
Abstract
Purpose
Long noncoding RNA (lncRNA) have been reported to be involved in the development of multiple cancers. The aim of this study was to report the identification of lncRNA-CTD-2108O9.1, which we have named lncRNA low expressed in gastric cancer (lncRNA-LOWEG), and investigate its role in cancer development.
Methods
Total RNA was extracted from the tissues of 94 patients with GC, one normal gastric epithelial cell line and four GC cell lines. Expression levels of lncRNA-LOWEG were determined by real-time PCR. Moreover, CCK-8 proliferation assay, transwell cell invasion assay and flow cytometry were performed to study the effects of lncRNA-LOWEG on SGC-7901 cell proliferation, cell invasion and cell cycle progression. Lastly, western blot and real-time PCR were used to verify the potential target genes of lncRNA-LOWEG.
Results
Significantly reduced expression of lncRNA-LOWEG was found in gastric cancer tissues and cell lines (SGC-7901, AGS, BGC-823 and HG-27) compared with patient-matched nontumorous adjacent tissues (P < 0.01) or the normal gastric cell line GES-1 (P < 0.05). Moreover, the transwell assay showed that the number of cells capable of passing through the Matrigel was significantly reduced after lncRNA-LOWEG transfection (P < 0.05). However, lncRNA-LOWEG overexpression did not significantly influence cell proliferation (P > 0.05) and cell cycle progression (P > 0.05). Lastly, western blot and real-time PCR analysis suggested that lncRNA-LOWEG is positively correlated with the expression of leukemia inhibitory factor receptor (LIFR) gene at the translational level.
Conclusions
LncRNA-LOWEG is a tumor suppressor that inhibits GC cell invasion. And LIFR gene is up-regulated by lncRNA-LOWEG.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is the one of the leading causes of death (Murray et al. 2012), and gastric cancer (GC) is the second leading cancer-related cause of death worldwide (Jemal et al. 2011). Fast development and metastasis of GC contribute greatly to its poor prognosis (Hohenberger and Gretschel 2003; Luebeck et al. 2013). In order to improve early diagnosis and targeted therapy of GC, there is a pressing need to understand the underlying molecular mechanism as well as identify new prognostic biomarkers and therapeutic targets that can be utilized to effectively treat this disease (Kong et al. 2013).
Long noncoding RNAs (lncRNAs) are a class of transcripts longer than 200 nucleotides that do not code for any proteins (Mattick and Makunin 2006; Ponting et al. 2009). Recently, many studies have demonstrated that lncRNAs are involved in various diseases and have multiple functions in a wide range of biological processes (Gupta et al. 2010; Yuan et al. 2014). Furthermore, there is growing evidence suggesting that lncRNAs can function as an oncogene or tumor suppressor (Hu et al. 2014b; Yang et al. 2013a), making lncRNA an ideal candidate for better understanding the development of cancers, including GC. For instance, lncRNA-GAPLINC, which can regulate CD44-dependent cell invasiveness, associates with the poor prognosis of GC and can be utilized as a means of diagnosing this disease (Hu et al. 2014b).
Even though lncRNAs have been found to be involved in many diseases, the mechanisms by which lncRNAs regulate cancer progression remain largely unknown. LncRNAs are capable of regulating different cancer pathways at various levels (i.e., transcriptionally, post-transcriptionally and translationally) (Di Gesualdo et al. 2014). At the same time, a limited number of studies have investigated the role of lncRNAs in GC (Fang et al. 2015; Han et al. 2014; Hu et al. 2014b; Li et al. 2014). And so it is necessary to not only identify new lncRNAs, but also elucidate their biological effects on GC. In our study, we report the identification of lncRNA-CTD-2108O9.1 (ENST00000508986), which we have named lncRNA low expressed in GC (lncRNA-LOWEG). Here, we focus on the expression level of lncRNA-LOWEG in GC and the role of lncRNA-LOWEG in inhibiting cell invasiveness and regulating leukemia inhibitory factor receptor (LIFR) gene.
Material and method
Human tissue samples
Ninety-four gastric tissue samples were obtained from patients who were diagnosed with GC and underwent surgical resection at the First Hospital of China Medical University, between 2007 and 2009. Ninety-four samples of matched nontumorous adjacent tissue (NAT) were also collected from parts of the resected specimens that were furthest from the tumor (>5 cm). The samples were snap-frozen in liquid nitrogen and stored at −80 °C. No previous local or systemic treatment had been conducted on these patients before the operation. Tumor histological grades were assessed according to the criteria set by the World Health Organization using the 7th edition TNM staging of the International Union Against Cancer (UICC). Patient follow-ups were performed every 3–6 months after surgery until February 2014. Overall survival (OS) was defined as the length of time between surgery and death or the last follow-up examination. Disease-free survival (DFS) was defined as the length of time between surgery and disease recovery or the last follow-up examination.
Ethical approval of the study protocol
This study was conducted according to the principles expressed in the Declaration of Helsinki. Tissue specimens were collected after obtaining the informed consent of patients in accordance with institutional ethical guidelines, which were reviewed and approved by the Research Ethics Committee of China Medical University (Shenyang, China).
Cell culture
Human GC cell lines (SGC-7901, AGS, BGC-823 and HG-27) and one normal gastric epithelial cell line (GES-1) were purchased from the Institute of Biochemistry and Cell Biology at the Chinese Academy of Sciences (Shanghai, China). SGC-7901, HG-27 and BGC-823 were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA), AGS was cultured in F-12 medium (Invitrogen), and GES-1 was cultured in Dulbecco’s Modified Eagle medium (Invitrogen). All cell culture media were supplemented with 10 % fetal bovine serum at 37 °C and 5 % CO2.
Plasmids, SiRNAs and transfection
For construction of eukaryotic expression vectors, the full-length lncRNA-LOWEG cDNA was synthesized and cloned into pEX-2(pGCMV/MCS/IRES/EGFP/Neo) plasmid (GenePharma). A pEX-2 negative control plasmid, with no functional cDNA, was also constructed. DNA sequencing was used to verify nucleotide sequences. Transfections were performed using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer’s protocol. A final concentration of 0.2 µg/ml plasmid was used for each transfection in a 6-well plate with 2.5 ml culture medium. The lncRNA-LOWEG transfected cells, the negative control plasmids transfected cells and SGC-7901 cells were named as LOWEG group, negative control (NC) group and SGC-7901 control (SGC-7901) group, respectively. SiRNA duplexes specific for lncRNA-LOWEG were synthesized (RIBOBIO) with a sequence 5′-GGCCGAAAUGUGAUCUCAA dTdT-3′. A control nonspecific siRNA was also provided by RIBOBIO and used as negative control. Transfections were performed using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer’s protocol. A final concentration of 100 nM was used for each transfection in a 6-well plate with 2 ml culture medium. The siRNA for lncRNA-LOWEG transfected cells and negative control siRNA transfected cells were named as Si-LOWEG group and Si-NC group, respectively. Transfection efficiency was monitored by real-time RT-PCR.
RNA isolation and real-time RT-PCR
Total RNA from the specimens and cultured cells was isolated using the Trizol reagent (Invitrogen) according to the manufacturer’s instructions. The concentration and purity of RNA were controlled by UV spectrophotometry (A260/A280 > 1.9) using a nano-photometer UV/Vis spectrophotometer (Implen). The first strand of cDNA was synthesized using the PrimeScript RT reagent Kit (Takara) according to the manufacturer’s instructions (Invitrogen). For real-time PCR, 2 μL diluted RT products were mixed with 12.5 μL SYBR Premix Ex Taq II (Takara), 0.5 μL forward and reverse primers (10 μM) and 9.5 μL nuclease-free water in a final volume of 25 μL, according to the manufacturer’s instructions. All reactions were run using the following protocol: one cycle at 95 °C for 3 min, 40 cycles of 95 °C for 15 s and 60 °C for 60 s. The specificity of the PCR amplification was validated by the presence of a single peak in the melting curve analysis. The expression of target RNA was calculated relative to an endogenous reference (GAPDH RNA). The primers used are as follows: GAPDH primer: 5′-CGGATTTGGTCGTATTGGG-3′ (forward) and 5′-CTGGAAGATGGTGATGGGATT-3′ (reverse); lncRNA-LOWEG primer: 5′-CCCAGAAGTTTTCAGCCCTCA-3′ (forward) and 5′-GACACTTTCCTTCATGGAATTTG-3′ (reverse); and LIFR primer: 5′-TCCAATCAGAGCCATCCCTA-3′ (forward) and 5′-GAACAAGGCAGGAGTTGAGG-3′ (reverse).
Cell counting kit 8 proliferation assay
The capacity for cellular proliferation was measured with the cell counting kit 8 (CCK-8, Dojindo). Twenty-four hours after vectors transfection, cells (approximately 2.5 × 103) were seeded into 96-well culture plates for 24, 48, 72 and 96 h. The cells were then incubated with 10 μL of CCK-8 for 1 h at 37 °C. The optical density was determined with a spectrophotometer (Spectra Max plus384, Molecular Devices) at a wavelength of 450 nm.
Transwell invasion assay
The invasive potential of SGC-7091 cells was assessed using a transwell invasion assay, which was conducted in 24-well plates using the transwell device containing microporous 8-μm membranes (Corning). Each membrane was coated with 50 μg of Matrigel (BD Biosciences), which was rehydrated by adding 45 μL of serum-free medium. After incubating at 37 °C for 4 h, the Matrigel solidified and served as the extracellular matrix for analysis of tumor cell invasion. Cells (approximately 5 × 104), in 200 μL of serum-free RPMI-1640 medium, were added into the upper compartment of the chamber. A total of 600 μL of RPMI-1640 medium with 10 % fetal bovine serum were placed in the bottom compartment of the chamber. After 24 h of incubation at 37 °C with 5 % CO2, the medium was removed from the upper chamber. The noninvaded cells on the upper side of the chamber were scraped off with a cotton swab. Cells that had migrated from the Matrigel into the pores of the inserted filter were fixed with 100 % methanol and stained with hematoxylin and eosin. The number of cells invading through the Matrigel was counted in ten randomly selected visual fields from the central and peripheral portion of the filter using an inverted microscope (Leica DMI300B).
Cell cycle detection
At 48 h after transfection, the adhered cells were obtained by trypsinization and pooled with the floating cells and centrifuged at 2000 rpm for 5 min. Then, the cells were treated by cell cycle detection kit (KeyGene) according to the manufacturer’s instructions. Finally, the samples were analyzed using the FACScalibur flow cytometer (BD Bioscience) and BD CellQuest software.
Western blot
Western blot assays were performed to examine the expressions of LIFR proteins. Briefly, 48 h after transfection, whole cell extracts were prepared using a Protein Extraction Kit (KeyGene). Proteins were separated by SDS-PAGE and blotted onto a polyvinylidene difluoride (PVDF) membrane (Millipore). Membranes were probed with the specific primary antibodies and then with peroxidase-conjugated secondary antibodies. The bands were visualized by chemiluminescence (Thermo Fisher) and analyzed by Microchemi 4.2 (Dnr). The β-actin antibody was used as a control for total protein input. The western blotting analysis was repeated at least three times. The following antibodies were used: LIFR antibody (1:1000, Santa Cruz Biotechnology) and β-actin antibody (1:4000, Sigma Aldrich).
Statistical analysis
RNA expression levels in GC tissues, relative to control tissues, were calculated using the formula: 2−ΔΔCT, where ΔΔC T is the difference in the ΔC T values between the treatment tumor cells and the control (ΔΔC T = ΔC T tumor lncRNA − ΔC T NATs lncRNA) and ΔC T is the difference in the C T value between the target and endogenous reference (GAPDH; ΔC T = C T lncRNA − C T GAPDH). Survival analysis was performed by cox proportional hazards model and log-rank test. For experiments, data are presented as mean ± 1 standard deviation (SD) of the mean from at least three separate experiments. Statistical analysis was performed using Student’s t test and nonparametric tests (Mann–Whitney U test between two groups and Kruskall–Wallis test for three or more groups). Statistical analysis was performed using SPSS 17.0 computer software (SPSS Inc., Chicago, IL, USA), and P < 0.05 was considered statistically significant.
Results
Expression of LncRNA-LOWEG is down-regulated in GC
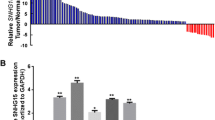
Among 94 patients with GC, 56 (59.6 %) showed lower expression of lncRNA-LOWEG in GC compared with their NATs (Fig. 1a). The median fold change was 0.69. Moreover, the ΔC T of lncRNA-LOWEG in cancer tissues and NATs were 5.63 ± 3.09 and 4.79 ± 2.55, respectively (Fig. 1b). There was a significantly lower expression of lncRNA-LOWEG in GC tissues in comparison with matched NATs (P < 0.01). Furthermore, compared with normal gastric cell line GES-1, lncRNA-LOWEG expression was also reduced by different degrees in SGC-7901 (P < 0.05), BGC-823 (P < 0.01), HG-27 (P < 0.05) and AGS (P < 0.01) cell lines (Fig. 1c).

Expression of lncRNA-LOWEG in gastric cancer. a Expression level of lncRNA-LOWEG in 94 gastric cancer patients. Expression of lncRNA-LOWEG was quantified by real-time RT-PCR. Data are presented as log base 2 of the fold change in gastric cancer relative to its NAT. In each case, experiments were conducted in triplicate and repeated three times. b ΔC T of lncRNA-LOWEG in cancer tissues and NATs. LncRNA-LOWEG was normalized to GAPDH mRNA levels. ΔC T = CT lncRNA-LOWEG − CT GAPDHRNA. c Expression of lncRNA-LOWEG in four gastric cancer cell lines. Levels of lncRNA-LOWEG in gastric cancer cell lines, BGC-823, SGC-7901, AGS and HG-27 are lower than those in human normal gastric epithelial cell line GES-1 (*P < 0.05, **P < 0.01). Data are presented as mean ± SD. Experiments were independently conducted three times to obtain the presented data
We next assessed the correlation between lncRNA-LOWEG expression and clinical characteristics. There was no statistically significant association between lncRNA-LOWEG expression and clinical pathological features including gender, age, tumor size, histologic grade, tumor stage, depth of tumor, lymphatic metastasis or lymphatic vessel invasion (P > 0.05, Table S1). At the same time, we also assessed the correlation between LOWEG expression and patient survival. Analysis on OS (HR 0.97, 95 % CI 0.58–1.63, P > 0.05) and DFS (HR = 1.00, 95 % Cl 0.59–1.67, P > 0.05) both demonstrated no significant difference in prognosis for patients with lncRNA-LOWEG expression higher than 1 compared with that for patients with an expression less than this relative value.
The effect of LncRNA-LOWEG on cell invasiveness, cell proliferation and cell cycle
First, we detected expression of lncRNA-LOWEG by real-time RT-PCR at 48 h after transfection in LOWEG group, NC group and SGC-7901 group. The expression level of lncRNA-LOWEG in LOWEG group was dramatically higher than other groups (42.94 ± 2.93-fold, compared with NC group, P < 0.01, Fig. 2a). To determine the effects of lncRNA-LOWEG on cell invasion, a transwell assay was performed. The invasive potential of tumor cells in the transwell assay was determined by the ability of these cells to invade a matrix barrier. After the cells were incubated for 24 h, the cells that had migrated to the basal side of the membrane were captured and counted. Representative micrographs of transwell filters are shown in Fig. 3a. The lncRNA-LOWEG transfected cells, the negative control plasmids transfected cells and SGC-7901 cells were named as LOWEG group, negative control (NC) group and SGC-7901 control (SGC-7901) group, respectively. The number of cells passing through the Matrigel in the LOWEG group (8.49 ± 2.77, P < 0.05) was significantly lower than in the NC (29.63 ± 8.18) and SGC-7901 (29.44 ± 10.58) groups (Fig. 3b). Also, there was no significant difference between the NC and SGC-7901 groups (P = 0.96).

Indication of lncRNA-LOWEG expression in transfected cells. Forty-eight hours after transfection, the efficiency of transfection with plasmids or siRNA was monitored by real-time RT-PCR. a The relative expression of lncRNA-LOWEG in LOWEG group was very high (42.94 ± 2.93-fold, compared with NC group, **P < 0.01). b The relative expression of lncRNA-LOWEG in Si-LOWEG group was very low (0.18 ± 0.04-fold, compared with Si-NC group, **P < 0.01)

Effect of lncRNA-LOWEG on cell invasion. A transwell assay was used to determine cell invasion. Cell invasive potential was markedly inhibited in the LOWEG group compared with the NC and SGC-7901 groups. a Representative micrographs of transwell filters, scale bars: 50 μm. b Statistical results are based on four independent experiments (mean ± SD, *P < 0.05)
To determine the effect of lncRNA-LOWEG on cell proliferation, a CCK-8 assay was performed. As determined by a CCK-8 assay, no significant differences were observed between lncRNA-LOWEG and NC or SGC-7901 groups (P > 0.05, Figure S1A). As in cell cycle detection, exogenous expression of lncRNA-LOWEG also did not cause significant differences, relative to the other two groups, in G0/G1, G2/M and S phase changes (P > 0.05, Figure S1C). Representative results of the cell cycle assay can be seen in Figure S1B.
LIFR is regulated by LncRNA-LOWEG in SGC-7901 cells
It is widely accepted that lncRNA can regulate its neighbor protein-coding genes7. LncRNA-LOWEG is located on the reverse strand of chromosome 5p13.2. The LIFR gene is located less than 50 kbp downstream of lncRNA-LOWEG. Furthermore, some studies have shown that LIFR is related to cancer and cancer metastasis (Chen et al. 2012; Okamura et al. 2010). Thus, we hypothesized that LIFR may be regulated by lncRNA-LOWEG. To test this hypothesis, we performed real-time RT-PCR and western blot to detect the transcription and protein level of LIFR upon lncRNA-LOWEG overexpression. Forty-eight hours after transfection, we examined the mRNA level of LIFR in cells and we did not find significant variation (P > 0.05, Fig. 4a). However, the protein level of LIFR was significantly increased by lncRNA-LOWEG overexpression at 48 h after transfection. As shown in Fig. 4b, the LIFR protein levels in the lncRNA-LOWEG group were dramatically higher than that of the NC or SGC-7901 groups. To provide further evidence that lncRNA-LOWEG could regulate LIFR, we knocked down the expression of lncRNA-LOWEG by siRNA which showed a perfect transfection efficiency (Fig. 2b). Similarly, the mRNA level of LIFR in Si-LOWEG group and Si-NC group did not show significant difference (P > 0.05, Fig. 4a). The protein level of LIFR was significantly decreased upon knocking down lncRNA-LOWEG expression. As shown in Fig. 4b, the LIFR protein levels in the Si-LOWEG group were dramatically lower than that of the Si-NC group. Taken together, our results suggest that LIFR is up-regulated by lncRNA-LOWEG at the translational level.

LIFR is regulated by lncRNA-LOWEG in SGC-7901 cells. a The results of real-time PCR revealed that lncRNA-LOWEG and knocking down lncRNA-LOWEG both had no effect on LIFR mRNA level (mean ± SD presented represents four independent experiments). b Western blot analysis shows that lncRNA-LOWEG interacted with LIFR and positively regulates its expression at translational level
Discussion
Growing evidence shows that aberrant expression of lncRNAs can play an important role during the initiation and development of human cancers. LncRNAs can act in oncogenic or tumor-suppressive roles in multiple cancers, including GC (Fang et al. 2015; Ponting et al. 2009). For instance, lncRNA-GAPLINC (Hu et al. 2014b) and lncRNA-GHET1 (Yang et al. 2014) were both highly expressed in GC as they promote cell proliferation and invasiveness. Moreover, lncRNA-LEIGC, a newly identified lncRNA, was lowly expressed in GC and could inhibit epithelial to mesenchymal transition, thus acting in a tumor-suppressive role (Han et al. 2014). In our study, we discovered a new lncRNA (lncRNA-LOWEG) that was expressed at a low level in GC. To our knowledge, our study is the largest one to date that assesses the expression level of a newly identified GC-associated lncRNA by real-time PCR. In our study, we found significantly lower levels of lncRNA-LOWEG expressed in primary GC tissues, relative to patient-matched normal tissue samples. At the same time, dramatically reduced expression of lncRNA-LOWEG was also found in GC cell lines. Taken together, these consistent results in tissue and cell lines suggest that low expression of lncRNA-LOWEG is universal in GC and that lncRNA-LOWEG may have a tumor-suppressive role. Expression of lncRNAs could be regulated by many factors. A recent study has shown that Ezh2-mediated H3K27 methylation plays an important role in the regulation of lncRNA expression (Wu et al. 2010). Moreover, Yang et al. (2013b) found that c-Myc could directly bind to the E-box element in the promoter region of lncRNA-CCAT1 and regulate its expression. However, the exact mechanism for the aberrant expression of lncRNA-LOWEG in GC remains to be elucidated, and this may be a new direction for our group in the future.
Invasion and metastasis are very important biological characteristics of malignant tumors (Cheng et al. 2013). Many recent studies have demonstrated that lncRNAs are involved in these processes and could influence the invasiveness of GC cells. For example, lncRNA-HOTAIR and lncRNA-H19 are classic oncogenic lncRNAs (Berteaux et al. 2005; Byun et al. 2007; Gupta et al. 2010; Kim et al. 2013). Xu et al. (2013) found that inhibition of lncRNA-HOTAIR could reduce invasiveness of AGS cells. Also Li et al. (2014) suggested that lncRNA-H19 could promote cell invasion in SGC-7901 and MKN-45 cells. In addition, lncRNA-GAPLINC, a newly identified GC during 2014, was found to regulate CD44-dependent cell invasiveness by forming a molecular decoy for miR211-3p (Hu et al. 2014b). As demonstrated by our transwell assay experiment, there was a dramatic reduction in the number of migrating cells after lncRNA-LOWEG transfection, relative to controls, which suggests that overexpression of lncRNA-LOWEG may inhibit invasiveness of GC cells. Overall, our findings suggest that lncRNA-LOWEG potentially inhibits cell invasion and GC progression.
The regulation of lncRNA on the expression of genes in close genomic proximity is called cis-acting regulation (Ponting et al. 2009), which is a common regulatory mechanism of lncRNAs (Faghihi et al. 2008; Katayama et al. 2005; Mercer et al. 2008). LncRNA-LOWEG is located on chromosome 5. Thus, we searched the neighbor protein-coding genes near lncRNA-LOWEG on chromosome 5 to find potential target genes which lncRNA-LOWEG might influence. Firstly, we found the EGFLAM gene which is located at the antisense of LOWEG. However, EGFLAM is related to ribbon synapse formation (Sato et al. 2008). Until now, no studies have demonstrated its involvement in cancer progression and development. Then, we found that the LIFR gene was located on the same strand within 50 kbp downstream of lncRNA-LOWEG. LIFR is an integral component of the glycoprotein 130–LIFR signaling complex (Kishimoto et al. 1994). Reduced expressions of LIFR have been found in many cancers such as colorectal cancer (Cho et al. 2011), breast cancer (Chen et al. 2012) and hepatocellular carcinoma (Okamura et al. 2010). de Iglesia et al. (2008) have previously demonstrated that LIFR had the tumor-suppressive function in the PTEN pathway. Moreover, Chen et al. (2012) suggested that LIFR is a metastasis suppressor that functions through the Hippo-YAP pathway. All of these findings strongly suggest that LIFR has similar tumor-suppressive role compared with lncRNA-LOWEG. In our study, dramatically enhanced expression of LIFR protein was found by western blot at 48 h after lncRNA-LOWEG transfection. Considering that no significant change in mRNA level was observed, the results imply that LIFR is regulated by lncRNA-LOWEG and that its regulation is likely at the translational level. To examine this phenomenon in more detail, we knocked down the expression of lncRNA-LOWEG by siRNA. Also with no significant change in mRNA level, dramatically decreased expression of LIFR protein was found by western blot. This finding could provide further evidence for the conclusion that lncRNA-LOWEG can regulate LIFR at translational level and makes it more solid. Many studies have shown that lncRNAs can regulate translation process through multiple mechanisms, such as forming ceRNA with miRNA (Tang et al. 2014), forming lncRNA-mRNA complexes(Carrieri et al. 2012) and influencing eukaryotic translation initiation factor-4E (Hu et al. 2014a). However, the specific biological mechanism by which lncRNA-LOWEG regulates LIFR still needs to be further investigated. Presented data have shown correlations among lncRNA-LOWEG, LIFR gene and metastatic procedures. So in the future, to clarify the concrete relationship between them can be another important task for our team.
To our knowledge, we have performed the largest study and follow-up assessing the expression level of a newly identified lncRNA in GC by real-time PCR to date. Significantly reduced expression of lncRNA-LOWEG was found in cancerous tissues relative to noncancerous tissues from the same patients. Moreover, a cell invasion assay suggested the potential role of lncRNA-LOWEG in tumor invasion and the progression of gastric cancer. Additionally, the results of real-time PCR and western blot highlighted that lncRNA-LOWEG interacts with LIFR and enhances its expression at the translational level.
Conclusions
LncRNA-LOWEG is a tumor suppressor that inhibits GC cell invasion. And LIFR gene is up-regulated by lncRNA-LOWEG. Based on our study, lncRNA-LOWEG is a potential biomarker and therapeutic target for the treatment of GC.
References
Berteaux N, Lottin S, Monte D, Pinte S, Quatannens B, Coll J, Hondermarck H, Curgy JJ, Dugimont T, Adriaenssens E (2005) H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J Biol Chem 280(33):29625–29636. doi:10.1074/jbc.M504033200
Byun HM, Wong HL, Birnstein EA, Wolff EM, Liang G, Yang AS (2007) Examination of IGF2 and H19 loss of imprinting in bladder cancer. Cancer Res 67(22):10753–10758. doi:10.1158/0008-5472.CAN-07-0329
Carrieri C, Cimatti L, Biagioli M, Beugnet A, Zucchelli S, Fedele S, Pesce E, Ferrer I, Collavin L, Santoro C, Forrest AR, Carninci P, Biffo S, Stupka E, Gustincich S (2012) Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 491(7424):454–457. doi:10.1038/nature11508
Chen D, Sun Y, Wei Y, Zhang P, Rezaeian AH, Teruya-Feldstein J, Gupta S, Liang H, Lin HK, Hung MC, Ma L (2012) LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat Med 18(10):1511–1517. doi:10.1038/nm.2940
Cheng JC, Klausen C, Leung PC (2013) Hypoxia-inducible factor 1 alpha mediates epidermal growth factor-induced down-regulation of E-cadherin expression and cell invasion in human ovarian cancer cells. Cancer Lett 329(2):197–206. doi:10.1016/j.canlet.2012.10.029
Cho YG, Chang X, Park IS, Yamashita K, Shao C, Ha PK, Pai SI, Sidransky D, Kim MS (2011) Promoter methylation of leukemia inhibitory factor receptor gene in colorectal carcinoma. Int J Oncol 39(2):337–344. doi:10.3892/ijo.2011.1050
de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, Levy DE, Depinho RA, Bonni A (2008) Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev 22(4):449–462. doi:10.1101/gad.1606508
Di Gesualdo F, Capaccioli S, Lulli M (2014) A pathophysiological view of the long non-coding RNA world. Oncotarget 5(22):10976–10996
Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G 3rd, Kenny PJ, Wahlestedt C (2008) Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med 14(7):723–730. doi:10.1038/nm1784
Fang XY, Pan HF, Leng RX, Ye DQ (2015) Long noncoding RNAs: Novel insights into gastric cancer. Cancer letters 356((2 Pt B)):357–366. doi:10.1016/j.canlet.2014.11.005
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464(7291):1071–1076. doi:10.1038/nature08975
Han Y, Ye J, Wu D, Wu P, Chen Z, Chen J, Gao S, Huang J (2014) LEIGC long non-coding RNA acts as a tumor suppressor in gastric carcinoma by inhibiting the epithelial-to-mesenchymal transition. BMC Cancer 14(1):932. doi:10.1186/1471-2407-14-932
Hohenberger P, Gretschel S (2003) Gastric cancer. Lancet 362(9380):305–315
Hu G, Lou Z, Gupta M (2014a) The long non-coding RNA GAS5 cooperates with the eukaryotic translation initiation factor 4E to regulate c-Myc translation. PLoS One 9(9):e107016. doi:10.1371/journal.pone.0107016
Hu Y, Wang J, Qian J, Kong X, Tang J, Wang Y, Chen H, Hong J, Zou W, Chen Y, Xu J, Fang JY (2014b) Long Noncoding RNA GAPLINC Regulates CD44-Dependent Cell Invasiveness and Associates with Poor Prognosis of Gastric Cancer. Cancer Res 74(23):6890–6902. doi:10.1158/0008-5472.CAN-14-0686
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61(2):69–90. doi:10.3322/caac.20107
Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, Suzuki H, Carninci P, Hayashizaki Y, Wells C, Frith M, Ravasi T, Pang KC, Hallinan J, Mattick J, Hume DA, Lipovich L, Batalov S, Engstrom PG, Mizuno Y, Faghihi MA, Sandelin A, Chalk AM, Mottagui-Tabar S, Liang Z, Lenhard B, Wahlestedt C, Group RGER, Genome Science G, Consortium F (2005) Antisense transcription in the mammalian transcriptome. Science 309(5740):1564–1566. doi:10.1126/science.1112009
Kim K, Jutooru I, Chadalapaka G, Johnson G, Frank J, Burghardt R, Kim S, Safe S (2013) HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 32(13):1616–1625. doi:10.1038/onc.2012.193
Kishimoto T, Taga T, Akira S (1994) Cytokine signal transduction. Cell 76(2):253–262
Kong X, Qian J, Chen LS, Wang YC, Wang JL, Chen H, Weng YR, Zhao SL, Hong J, Chen YX, Zou W, Xu J, Fang JY (2013) Synbindin in extracellular signal-regulated protein kinase spatial regulation and gastric cancer aggressiveness. J Natl Cancer Inst 105(22):1738–1749. doi:10.1093/jnci/djt271
Li H, Yu B, Li J, Su L, Yan M, Zhu Z, Liu B (2014) Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget 5(8):2318–2329
Luebeck EG, Curtius K, Jeon J, Hazelton WD (2013) Impact of tumor progression on cancer incidence curves. Cancer Res 73(3):1086–1096. doi:10.1158/0008-5472.CAN-12-2198
Mattick JS, Makunin IV (2006) Non-coding RNA. Human molecular genetics 15 Spec No 1:R17-29 doi:10.1093/hmg/ddl046
Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS (2008) Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci USA 105(2):716–721. doi:10.1073/pnas.0706729105
Murray CJ, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, Aboyans V, Abraham J, Ackerman I, Aggarwal R, Ahn SY, Ali MK, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Bahalim AN, Barker-Collo S, Barrero LH, Bartels DH, Basanez MG, Baxter A, Bell ML, Benjamin EJ, Bennett D, Bernabe E, Bhalla K, Bhandari B, Bikbov B, Bin Abdulhak A, Birbeck G, Black JA, Blencowe H, Blore JD, Blyth F, Bolliger I, Bonaventure A, Boufous S, Bourne R, Boussinesq M, Braithwaite T, Brayne C, Bridgett L, Brooker S, Brooks P, Brugha TS, Bryan-Hancock C, Bucello C, Buchbinder R, Buckle G, Budke CM, Burch M, Burney P, Burstein R, Calabria B, Campbell B, Canter CE, Carabin H, Carapetis J, Carmona L, Cella C, Charlson F, Chen H, Cheng AT, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahiya M, Dahodwala N, Damsere-Derry J, Danaei G, Davis A, De Leo D, Degenhardt L, Dellavalle R, Delossantos A, Denenberg J, Derrett S, Des Jarlais DC, Dharmaratne SD, Dherani M, Diaz-Torne C, Dolk H, Dorsey ER, Driscoll T, Duber H, Ebel B, Edmond K, Elbaz A, Ali SE, Erskine H, Erwin PJ, Espindola P, Ewoigbokhan SE, Farzadfar F, Feigin V, Felson DT, Ferrari A, Ferri CP, Fevre EM, Finucane MM, Flaxman S, Flood L, Foreman K, Forouzanfar MH, Fowkes FG, Fransen M, Freeman MK, Gabbe BJ, Gabriel SE, Gakidou E, Ganatra HA, Garcia B, Gaspari F, Gillum RF, Gmel G, Gonzalez-Medina D, Gosselin R, Grainger R, Grant B, Groeger J, Guillemin F, Gunnell D, Gupta R, Haagsma J, Hagan H, Halasa YA, Hall W, Haring D, Haro JM, Harrison JE, Havmoeller R, Hay RJ, Higashi H, Hill C, Hoen B, Hoffman H, Hotez PJ, Hoy D, Huang JJ, Ibeanusi SE, Jacobsen KH, James SL, Jarvis D, Jasrasaria R, Jayaraman S, Johns N, Jonas JB, Karthikeyan G, Kassebaum N, Kawakami N, Keren A, Khoo JP, King CH, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Laden F, Lalloo R, Laslett LL, Lathlean T, Leasher JL, Lee YY, Leigh J, Levinson D, Lim SS, Limb E, Lin JK, Lipnick M, Lipshultz SE, Liu W, Loane M, Ohno SL, Lyons R, Mabweijano J, MacIntyre MF, Malekzadeh R, Mallinger L, Manivannan S, Marcenes W, March L, Margolis DJ, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGill N, McGrath J, Medina-Mora ME, Meltzer M, Mensah GA, Merriman TR, Meyer AC, Miglioli V, Miller M, Miller TR, Mitchell PB, Mock C, Mocumbi AO, Moffitt TE, Mokdad AA, Monasta L, Montico M, Moradi-Lakeh M, Moran A, Morawska L, Mori R, Murdoch ME, Mwaniki MK, Naidoo K, Nair MN, Naldi L, Narayan KM, Nelson PK, Nelson RG, Nevitt MC, Newton CR, Nolte S, Norman P, Norman R, O’Donnell M, O’Hanlon S, Olives C, Omer SB, Ortblad K, Osborne R, Ozgediz D, Page A, Pahari B, Pandian JD, Rivero AP, Patten SB, Pearce N, Padilla RP, Perez-Ruiz F, Perico N, Pesudovs K, Phillips D, Phillips MR, Pierce K, Pion S, Polanczyk GV, Polinder S, Pope CA 3rd, Popova S, Porrini E, Pourmalek F, Prince M, Pullan RL, Ramaiah KD, Ranganathan D, Razavi H, Regan M, Rehm JT, Rein DB, Remuzzi G, Richardson K, Rivara FP, Roberts T, Robinson C, De Leon FR, Ronfani L, Room R, Rosenfeld LC, Rushton L, Sacco RL, Saha S, Sampson U, Sanchez-Riera L, Sanman E, Schwebel DC, Scott JG, Segui-Gomez M, Shahraz S, Shepard DS, Shin H, Shivakoti R, Singh D, Singh GM, Singh JA, Singleton J, Sleet DA, Sliwa K, Smith E, Smith JL, Stapelberg NJ, Steer A, Steiner T, Stolk WA, Stovner LJ, Sudfeld C, Syed S, Tamburlini G, Tavakkoli M, Taylor HR, Taylor JA, Taylor WJ, Thomas B, Thomson WM, Thurston GD, Tleyjeh IM, Tonelli M, Towbin JA, Truelsen T, Tsilimbaris MK, Ubeda C, Undurraga EA, van der Werf MJ, van Os J, Vavilala MS, Venketasubramanian N, Wang M, Wang W, Watt K, Weatherall DJ, Weinstock MA, Weintraub R, Weisskopf MG, Weissman MM, White RA, Whiteford H, Wiebe N, Wiersma ST, Wilkinson JD, Williams HC, Williams SR, Witt E, Wolfe F, Woolf AD, Wulf S, Yeh PH, Zaidi AK, Zheng ZJ, Zonies D, Lopez AD, AlMazroa MA, Memish ZA (2012) Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380(9859):2197–2223. doi:10.1016/S0140-6736(12)61689-4
Okamura Y, Nomoto S, Kanda M, Li Q, Nishikawa Y, Sugimoto H, Kanazumi N, Takeda S, Nakao A (2010) Leukemia inhibitory factor receptor (LIFR) is detected as a novel suppressor gene of hepatocellular carcinoma using double-combination array. Cancer Lett 289(2):170–177. doi:10.1016/j.canlet.2009.08.013
Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136(4):629–641. doi:10.1016/j.cell.2009.02.006
Sato S, Omori Y, Katoh K, Kondo M, Kanagawa M, Miyata K, Funabiki K, Koyasu T, Kajimura N, Miyoshi T, Sawai H, Kobayashi K, Tani A, Toda T, Usukura J, Tano Y, Fujikado T, Furukawa T (2008) Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat Neurosci 11(8):923–931. doi:10.1038/nn.2160
Tang J, Zhuo H, Zhang X, Jiang R, Ji J, Deng L, Qian X, Zhang F, Sun B (2014) A novel biomarker Linc00974 interacting with KRT19 promotes proliferation and metastasis in hepatocellular carcinoma. Cell Death Dis 5:e1549. doi:10.1038/cddis.2014.518
Wu SC, Kallin EM, Zhang Y (2010) Role of H3K27 methylation in the regulation of lncRNA expression. Cell Res 20(10):1109–1116. doi:10.1038/cr.2010.114
Xu ZY, Yu QM, Du YA, Yang LT, Dong RZ, Huang L, Yu PF, Cheng XD (2013) Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial-mesenchymal transition in gastric cancer. Int J Biol Sci 9(6):587–597. doi:10.7150/ijbs.6339
Yang F, Huo XS, Yuan SX, Zhang L, Zhou WP, Wang F, Sun SH (2013a) Repression of the long noncoding RNA-LET by histone deacetylase 3 contributes to hypoxia-mediated metastasis. Mol Cell 49(6):1083–1096. doi:10.1016/j.molcel.2013.01.010
Yang F, Xue X, Bi J, Zheng L, Zhi K, Gu Y, Fang G (2013b) Long noncoding RNA CCAT1, which could be activated by c-Myc, promotes the progression of gastric carcinoma. J Cancer Res Clin Oncol 139(3):437–445. doi:10.1007/s00432-012-1324-x
Yang F, Xue X, Zheng L, Bi J, Zhou Y, Zhi K, Gu Y, Fang G (2014) Long non-coding RNA GHET1 promotes gastric carcinoma cell proliferation by increasing c-Myc mRNA stability. FEBS J 281(3):802–813. doi:10.1111/febs.12625
Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT, Zhou CC, Wang SB, Wang YZ, Yang Y, Yang N, Zhou WP, Yang GS, Sun SH (2014) A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 25(5):666–681. doi:10.1016/j.ccr.2014.03.010
Acknowledgments
This work was supported by National Science Foundation of China (Nos. 81201888, 81372549 and No. 81172370), Specialized Research Fund for the Doctoral Program of Higher Education (No. 20122104110009) and Natural Science Foundation of Liaoning Province (No. 2014029201), Program of Education Department of Liaoning Province (L2014307).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical standards
This study was conducted according to the principles expressed in the Declaration of Helsinki. Tissue specimens were collected after obtaining the informed consent of patients in accordance with institutional ethical guidelines, which were reviewed and approved by the Research Ethics Committee of China Medical University (Shenyang, China).
Additional information
Jun-hua Zhao and Jing-xu Sun have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
The effect of lncRNA-LOWEG on cell proliferation and the cell cycle. Overexpression of lncRNA-LOWEG did not cause significant differences in cell proliferation and cell cycle detection. (A) Twenty-four hours after transfection, cells were plated in 96-well plates. After 24, 48, 72 and 96 h, cell proliferation was measured using a CCK-8 assay. Data are presented as mean ± SD. Experiments were independently conducted three times to obtain the presented data. (B) Representative results of a cell cycle assay by flow cytometry 48 h after transfection. (C) Statistical results are based on four independent experiments (Mean ± SD) of cell cycle detection. (TIFF 4871 kb)
Rights and permissions
About this article

Cite this article
Zhao, Jh., Sun, Jx., Song, Yx. et al. A novel long noncoding RNA-LOWEG is low expressed in gastric cancer and acts as a tumor suppressor by inhibiting cell invasion. J Cancer Res Clin Oncol 142, 601–609 (2016). https://doi.org/10.1007/s00432-015-2071-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-015-2071-6