
Abstract
Purpose
We performed deep sequencing of target genes in head and neck squamous cell carcinoma (HNSCC) tumors to identify somatic mutations that are associated with induction chemotherapy (IC) response.
Methods
Patients who were diagnosed with HNSCC were retrospectively identified. Patients who were treated with IC were divided into two groups: good responders and poor responders by tumor response and progression-free survival. Targeted gene sequencing for 2404 somatic mutations of 44 genes was performed on HNSCC tissues. Mutations with total coverage of <500 were excluded, and the cutoff for altered allele frequency was >10 %.
Results
Of the 71 patients, 45 were treated upfront with IC. Mean total coverage was 1941 per locus, and 42.2 % of tumors had TP53 mutations. Thirty-three mutations in TP53, NOTCH3, FGFR2, FGFR3, ATM, EGFR, MET, PTEN, FBXW7, SYNE1, and SUFU were frequently altered in poor responders. Among the patients who were treated with IC, those with unfavorable genomic profiles had significantly poorer overall survival than those without unfavorable genomic profiles (hazard ratio 6.45, 95 % confidence interval 2.07–20.10, P < 0.001).
Conclusions
Comprehensive analysis of mutation frequencies identified unfavorable genomic profiles, and the patients without unfavorable genomic profiles can obtain clinical benefits from IC in patients with HNSCC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Although induction chemotherapy (IC), defined by a short course of chemotherapy before definitive treatment such as operation or radiation, has been shown to be an effective strategy for organ preservation and reduction in distant metastasis (Lefebvre et al. 1996; Wolf et al. 1991), the overall survival benefit from IC in HNSCC patients remains controversial. While three randomized phase III trials (Cohen et al. 2014; Haddad et al. 2013; Hitt et al. 2014) failed to demonstrate an overall survival benefit from IC, a recent study showed a significant gain in survival in patients treated with IC and concurrent chemoradiotherapy (CCRT) compared to CCRT alone (Ghi et al. 2014). We have recently demonstrated that patients with bulky N stage benefit from IC in terms of survival (Ock et al. 2014) and identifying additional factors associated with survival benefit from IC would be valuable for improving the treatment outcomes of HNSCC patients. However, to date, predictive factors to decide which patients might benefit from IC are not clear.
One such potential factor includes genomic mutations. The mutational landscape of HNSCC has been characterized using advanced genomic approaches led by The Cancer Genome Atlas (TCGA) project (Agrawal et al. 2011; Hoadley et al. 2014; Keck et al. 2015; Lechner et al. 2013; Seiwert et al. 2015; Stransky et al. 2011). Besides identifying well-known mutations of TP53 and incorporation of human papilloma virus (HPV), which induces cell cycle dysregulation, recent findings showed frequent mutations in NOTCH family members, suggesting a role in oncogenesis or progression of HNSCC. However, the prognostic significance as well as the predictive value of each frequently mutated locus remains unknown for HNSCC patients.
Therefore, in this study, we performed deep sequencing of target genes in HNSCC tumor samples. We aimed to identify somatic mutations that are associated with clinical outcomes, especially IC response.
Methods
Study population
Medical records were retrospectively reviewed for patients diagnosed with HNSCC who were treated at Seoul National University Hospital from December 2004 to November 2012. Patients with paraffin-embedded tumor samples that were obtained prior to treatment and had enough material for targeted gene sequencing were included.
DNA extraction
Genomic DNA was extracted from 10-micrometer-thick sections of 10 % neutral formalin-fixed, paraffin-embedded (FFPE) tumor tissue blocks using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The concentration and purity of the extracted DNA were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, USA) and Qubit fluorometric quantitation (Life Technologies, Grand Island, NY, USA). The extracted DNA was stored at −20 °C until use.
Detection of cancer hot spot variants by targeted gene sequencing and bioinformatic analysis
Targeted gene sequencing was performed as previously described (Han et al. 2014). Ten nanograms of DNA was used for multiplex PCR of a panel covering hot spot mutations in the following 44 head and neck cancer-related genes: AKT1, ATM, BCL2L1, BRAF, CASP8, CCND1, CCND3, CDKN2A, CDKN2B, EGFR, ERBB2, ERBB4, FAT1, FBXW7, FGFR1, FGFR2, FGFR3, HRAS, JAK2, JAK3, KIT, KRAS, MDM2, MET, MLL2, MPL, MYC, NOTCH1, NOTCH2, NOTCH3, NRAS, PDGFRA, PIK3AP1, PIK3CA, PTEN, RASA1, RB1, RIMBP2, SMAD4, SOX2, SUFU, SYNE1, TP53, and TP63 (Ion AmpliSeq Customized Panel, Life Technologies, Grand Island, NY, USA) (Singh et al. 2013). We designed targeted gene sequencing panel based on previous results of whole exome sequencing and targeted gene sequencing of head and neck squamous cell carcinoma, including genomic mutations of the biologic significance and high frequency (Agrawal et al. 2011; Lechner et al. 2013; Stransky et al. 2011). Fragment libraries were constructed by DNA fragmentation, barcode and adaptor ligation, and library amplification using the Ion DNA Barcoding kit (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s instructions. The size distribution of the DNA fragments was analyzed on the Agilent Bioanalyzer using the High Sensitivity Kit (Agilent, Santa Clara, CA, USA). Template preparation, emulsion PCR, and Ion Sphere Particle (ISP) enrichment were performed using the Ion Xpress Template kit (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s instructions. The ISPs were loaded onto a P1 chip and sequenced using an Ion P1 Sequencing 200 kit (Life Technologies, Grand Island, NY, USA). Ion Torrent platform-specific pipeline software (Torrent Suite v2.0) was used to separate the barcoded reads, generate sequence alignment with the hg19 human genome reference, perform target-region coverage analysis, and filter and remove poor signal reads. The alignment file from Torrent Suite was transferred to Ion Reporter (Ion Reporter v4.0) for variant file generation using default parameters.
Bioinformatic analysis was performed as previously described with modification (Han et al. 2014). After a successful sequencing reaction, the raw signal data were analyzed using Torrent Suite v3.4.2. The pipeline includes signal processing, base calling, quality score assignment, adapter trimming, read alignment to human genome 19 reference, mapping quality QC, coverage analysis, and variant calling. After completion of the primary data analysis, lists of detected sequence variants (SNVs and INDELs) were compiled in a VCF (variant call file) format. For downstream analysis, variants with minimum coverage of 500 reads containing at least 10 % of the altered allele per total allele were selected. Variant calls were further analyzed using internally developed software that allows variant filtering and annotation using refGene in UCSC, COSMIC v.67, dbSNP build 138. To minimize false positives, variants were filtered with a normal population variant database, The Korean Personal Genomes Project (http://opengenome.net/) (Zhang et al. 2014). Reported loci in dbSNP were included in the analysis since filtering out dbSNP loci may cause loss of true reliable genomic alterations (Hudson et al. 2014). A total of 2404 genomic alterations of 44 genes including 482 cosmic alterations were analyzed. Supplementary Table 1A lists general information of the loci included in this study.
Treatment
The decision to treat patients with IC was determined by a multidisciplinary team (Lim et al. 2013; Ock et al. 2014). Patients were treated upfront with IC for two or three cycles every 3 weeks, followed by definitive CCRT or radical surgery (including primary tumor and regional lymph node dissection). IC regimens included docetaxel, cisplatin, or 5-fluorouracil. CCRT regimens consisted of cisplatin. Radiation therapy was given standard fractionated dose of more than 60 Gy for primary tumors and regional lymph nodes, with concurrent chemotherapy.
Response evaluation of induction chemotherapy
Patients were assessed following complete physical examination and MRI or CT of the head and neck. Follow-up imaging was performed after two cycles of IC. IC response was evaluated according to RECIST v1.1 (Eisenhauer et al. 2009). Patients treated with IC were divided retrospectively into two groups: good responders and poor responders. Patients with complete remission (100 % reduction) or partial response (more than 30 % reduction) by RECIST 1.1 with progression-free survival (PFS) more than 18 months were categorized as good responders. All other patients were classified as poor responders.
Immunohistochemistry of p16
Representative formalin-fixed paraffin-embedded tissue blocks from each case were submitted for immunohistochemistry (IHC) using mouse anti-p16 (E6H4) monoclonal antibody (mAb) (Roche/MTM/Ventana Laboratories, Tucson, AZ). IHC was performed using the Ventana Benchmark XT system (Ventana Medical Systems, Tucson, AZ). p16 was considered as positive when IHC study showed diffuse and strong nuclear and cytoplasmic staining in ≥70 % of the tumor cells (Gronhoj Larsen et al. 2014).
Statistical analysis
Each gene was considered positive for a somatic mutation if any locus in the gene was significantly altered (more than 10 % of the alleles altered with total coverage of more than 500). The frequency of each mutation was compared to clinicopathologic characteristics such as smoking, location of tumor, and response to induction chemotherapy as well as TP53 mutation. Fisher’s exact test was performed to assess significant differences in frequencies. To identify somatic mutations associated with response to induction chemotherapy, frequencies of mutated loci were compared between good responders and poor responders. Genomic loci that were only altered at a high frequency (more than 10 %) in poor responders were defined as being part of a high-risk genomic profile.
PFS was calculated from the diagnosis date to the date of disease progression, as confirmed by imaging, death, or the last follow-up date if censored. Overall survival (OS) was measured from the diagnosis date until death or the last follow-up date, if censored. Survival analyses were carried out according to the Kaplan–Meier method with log-rank testing to assess differences between the groups. Subgroup analysis was carried out using the Cox proportional hazards regression model, summarizing the hazard ratio (HR) and 95 % confidence interval (CI) of each group. All reported P values are two-sided and considered significant if P < 0.05. In order to reduce false-positive values of P, q value by Benjamini and Hochberg’s false discovery rate (FDR) test were performed, which were also considered significant if q < 0.05. All statistical analyses were carried out using R version 3.1.2 (http://www.r-project.org).
Results
Patient characteristics by treatment type and response
Of the 71 patients included in this study, 45 were treated upfront with IC followed by definitive treatment and 26 received definitive treatment without IC. Of the 45 patients who were treated with IC, 23 were good responders and 22 were poor responders as defined above (Supplementary Fig. 1 and Supplementary Table 1B). Clinicopathologic characteristics of the three groups (good responders to IC, poor responders to IC, and no IC) are listed in Table 1. Although clinicopathologic features were generally balanced among the three groups, patients treated with IC had tumors with higher N stage (P < 0.001) and were significantly more likely to have undergone definitive treatment with concurrent chemoradiotherapy rather than radical surgery (P < 0.001). Among the patients treated with IC, oropharyngeal tumors occurred were more frequently in good responders than in poor responders (56.5 vs. 22.7 %, P = 0.028).
Among the 26 patients who were not treated with IC, 6 patients received definitive concurrent chemoradiotherapy only, whereas 20 patients were treated with surgical resection of tumor. Among 20 patients who undergone surgery, 7 patients received adjuvant radiotherapy and 10 patients received adjuvant chemoradiotherapy, but 3 patients did not receive adjuvant treatment (Table 1).
Genomic profiles according to clinicopathologic characteristics and TP53 mutation
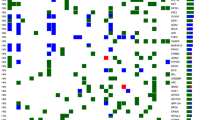
Mean total coverage was 1941 reads per locus, and 42.2 % of samples had TP53 mutations (Fig. 1); however, mutations of MPL, CCND1, CDKN2A, CDKN2B, PIK3AP1, RASA1, RIMBP2, and SOX2 were not found. Somatic mutation frequencies of HRAS, FBXW7, NOTCH1, EGFR, MET, FGFR3, MYC, AKT1, and BCL2L1 were significantly higher in tumors with TP53 mutations (q < 0.05) (Table 2). Frequencies of FBXW7, FGFR3, and CCND3 mutations were significantly higher in patients with a history of smoking (P = 0.013, 0.025, and 0.014, respectively), although their q values were not significant. CCND3 mutations occurred more frequently in oropharyngeal tumors than in other tumor locations (P = 0.0425, q = 0.983). Among patients who were treated with IC, good responders had PIK3CA mutations more frequently than poor responders (47.8 vs. 13.6 %, P = 0.023, q = 0.830). However, other than PIK3CA mutation, there were no significant differences of the frequency of gene-level mutations in good or poor responders (TP53 mutation in good responder: 56.5 % vs. in poor responder: 50.0 %, P = 0.768, Table 2). Moreover, multivariate analysis including other clinicopathologic factors showed that the prognostic impact of PIK3CA was insignificant (P = 0.998, Supplementary Table 3). Table 2 lists all frequencies of somatic mutation according to TP53 mutation and clinicopathologic characteristics.

Mutation events sorted by response to induction chemotherapy. Relevant clinicopathologic characteristics are described above the heat map of genomic changes. See the key for explanation of the color coding of the observed changes and patient characteristics
Survival analysis according to response to induction chemotherapy and PIK3CA mutation
During a median follow-up duration of 70.0 months (range 21.7–117.7 months), the 3-year OS rate was 70.5 % in all patients. Although there was no difference in the 3-year OS rate between patients who received IC and those who did not (69.3 vs. 74.7 %, P = 0.936, Supplementary Fig. 2), and the survival outcome was not influenced by the regimen of IC as well (Supplementary Fig. 3), the 3-year OS rate of good responders to IC was significantly higher than that of poor responders (95.0 vs. 42.3 %, P = 0.002) and of patients that did not receive IC treatment (P = 0.009, Fig. 2a). In all enrolled patients (N = 71), patients with PIK3CA mutation had significantly favorable overall survival (HR 0.09, 95 % CI 0.01–0.68, P = 0.019). Moreover, multivariate analysis showed that PIK3CA mutation was an independent prognostic factor along with tumor location of oropharynx [adjusted HR 0.10 (0.01–0.77), P = 0.027, Supplementary Table 3]. Among patients that received IC, those with tumors with PIK3CA mutations had significantly more favorable OS than those with tumors without PIK3CA mutations (HR 0.09, 95 % CI 0.01–0.71, P = 0.004). However, this survival benefit was not significantly affected by IC treatment (with PIK3CA mutations: P = 0.515, without PIK3CA mutations: P = 0.705, Fig. 2b).

Overall survival according to induction chemotherapy and PIK3CA mutation. Kaplan–Meier curves for overall survival of three groups: patients with good response or poor response to induction chemotherapy and patients that did not receive IC treatment (a). Kaplan–Meier curves for overall survival of four groups based on whether the patient had PIK3CA mutations (+: mutation, −: wild type) and whether the patient received IC (b). HR hazard ratio, CI confidence interval
Unfavorable genomic profiles are associated with poor clinical outcomes of induction chemotherapy
To identify somatic mutations that might influence response to induction chemotherapy, we compared the frequencies of all 2404 mutations between patients that responded well to induction chemotherapy and those that responded poorly. Somatic mutations that were only altered with high frequency (more than 10 %) in poor responders were defined as part of a unfavorable genomic profile. Thirty-two mutations in TP53, NOTCH3, FGFR2, FGFR3, ATM, EGFR, MET, FBXW7, SYNE1, PTEN, and SUFU were altered only and frequently in poor responders (detailed genomic information of 32 mutations is listed in Table 3). In all patients (N = 71), unfavorable genomic profile conferred the overall poor prognostic impact (HR 2.49, 95 % CI 1.09–5.68, P = 0.026). Among the patients who were treated with IC, those with unfavorable genomic profiles had significantly poorer OS than those without unfavorable genomic profiles (HR 6.45, 95 % CI 2.07–20.1, P < 0.001, Supplementary Fig. 3A). However, the unfavorable genomic profiles did not confer any prognostic impact in patients who were not treated with IC (HR 0.68, 95 % CI 0.32–1.43, P = 0.297). While induction chemotherapy did not confer any survival benefit when all patients that received IC were compared to all patients that did not receive IC (HR 0.96, P = 0.936, Supplementary Fig. 2), among patients with unfavorable genomic profiles, IC contributed to significantly worse overall survival (HR 3.77, 95 % CI 1.05–13.6, P = 0.030, Fig. 3b). However, patients without unfavorable genomic profiles significantly benefited from IC (HR 0.26, 95 % CI 0.07–0.99, P = 0.035, Fig. 3c). Multivariate analysis showed that unfavorable genomic profiles independently associated with worse overall survival in patients treated with IC (adjusted HR 5.09, 95 % CI 1.58–16.4, P = 0.006, Supplementary Table 4).

Overall survival according to unfavorable genomic profiles. Kaplan–Meier curves for overall survival of patients that received IC treatment by the presence or absence of unfavorable genomic profiles. HR hazard ratio, CI confidence interval
Discussion
In this study, we performed deep sequencing of target genes in HNSCC tumors and identified a subset of genomic mutations that are associated with clinical benefit from IC. Patients with tumors that contained unfavorable genomic profiles had poorer clinical outcomes when treated with IC; inversely, those who did not have unfavorable genomic profiles had a prolonged overall survival from IC.
The decision of whether to include IC in the treatment of patients with HNSCC is challenging. Although the IC can result in organ preservation and reduction in distant metastasis, a survival benefit from IC has been inconsistently demonstrated in previous trials (Cohen et al. 2014; Ghi et al. 2014; Haddad et al. 2013; Hitt et al. 2014). We performed this study in order to identify any genomic mutations that are associated with clinical outcome of IC treatment in patients with HNSCC.
We found somatic mutations in 44 genes in HNSCC patients by deep sequencing of target genes. Patients with tumors containing TP53 mutations had significantly higher mutation frequencies in HRAS, FBXW7, NOTCH1, EGFR, MET, FGFR3, MYC, AKT1, and BCL2L1. Since one of the main roles of TP53 is in DNA repair, loss-of-function mutations may contribute to increased frequencies of mutations of other genes. In addition, smoking is associated with TP53 mutations and also contributes to increased mutagenesis (Vrieling et al. 1992; Wittekindt et al. 2012), resulting in higher mutations in FBXW7, FGFR3, and CCND3. Previous results have revealed frequent TP53 mutations (81–100 %) and high mutation rates of genes in HPV-negative HNSCC (Lechner et al. 2013; Seiwert et al. 2015). Although we did not perform HPV genotyping or p16 immunohistochemistry in this study, it is possible that the tumors with TP53 mutations and increased rates of mutations may also be HPV negative. Although oropharyngeal tumors were more likely to have mutations of CCND3, the biologic significance is unclear and may be a false-positive result (P = 0.0425, FDR q = 0.9830).
The frequency of somatic mutations in the present study was generally higher than previously reported (Agrawal et al. 2011; Hoadley et al. 2014; Stransky et al. 2011), even though copy number variation analysis was not included. Furthermore, there are large discrepancies in mutation frequencies among previous reports (Agrawal et al. 2011; Hoadley et al. 2014; India Project Team of the International Cancer Genome 2013; Pickering et al. 2013; Stransky et al. 2011). For example, reported mutation frequency of CDKN2A varies from 0 to 12 % of FAT1 from 0 to 40 %. These variations may be due to differences in bioinformatics techniques of mutation calling or differences in sequencing coverage (Riaz et al. 2014). Moreover, expression of p16, which is encoded by CDKN2A, was lost in 60 % of assessed samples, which implied that the genomic alterations of CDKN2A would be more of deep deletion of copy number variation rather than single nucleotide variations in the current study. Of particular note, in this study, NOTCH2 and EGFR were much more frequently altered compared to previous reports (42 vs. 0–5 % and 23.9 vs. 3–5 %, respectively). Although deep coverage may partially contribute to the higher mutation frequencies found in this study, the discordance could also be due to contamination of germ line mutations since we did not filter by comparison with genomic DNA from normal tissue. Moreover, as we used amplicon-based sequencing, INDEL mutations would be not falsely called, which should be also considered to be cautious to interpret this result.
In the current study, good responders to IC were significantly more likely to have PIK3CA mutations than poor responders. Moreover, we showed that mutation of PIK3CA was associated with more favorable overall survival in HNSCC irrespective of IC treatment. Previous reports revealed that the PIK3CA mutation rate was higher in HPV-positive HNSCC (Lechner et al. 2013; Seiwert et al. 2015), and HPV positivity or p16 positivity is a well-known good prognostic factor in HNSCC, irrespective of the tumor origin (Chung et al. 2014; Rischin et al. 2010). Interestingly, the prognostic impact of PIK3CA mutation was still significant even adjusted by p16 status in the current analysis, which would warrant a concise clinical study to find out whether HPV/p16 status or PIK3CA mutation would be biologically more important. Although PIK3CA mutations were associated with favorable prognosis in the current study, PIK3CA mutations were not predictive of benefit from IC since patients did not benefit from IC whether they had PIK3CA mutations or not.
We then broadened our scope to include all analyzed somatic mutations in order to generate a finely tuned genomic model to predict survival outcome according to IC. When the mutation frequency of each genomic locus was compared between good responders and poor responders, we identified a total of 32 unfavorable genomic profiles based on alterations in 11 genes including TP53, NOTCH3, FGFR2, FGFR3, ATM, EGFR, MET, PTEN, FBXW7, SYNE1, and SUFU. These genes were frequently altered in poor responders but not altered in good responders. Patients who had unfavorable genomic profiles had poorer survival if treated with IC. Conversely, patients who did not have any of unfavorable genomic profiles had a survival benefit if treated with IC. The mutations of genes included in the unfavorable genomic profiles have been known to play a crucial role in cancer progression. Firstly, the functional alteration of FGFR2, FGFR3, EGFR, and MET would be associated with epithelial–mesenchymal transition which would confer resistance to chemotherapy (Azuma et al. 2014; Byers et al. 2013). Besides, FBXW7 is a member of the F-box protein family which acts as a tumor suppressor mediated by its major targets of NOTCH family (Agrawal et al. 2011), which could be related to a poor prognosis. Although TP53 gene-level mutation was not associated with poor outcomes, certain mutations of TP53 were only significantly associated with the worse survival, which would imply the biologic significance of gene mutations should be analyzed in a locus-specific manner. Taken together, these results suggest that a subset of patients may benefit from IC and external validation of these unfavorable genomic profiles in an independent cohort should be performed. In addition, further studies are needed to understand the biologic implications of these unfavorable genomic profiles.
There are several limitations of this study. Due to the retrospective design, treatment bias may have resulted in confounding factors that contributed to treatment outcomes. The patients that decided to receive IC were significantly more likely to have advanced N stage tumors, and they tended to undergo CCRT as definitive treatment. Since 82.2 % patients who were treated with IC achieved more than a partial response, their clinicians may have decided that CCRT was warranted. However, no significant survival differences were observed between patients that had IC treatment and those that did not (Supplementary Fig. 2) or between those that had definitive treatment as CCRT or not (data not shown, log rank P = 0.94). In addition, only a small number of patients were included in the survival analysis; therefore, the survival outcome of IC treatment might be inconsistent with the previous study which has proven the survival benefit from IC in especially N2 or N3 stage (Ock et al. 2014). Moreover, the proportion of patients with N2 or N3 stage in the current study was lower than that of the previous report (56.3 vs. 62.1 %), which would also account for the inconsistent results of the survival outcome of IC. Finally, this study did not evaluate HPV and p16 status in all patients, which are significant prognostic factors (Chung et al. 2014; Rischin et al. 2010). In order to complete the prediction model of survival outcome, HPV/p16 status in all patients should be adjusted. However, the underlying question of why HPV/p16 negative is associated with poor prognosis remains unclear. Considering the widespread mutations in HPV/p16-negative tumors, the unfavorable genomic profiles reported in this study may be informative in terms of identifying mutations that contribute to tumorigenesis in HNSCC.
Deep sequencing of target genes in HNSCC and survival analysis revealed that PIK3CA mutations conferred favorable clinical outcome regardless of IC. Comparative analysis of mutation frequencies between good responders and poor responders to IC has identified unfavorable genomic profiles that are significantly associated with clinical benefits from IC in patients with HNSCC. External validation of these genomic profiles is warranted.
References
Agrawal N et al (2011) Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333:1154–1157. doi:10.1126/science.1206923
Azuma K et al (2014) Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol 25:1935–1940. doi:10.1093/annonc/mdu242
Byers LA et al (2013) An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3 K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 19:279–290. doi:10.1158/1078-0432.CCR-12-1558
Chung CH et al (2014) p16 Protein expression and human papillomavirus status as prognostic biomarkers of nonoropharyngeal head and neck squamous cell carcinoma. J Clin Oncol 32:3930–3938. doi:10.1200/JCO.2013.54.5228
Cohen EE et al (2014) Phase III randomized trial of induction chemotherapy in patients with N2 or N3 locally advanced head and neck cancer. J Clin Oncol 32:2735–2743. doi:10.1200/JCO.2013.54.6309
Eisenhauer EA et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247. doi:10.1016/j.ejca.2008.10.026
Ghi MG et al (2014) Concomitant chemoradiation (CRT) or cetuximab/RT (CET/RT) versus induction Docetaxel/Cisplatin/5-Fluorouracil (TPF) followed by CRT or CET/RT in patients with Locally Advanced Squamous Cell Carcinoma of Head and Neck (LASCCHN). A randomized phase III factorial study (NCT01086826). J Clin Oncol 32:(suppl; abstr 6004)
Gronhoj Larsen C et al (2014) Correlation between human papillomavirus and p16 overexpression in oropharyngeal tumours: a systematic review. Br J Cancer 110:1587–1594. doi:10.1038/bjc.2014.42
Haddad R et al (2013) Induction chemotherapy followed by concurrent chemoradiotherapy (sequential chemoradiotherapy) versus concurrent chemoradiotherapy alone in locally advanced head and neck cancer (PARADIGM): a randomised phase 3 trial. Lancet Oncol 14:257–264. doi:10.1016/S1470-2045(13)70011-1
Han JY et al (2014) Comparison of targeted next-generation sequencing with conventional sequencing for predicting the responsiveness to epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) therapy in never-smokers with lung adenocarcinoma. Lung Cancer 85:161–167. doi:10.1016/j.lungcan.2014.04.009
Hitt R et al (2014) A randomized phase III trial comparing induction chemotherapy followed by chemoradiotherapy versus chemoradiotherapy alone as treatment of unresectable head and neck cancer. Ann Oncol 25:216–225. doi:10.1093/annonc/mdt461
Hoadley KA et al (2014) Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158:929–944. doi:10.1016/j.cell.2014.06.049
Hudson AM et al (2014) Discrepancies in cancer genomic sequencing highlight opportunities for driver mutation discovery. Cancer Res 74:6390–6396. doi:10.1158/0008-5472.CAN-14-1020
India Project Team of the International Cancer Genome C (2013) Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat Commun 4:2873. doi:10.1038/ncomms3873
Keck MK et al (2015) Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin Cancer Res 21:870–881. doi:10.1158/1078-0432.CCR-14-2481
Lechner M et al (2013) Targeted next-generation sequencing of head and neck squamous cell carcinoma identifies novel genetic alterations in HPV+ and HPV− tumors. Genome Med 5:49. doi:10.1186/gm453
Lefebvre JL, Chevalier D, Luboinski B, Kirkpatrick A, Collette L, Sahmoud T (1996) Larynx preservation in pyriform sinus cancer: preliminary results of a European Organization for Research and Treatment of Cancer phase III trial. EORTC Head and Neck Cancer Cooperative Group. J Natl Cancer Inst 88:890–899
Lim Y et al (2013) Clinical outcomes of radiation-based locoregional therapy in locally advanced head and neck squamous cell carcinoma patients not responding to induction chemotherapy. Oral Surg Oral Med Oral Pathol Oral Radiol 116:55–60. doi:10.1016/j.oooo.2013.02.007
Ock CY et al (2014) Effect of induction chemotherapy on survival in locally advanced head and neck squamous cell carcinoma treated with concurrent chemoradiotherapy: single center experience. Head Neck. doi:10.1002/hed.23891
Pickering CR et al (2013) Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov 3:770–781. doi:10.1158/2159-8290.CD-12-0537
Riaz N, Morris LG, Lee W, Chan TA (2014) Unraveling the molecular genetics of head and neck cancer through genome-wide approaches. Genes Dis 1:75–86. doi:10.1016/j.gendis.2014.07.002
Rischin D et al (2010) Prognostic significance of p16INK4A and human papillomavirus in patients with oropharyngeal cancer treated on TROG 02.02 phase III trial. J Clin Oncol 28:4142–4148. doi:10.1200/JCO.2010.29.2904
Seiwert TY et al (2015) Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res 21:632–641. doi:10.1158/1078-0432.CCR-13-3310
Singh RR et al (2013) Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn JMD 15:607–622. doi:10.1016/j.jmoldx.2013.05.003
Stransky N et al (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333:1157–1160. doi:10.1126/science.1208130
Vrieling H, Thijssen JC, Rossi AM, van Dam FJ, Natarajan AT, Tates AD, van Zeeland AA (1992) Enhanced hprt mutant frequency but no significant difference in mutation spectrum between a smoking and a non-smoking human population. Carcinogenesis 13:1625–1631
Wittekindt C, Wagner S, Mayer CS, Klussmann JP (2012) Basics of tumor development and importance of human papilloma virus (HPV) for head and neck cancer. GMS Curr Top Otorhinolaryngol Head Neck Surg. doi:10.3205/cto000091
Wolf GT, Hong WK, Fisher SG (1991) Induction chemotherapy plus radiation compared with surgery plus radiation in patients with advanced laryngeal cancer. The Department of Veterans Affairs Laryngeal Cancer Study Group. N Engl J Med 324:1685–1690. doi:10.1056/NEJM199106133242402
Zhang W et al (2014) Whole genome sequencing of 35 individuals provides insights into the genetic architecture of Korean population. BMC Bioinform 15(Suppl 11):S6. doi:10.1186/1471-2105-15-S11-S6
Acknowledgments
We thank the patients and their families who generously donated valuable tissue samples.
Funding
This study was supported by SNUH Research Fund (Grant Nos. 04-2013-0760 and 30-2013-0070).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study protocol was approved by the Institutional Review Board of Seoul National University Hospital (approval number: H-1307-051-504) and was conducted in accordance with the principles of the Declaration of Helsinki.
Informed consent
For the patients diagnosed before January 31, 2013, the institutional review board approved the waiver of informed consent for this retrospective analysis according to Bioethics and Safety Act of Korea. For the patients diagnosed after February 1, 2013, we obtained informed consents from the patients.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ock, CY., Son, B., Keam, B. et al. Identification of genomic mutations associated with clinical outcomes of induction chemotherapy in patients with head and neck squamous cell carcinoma. J Cancer Res Clin Oncol 142, 873–883 (2016). https://doi.org/10.1007/s00432-015-2083-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-015-2083-2