variations and ALK/ROS1 fusions (Table 1). Previous studies have shown that multiple mutations are usually caused by tumor heterogeneity (Cai et al. 2015; Fan et al. 2018). Currently, there is no consensus on how to treat patients with multiple mutations. Patients treated with EGFR TKI and ALK TKI go into remission (Recondo et al. 2018); however, the right TKI combination that can demonstrate the best efficacy needs to be investigated.
Few of the study limitations include its retrospective design. Since our NGS panel is designed to detect single-nucleotide variants, insertions, deletions and fusions, other mutaions such as copy number variants and structural variants are unable to be detected in this study. Therefore, the data acquired may underestimate the mutation burden for the cases.
We have paid more attentions and set strict quality control processes to ensure the true positive mutations we have detected when analyzing FFPE material for the introduction of low frequent variant detection (Ye et al. 2013). The process of generating raw sequencing data by NGS technology is subject to strict quality control to filter the possible false-positive results. We have eliminated the oxidized sample which might be generated by formaldehyde, a component of formalin. The mean sequencing depth was 2000 and called variants were only accepted if the allele frequency (AF) was ≥ 1%. That means the sum of the support reads of both plus and minus chains of mutation and wild-type was 2000, and at least 20 reads were needed to determine positive mutants. In addition, the cohorts in our study were enrolled to the clinical trial of the Human EGFR, KRAS, BRAF, PIK3CA, ALK, ROS1 gene mutation detection kit (semiconductor sequencing method), which was approved by the National Medical Products Administration (NMPA). In the clinical trial, we compared the consistency of Sanger sequencing and NGS technology in mutation detection of FFPE samples. The results showed that the consistency of EGFR mutation between NGS and Sanger sequencing was up to 99% (Supplementary Table 5).
Sensitive detection methods are of great significance in the detection of primary drug-resistant mutations due to the de novo T790M mutation associated with worse clinical benefits in NSCLC patients treated with first and second EGFR TKI therapy but with great survival benefit treated with third EGFR TKI therapy (Liu et al. 2017; Zhang et al. 2018). The pretreatment T790M mutation widely exists in EGFR mutant NSCLC, ranging from 0.009% to 26.9% (Watanabe et al. 2015). Compared with conventional NGS (limit of detection, 1%), digital PCR and the unique molecular identifiers (UMI) labeled NGS technology are more sensitive methods to detect the rare variants. For patients with resistance to the first-line TKI treatment, ultra-sensitive methods such as digital PCR (LoD 0.01%) and UMI-labeled NGS technology (LoD 0.1%) can detect those rare T790M mutations through analysis of circulating tumor DNA, indicating the development of drug resistance (Oxnard et al. 2018; Vollbrecht et al. 2018; Watanabe et al. 2015). Compared with digital PCR, NGS technology has advantages in detecting novel resistant mutations with tissue biopsies for massively parallel sequence analysis, despite its lower sensitivity relatively (Ding et al. 2019; Xu et al. 2017). Therefore, this high-resolution mutational profiling of pretreatment T790M mutation using ultra-sensitive methods can open the possibility of developing strategies for personalized cancer therapies in NSCLC patients.
In conclusion, co-occurring genetic alterations might play an underlying role in resistance mechanisms, thereby potentially explaining the diversity of individual patient outcomes. Our results indicated that T790M was a common mutation in pre-TKI-treated NSCLC, and might be enriched following TKI treatment. In addition, differences in the MAF of EGFR T790M may also result in different responses to TKI therapy in patients harboring dual mutations.
Abbreviations
- MAF:
-
Mutant allele frequency
- NGS:
-
Next-generation sequencing
- FFPE:
-
Formalin-fixed paraffin-embedded
- NSCLC:
-
Non-small cell lung cancer
- TKI:
-
Tyrosine kinase inhibitors
- EGFR :
-
Epidermal growth factor receptor
- ALK :
-
Anaplastic lymphoma kinase
- ARMS:
-
Amplification refractory mutation system
- FISH:
-
Fluorescence in situ hybridization
- IHC:
-
Immunohistochemistry
- PFS:
-
Progression free survival
- OS:
-
Overall survival
References
Ahn MJ et al (2019) Osimertinib in patients with T790M mutation-positive, advanced non-small cell lung cancer: long-term follow-up from a pooled analysis of 2 phase 2 studies. Cancer 125:892–901. https://doi.org/10.1002/cncr.31891
Akamatsu H et al (2018) Osimertinib in Japanese patients with EGFR T790M mutation-positive advanced non-small-cell lung cancer: AURA3 trial. Cancer Sci 109:1930–1938. https://doi.org/10.1111/cas.13623
Blakely CM et al (2017) Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet 49:1693–1704. https://doi.org/10.1038/ng.3990
Butler KS, Young MY, Li Z, Elespuru RK, Wood SC (2016) Performance characteristics of the AmpliSeq cancer hotspot panel v2 in combination with the ion torrent next generation sequencing personal genome machine. Regul Toxicol Pharmacol 74:178–186. https://doi.org/10.1016/j.yrtph.2015.09.011
Cai W et al (2015) Intratumoral heterogeneity of ALK-rearranged and ALK/EGFR coaltered lung adenocarcinoma. J Clin Oncol 33:3701–3709. https://doi.org/10.1200/JCO.2014.58.8293
Canale M et al (2017) Impact of TP53 mutations on outcome in EGFR-mutated patients treated with first-line tyrosine kinase inhibitors. Clin Cancer Res 23:2195–2202. https://doi.org/10.1158/1078-0432.CCR-16-0966
Chen LY et al (2016) Coexistence of EGFR T790M mutation and common activating mutations in pretreatment non-small cell lung cancer: a systematic review and meta-analysis. Lung Cancer 94:46–53. https://doi.org/10.1016/j.lungcan.2016.01.019
Ding PN et al (2019) Plasma next generation sequencing and droplet digital PCR-based detection of epidermal growth factor receptor (EGFR) mutations in patients with advanced lung cancer treated with subsequent-line osimertinib. Thorac Cancer 10:1879–1884. https://doi.org/10.1111/1759-7714.13154
Drilon A et al (2015) Broad, hybrid capture-based next-generation sequencing identifies actionable genomic alterations in lung adenocarcinomas otherwise negative for such alterations by other genomic testing approaches. Clin Cancer Res 21:3631–3639. https://doi.org/10.1158/1078-0432.CCR-14-2683
Fan J, Dai X, Nie X (2018) Concomitant epidermal growth factor receptor mutation and EML4-alk fusion in a patient with multifocal lung adenocarcinomas. J Thorac Oncol 13:e45–e48. https://doi.org/10.1016/j.jtho.2017.11.130
Godin-Heymann N et al (2007) Oncogenic activity of epidermal growth factor receptor kinase mutant alleles is enhanced by the T790M drug resistance mutation. Cancer Res 67:7319–7326. https://doi.org/10.1158/0008-5472.CAN-06-4625
Grasso C et al (2015) Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data. J Mol Diagn 17:53–63. https://doi.org/10.1016/j.jmoldx.2014.09.008
Gregorc V, Lazzari C, Karachaliou N, Rosell R, Santarpia M (2018) Osimertinib in untreated epidermal growth factor receptor (EGFR)-mutated advanced non-small cell lung cancer. Transl Lung Cancer Res 7:S165–S170. https://doi.org/10.21037/tlcr.2018.03.19
Hirsch FR, Suda K, Wiens J, Bunn PA Jr (2016) New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet 388:1012–1024. https://doi.org/10.1016/S0140-6736(16)31473-8
Hong S, Gao F, Fu S, Wang Y, Fang W, Huang Y, Zhang L (2018) Concomitant genetic alterations with response to treatment and epidermal growth factor receptor tyrosine kinase inhibitors in patients with EGFR-mutant advanced non-small cell lung cancer. JAMA Oncol 4:739–742. https://doi.org/10.1001/jamaoncol.2018.0049
Inukai M et al (2006) Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res 66:7854–7858. https://doi.org/10.1158/0008-5472.CAN-06-1951
Jurgens J, Engel-Riedel W, Stoelben E, Schildgen V, Schildgen O, Brockmann M (2016) The (Con-) fusion in ALK diagnostics: when food and drug administration-approved algorithms fail. J Clin Oncol 34:1961–1962. https://doi.org/10.1200/JCO.2015.65.4871
Ke EE, Wu YL (2016) Afatinib in the first-line treatment of epidermal-growth-factor-receptor mutation-positive non-small cell lung cancer: a review of the clinical evidence. Ther Adv Respir Dis 10:256–264. https://doi.org/10.1177/1753465816634545
Lee CK et al (2015) Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in egfr-mutant lung cancer: a meta-analysis. J Clin Oncol 33:1958–1965. https://doi.org/10.1200/JCO.2014.58.1736
Liang H et al (2018) The alteration of T790M between 19 del and L858R in NSCLC in the course of EGFR-TKIs therapy: a literature-based pooled analysis. J Thorac Dis 10:2311–2320. https://doi.org/10.21037/jtd.2018.03.150
Lim SM et al (2016) Targeted sequencing identifies genetic alterations that confer primary resistance to EGFR tyrosine kinase inhibitor (Korean Lung Cancer Consortium). Oncotarget 7:36311–36320. https://doi.org/10.18632/oncotarget.8904
Liu Y, Sun L, Xiong ZC, Sun X, Zhang SL, Ma JT, Han CB (2017) Meta-analysis of the impact of de novo and acquired EGFR T790M mutations on the prognosis of patients with non-small cell lung cancer receiving EGFR-TKIs. Onco Targets Ther 10:2267–2279. https://doi.org/10.2147/OTT.S133082
Lusebrink J, Pieper M, Tillmann RL, Brockmann M, Schildgen O, Schildgen V (2018a) Detailed overview on the mutations detected by and the sensitivity of the GeneReader NGS sequencing platform. Data Brief 18:1962–1966. https://doi.org/10.1016/j.dib.2018.04.114
Lusebrink J, Pieper M, Tillmann RL, Brockmann M, Schildgen O, Schildgen V (2018b) Pre-clinical validation of a next generation sequencing testing panel. Exp Mol Pathol 104:170–174. https://doi.org/10.1016/j.yexmp.2018.04.001
Mok TS et al (2017) Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 376:629–640. https://doi.org/10.1056/NEJMoa1612674
Oxnard GR et al (2018) Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol 4:1527–1534. https://doi.org/10.1001/jamaoncol.2018.2969
Peng M, Weng YM, Liu HL, Yang GF, Yao Y, Han G, Song QB (2018) Clinical characteristics and survival outcomes for non-small-cell lung cancer patients with epidermal growth factor receptor double mutations. Biomed Res Int 2018:7181368. https://doi.org/10.1155/2018/7181368
Recondo G, Facchinetti F, Olaussen KA, Besse B, Friboulet L (2018) Making the first move in EGFR-driven or ALK-driven NSCLC: first-generation or next-generation TKI? Nat Rev Clin Oncol 15:694–708. https://doi.org/10.1038/s41571-018-0081-4
Rosell R et al (2012) Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13:239–246. https://doi.org/10.1016/S1470-2045(11)70393-X
Schildgen V, Schildgen O (2018) The lonely driver or the orchestra of mutations? How next generation sequencing datasets contradict the concept of single driver checkpoint mutations in solid tumours—NSCLC as a scholarly example. Semin Cancer Biol. https://doi.org/10.1016/j.semcancer.2018.11.005
Soejima K, Yasuda H, Hirano T (2017) Osimertinib for EGFR T790M mutation-positive non-small cell lung cancer. Expert Rev Clin Pharmacol 10:31–38. https://doi.org/10.1080/17512433.2017.1265446
Su KY et al (2012) Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol 30:433–440. https://doi.org/10.1200/JCO.2011.38.3224
Suda K, Mitsudomi T (2014) Successes and limitations of targeted cancer therapy in lung cancer Prog. Tumor Res 41:62–77. https://doi.org/10.1159/000355902
Vollbrecht C, Lehmann A, Lenze D, Hummel M (2018) Validation and comparison of two NGS assays for the detection of EGFR T790M resistance mutation in liquid biopsies of NSCLC patients. Oncotarget 9:18529–18539. https://doi.org/10.18632/oncotarget.24908
Wang WX et al (2018) TP53 mutations predict for poor survival in ALK rearrangement lung adenocarcinoma patients treated with crizotinib. J Thorac Dis 10:2991–2998. https://doi.org/10.21037/jtd.2018.04.98
Watanabe M et al (2015) Ultra-sensitive detection of the pretreatment EGFR T790M mutation in non-small cell lung cancer patients with an EGFR-activating mutation using droplet digital PCR. Clin Cancer Res 21:3552–3560. https://doi.org/10.1158/1078-0432.CCR-14-2151
Wu YL et al (2014) Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 15:213–222. https://doi.org/10.1016/S1470-2045(13)70604-1
Xu T et al (2017) Cross-platform comparison of four leading technologies for detecting EGFR mutations in circulating tumor DNA from non-small cell lung carcinoma patient plasma. Theranostics 7:1437–1446. https://doi.org/10.7150/thno.16558
Yang JC et al (2015) Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol 16:141–151. https://doi.org/10.1016/S1470-2045(14)71173-8
Ye X et al (2013) High T790M detection rate in TKI-naive NSCLC with EGFR sensitive mutation: truth or artifact? J Thorac Oncol 8:1118–1120. https://doi.org/10.1097/JTO.0b013e31829f691f
Youssef O, Knuuttila A, Piirila P, Bohling T, Sarhadi V, Knuutila S (2018) Hotspot mutations detectable by next-generation sequencing in exhaled breath condensates from patients with lung cancer. Anticancer Res 38:5627–5634. https://doi.org/10.21873/anticanres.12897
Yu HA et al (2018) Concurrent alterations in EGFR-mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of MTOR as a mediator of resistance. Clin Cancer Res 24:3108–3118. https://doi.org/10.1158/1078-0432.CCR-17-2961
Zhang Y et al (2014) Patients with exon 19 deletion were associated with longer progression-free survival compared to those with L858R mutation after first-line EGFR-TKIs for advanced non-small cell lung cancer: a meta-analysis. PLoS One 9:e107161. https://doi.org/10.1371/journal.pone.0107161
Zhang B et al (2018) Coexistence of sensitive and resistant epidermal growth factor receptor (EGFR) mutations in pretreatment non-small cell lung cancer (NSCLC) patients: First or third generation tyrosine kinase inhibitors (TKIs)? Lung Cancer 117:27–31. https://doi.org/10.1016/j.lungcan.2018.01.006
Zheng G et al (2016) Test feasibility of next-generation sequencing assays in clinical mutation detection of small biopsy and fine needle aspiration specimens. Am J Clin Pathol 145:696–702. https://doi.org/10.1093/ajcp/aqw043
Acknowledgements
We acknowledge all other studies that support our work and were not cited due to length limitations. We would like to thank all the patients who have participated in this study. We are also thankful to the doctors from departments of oncology, doctors from departments of pathology for technics and staff of the Novogene Bioinformatics.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest.
Ethics approval and consent to participate
This study was approved by the ethics committee of Peking University Cancer Hospital (2017TW06), and written informed consent was obtained from all patients.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tang, Y., Che, N., Yu, Y. et al. Co-occurring genetic alterations and primary EGFR T790M mutations detected by NGS in pre-TKI-treated NSCLCs. J Cancer Res Clin Oncol 146, 407–416 (2020). https://doi.org/10.1007/s00432-019-03065-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-019-03065-0