
Abstract
We report on a 6-year-old boy with nephrotic syndrome (NS) who developed Wernicke’s encephalopathy (WE) concomitantly with posterior reversible encephalopathy syndrome (PRES). In this case, the recurrence of encephalopathy with different causes made his clinical picture complex, and the follow-up findings of magnetic resonance imaging (MRI) were critically useful for the adequate diagnosis and timely management of the patient. This case suggests the need to consider WE as a possible serious complication in patients with NS, and also emphasizes the usefulness of MRI in the diagnosis of WE, especially in pediatric cases with complex clinical symptoms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wernicke’s encephalopathy (WE) is a serious neurological disorder characterized by ophthalmoplegia, ataxia, and confusion of acute or subacute onset. WE is caused by the deficiency of thiamine, a cofactor of several metabolic enzymes, which is sufficiently stored in the body for up to only 18 days [10]. Although WE historically and most commonly is associated with alcoholism, non-alcoholic WE has increasingly been described in conditions predisposing to poor nutrition [9]. On the other hand, reports of WE in infants and children are still rare, and the majority of pediatric cases were associated with malignancy and gastrointestinal disease [11]. We here report a case of WE that concomitantly occurred with posterior reversible encephalopathy syndrome (PRES) in a child with nephrotic syndrome (NS). This is the first report of WE that occurred in association with NS.
Case report
A 6-year-old boy, who had developed NS when he was 4 years of age, was admitted because of the relapse of NS. He had relapses of NS soon after the onset, under high-dose administration of oral corticosteroid, and after being treated repeatedly with intravenous pulse methylprednisolone during his first hospitalization, He had been kept in remission of NS under treatment with low-dose oral corticosteroid with concomitant use of cyclosporine and mycophenolate mofetil. Renal histological examination at the time of onset revealed minor glomerular abnormalities.
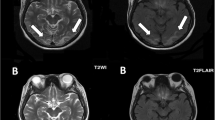
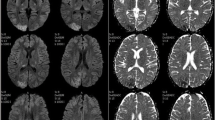
After admission with the relapse of NS, he was treated with intravenous methylprednisolone again, which was ineffective this time. He was administered tacrolimus and cyclosporine was stopped. Proteinuria decreased gradually thereafter; however, he developed herpes zoster rash two weeks after starting tacrolimus, and proteinuria began to increase once again. On day 27 after starting tacrolimus, he suddenly developed a generalized tonic convulsion. Computed tomography (CT) of the head revealed no abnormal findings at that time; however, magnetic resonance imaging (MRI), which was performed on the next day, showed multiple lesions with abnormal signals in the bilateral parietal and occipital regions (Fig. 1). PRES associated with the administration of tacrolimus was suspected, and tacrolimus was stopped. He soon became conscious and his convulsion was well-controlled with the administration of phenobarbital. However, massive proteinuria (30–50 g/day) continued, to which treatments with the intravenous administration of rituximab and plasmapheresis were ineffective. He became severely anasarcous, and suffered from serious nausea and recurrent vomiting. One month after his first convulsion, nystagmus and tremor in his upper limb and lip appeared and he became drowsy. Magnetic resonance (MR) examination, which was performed after two days of the onset of these symptoms, revealed newly appeared symmetrical, abnormal, high-intensity signals in the bilateral head of caudate nuclei, putamen, and medial thalami on diffusion-weighted imaging (DWI), and also in mamillary bodies on fluid-attenuated inversion recovery (FLAIR) sequence, although the abnormal signals that appeared in the parietal and occipital regions one month before were fading (Fig. 2). A diagnosis of WE was suspected on the basis of his clinical triad and the MRI lesions in the medial thalami and mamillary bodies. He was managed with intravenous total parenteral nutrition, and the daily administration of thiamine (one dose of 100 mg intravenous thiamine per day) was started. His nystagmus and tremor disappeared within a few days, and soon he became conscious. However, after a short duration of pyretic episode, he developed disseminated intravascular coagulation (DIC) syndrome of incomplete type. He also became hypertensive (blood pressure 170/90 mmHg), and 6 days after the second MR examination, he again became drowsy and developed a generalized convulsion. MR examination again revealed abnormal signals, suggesting the recurrence of PRES extensively in the bilateral occipital, parietal, temporal, and also in the frontal regions of the brain, although abnormal signals in the bilateral basal nuclei, medial thalami, and mamillary bodies that were observed in the previous examination were improving (Fig. 3). Following this episode, he recovered from DIC, his blood pressure was well controlled, and he became conscious within 10 days. His proteinuria began to decrease thereafter, and MR examination, which was performed 2 months later, showed improvements in all of the abnormal high-intensity signals seen previously, with improvement in his nephrotic status, although small high-intensity signals in T1-weighted (T1W) images still persisted. At present, 10 months after the last convulsion, his proteinuria has decreased to 1–3 g/day and he has no neurological symptoms.

Axial fluid-attenuated inversion recovery (FLAIR) (TE 100.0 ms/TR 8,000 ms/IR) (a) and diffusion-weighted imaging (DWI) (b) images at the level of basal ganglia on the first episode of encephalopathy, showing multiple lesions with abnormal signals in the bilateral occipital regions (arrows)

Axial FLAIR (TE 100.0 ms/TR 8000 ms/IR) images showing newly appeared symmetrical high-intensity signals in the bilateral medial thalami (a), bilateral head of caudate nuclei and putamen (b), and bilateral mamillary bodies (c) on the second episode of encephalopathy. The DWI image also showed abnormal high-intensity signals in the bilateral basal ganglia and medial thalami (d) (arrows)

Axial FLAIR (TE 100.0 ms/TR 8000 ms/IR) (a) and DWI (b) images showing the recurrence of abnormal signals in the bilateral occipital and temporal regions on the third episode of encephalopathy (arrows), although abnormal signals in the basal ganglia and medial thalami that were observed in the previous examination were fading
Discussion
Although WE has traditionally been observed in chronic malnourished alcoholics, WE may also occur in any clinical setting in which thiamine is not ingested or absorbed in adequate amounts. WE is rare in children, but has been reported in the context of chemotherapy, gastrointestinal disease, self-starvation [5, 6], prolonged intermittent vomiting [7], herpesvirus encephalitis [2], defective soy-based formula [4], and anorexia nervosa [9]. However, we can, thus far, find no literature on the PubMed database with the keywords “nephrotic syndrome” and “Wernicke’s encephalopathy”.
The clinical triad of WE is ophthalmoplegia, ataxia, and global confusional state. However, in a case series by Vasconcelos et al. [11], 13 of 31 (41.9%) pediatric patients with WE did not present with this classical triad, and were not diagnosed before death, suggesting the underdiagnosis of this clinical entity, especially in the pediatric population. In this context, several previous reports emphasized the usefulness of MR examination in the diagnosis of WE with incomplete clinical symptoms [3, 10, 12]. Also in our case, although the clinical picture was complex with various pathological factors, including nephrotic status, MR examinations of the brain were useful for the adequate diagnosis and timely management of the patient.
In regard to the MRI findings of WE, previous reports showed the diagnostic importance of symmetrical signal hyperintensity on DWI beneath standardized T2W imaging, FLAIR, and also enhancement on postcontrast T1W images within the posteromedial thalami, periventricular regions of the third ventricle, and the mamillary bodies [3, 10, 12]. DWI suggested that these hyperintense lesions on T2W imaging of the acute changes of WE include reversible vasogenic edema and are not caused by acute ischemia [8]. The pathophysiology of WE is not completely understood. The phosphoric esters of thiamine are involved in the function of excitable membranes, glucose metabolism, and neurotransmitter production. Because thiamine is important in maintaining osmotic gradients across cell membranes, thiamine deficiency leads to intracellular and extracellular edema [6]. Furthermore, in periventricular regions, the blood–brain barrier, where thiamine transport occurs through passive and active mechanisms, is defective and there is a high rate of thiamine-related glucose and oxidative metabolism [1]. This could explain the selective involvement in these regions. In our patient, abnormal symmetrical high-intensity signals were observed in the bilateral medial thalami and mamillary bodies, as well as in the basal nuclei on T2W imaging, FLAIR, and DWI. Although differential diagnosis such as newly appeared PRES, ischemic lesions, and viral encephalitis should have been considered, metabolic disorders such as WE were highly suspected on the basis of these MRI findings. Furthermore, these abnormal signals on MRI were faded in our case, along with the rapid improvement of his clinical triad following the administration of thiamine, which was consistent with the diagnosis of WE.
WE seems to be underdiagnosed in the pediatric population; however, WE can be a potentially fatal disorder if a prompt diagnosis and timely treatment are not provided. Our case suggests the need to consider WE as, although rare, a possible serious complication in patients with NS, and also emphasizes the usefulness of MRI in the diagnosis of WE, especially in pediatric cases with complex clinical symptoms.
References
Bae S-J, Lee HK, Lee J-H et al (2001) Wernicke’s encephalopathy: atypical manifestation at MR imaging. AJNR Am J Neuroradiol 22:1480–1482
Carvajal E, Verdeguer A, Fernández J-M et al (2001) Herpesvirus-6 encephalitis complicated by Wernicke-Korsakoff syndrome in a pediatric recipient of unrelated cord blood transplantation. J Pediatr Hematol Oncol 23:626–628. doi:10.1097/00043426-200112000-00016
Coe M, Carfagnini F, Tani G et al (2001) Wernicke’s encephalopathy in a child: case report and MR findings. Pediatr Radiol 31:167–168. doi:10.1007/s002470000396
Fattal-Valevski A, Kesler A, Sela B-A et al (2005) Outbreak of life-threatening thiamine deficiency in infants in Israel caused by a defective soy-based formula. Pediatrics 115:233–238. doi:10.1542/peds.2004-1255
Gropman AL, Gaillard WD, Campbell P et al (1998) Wernicke’s encephalopathy due to self starvation in a child. Lancet 351:1704–1705. doi:10.1016/S0140-6736(05)77742-4
Harter SB, Nokes SR (1995) Gadolinium-enhanced MR findings in a pediatric case of wernicke encephalopathy. AJNR Am J Neuroradiol 16:700–702
Ming X, Wang MM, Zee D et al (1998) Wernicke’s encephalopathy in a child with prolonged vomiting. J Child Neurol 13:187–189
Oka M, Terae S, Kobayashi R et al (2001) Diffusion-weighted MR findings in a reversible case of acute Wernicke encephalopathy. Acta Neurol Scand 104:178–181. doi:10.1034/j.1600-0404.2001.00098.x
Peters TE, Parvin M, Petersen C et al (2007) A case report of Wernicke’s encephalopathy in a pediatric patient with anorexia nervosa—restricting type. J Adolesc Health 40:376–383. doi:10.1016/j.jadohealth.2006.11.140
Unlu E, Cakir B, Asil T (2006) MRI findings of Wernicke encephalopathy revisited due to hunger strike. Eur J Radiol 57:43–53. doi:10.1016/j.ejrad.2005.07.002
Vasconcelos MM, Silva KP, Vidal G et al (1999) Early diagnosis of pediatric Wernicke’s encephalopathy. Pediatr Neurol 20:289–294. doi:10.1016/S0887-8994(98)00153-2
Weidauer S, Nichtweiss M, Lanfermann H et al (2003) Wernicke encephalopathy: MR findings and clinical presentation. Eur Radiol 13:1001–1009
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nishida, M., Sato, H., Kobayashi, N. et al. Wernicke’s encephalopathy in a patient with nephrotic syndrome. Eur J Pediatr 168, 731–734 (2009). https://doi.org/10.1007/s00431-008-0833-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-008-0833-8