
Abstract
Control of murine cytomegalovirus (mCMV) infection is mediated primarily by CD8 T cells, with four specificities dominating in BALB/c mice. Functional deletion of the respective immunodominant epitopes (IDEs) in mutant virus Δ4IDE revealed a still efficient control of infection. In a murine model of hematopoietic cell transplantation and infection with Δ4IDE, an mCMV-specific open reading frame (ORF) library screening assay indicated a strong CD8 T cell reactivity against the ORF-M54 product, the highly conserved and essential mCMV homolog of human CMV DNA polymerase UL54, which is a known inducer of in vivo protection against mCMV by DNA immunization. Applying bioinformatic algorithms for CD8 T cell epitope prediction, the top-scoring peptides were used to stimulate ex vivo-isolated CD8 T cells and to generate cytolytic T cell lines; yet, this approach failed to identify M54 epitope(s). As an alternative, a peptide library consisting of 549 10-mers with an offset of two amino acids (aa), covering the complete aa-sequence of the M54 protein, was synthesized and used for the stimulation. A region of 12 aa proved to encompass an epitope. An ‘alanine walk’ over this antigenic 12-mer and all possible 11-, 10- and 9-mers derived thereof revealed aa-residues critical for antigenicity, and terminal truncations identified the H-2Dd presented 8-mer M5483–90 as the optimal epitope. An increased frequency of the corresponding CD8 T cells in the absence of the 4 IDEs indicated immunodomination by the IDE-specific CD8 T cells as a mechanism by which the generation of M54-specific CD8 T cells is inhibited after infection with wild-type mCMV.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A major characteristic of cytomegaloviruses (CMV), of herpesviruses in general, is the establishment of latency. Worldwide, 50–100 % of the adult population are latently infected with human CMV (hCMV) that causes severe medical problems in the immunosuppressed [1, 2] or immunologically immature host [1, 3, 4]. Despite the morbidity and mortality associated with hCMV infections and the obvious need for a vaccine, and even though a number of candidate vaccines have been already tested clinically ([5], see also the article by S. Plotkin in this issue of MMI), there is no effective hCMV vaccine applicable to date.
CD8 T cells are the major effector cells for control of CMV infections, which makes them important targets for the development of T cell-based vaccines. In experimental adoptive transfer studies in a mouse model that mirrors the clinical situation of immunosuppressed patients with concurrent CMV infection [6], we have thoroughly studied the CD8 T cell-mediated control of murine CMV (mCMV) infection. We could show that most of the currently known CD8 T cell specificities defined in haplotype H-2d are highly protective in vivo [7–9], but exceptions exist in haplotypes H-2d [7] as well as H-2b [10]. In general, the protective potential of defined specificities, applied as cytolytic T cell (CTL) lines (CTLL), proved to be independent of their frequency in vivo, meaning that there is no correlation between the position in the immunodominance hierarchy and the efficacy in controlling infection (discussed in [7, 8]).
In search for a novel class of CD8 T cell targets for vaccine-mediated protection, essential non-structural proteins that are highly conserved among different CMV species were selected as potential candidates by D. H. Spector’s group [11]. Indeed, DNA immunization of mice with the mCMV homolog M54 of hCMV DNA polymerase UL54 was found to be protective in BALB/c (haplotype H-2d) mice [11]. Remarkably, immunization with the M54 DNA provided a high level of protection, similar to that elicited by the IE1 DNA coding for the first immunodominant CD8 T cell epitope identified for mCMV [12].
Here, we describe the identification of an M54-encoded CD8 T cell epitope whose existence was predicted by the DNA vaccination studies mentioned above. For this, we first applied bioinformatic algorithms that list and rank possible CD8 T cell epitope candidates. As we did not succeed with this approach, we used an overlapping peptide library spanning the whole M54 protein, and this eventually led to the identification of an atypical CD8 T cell epitope not disclosed by the currently available algorithms. Furthermore, we can show a strong increase in the frequency of cognate CD8 T cells in the absence of the four immunodominant CD8 T cell epitopes (IDEs) M105, m123/IE1, m145, and m164. These data support the conclusion of the M54 specificity being a promising candidate for implementation in an effective vaccine.
Materials and methods
mCMV mutant virus Δ4IDE-mut and generation of revertant virus Δ4IDE-rev
mCMV Δ4IDE-mut
The construction of this bacterial artificial chromosome (BAC)-derived mutant has been published previously [13], and high-titer stocks were prepared by sucrose gradient purification as described [14].
Shuttle plasmid for reverse mutation
The shuttle plasmid pST76K-M105 was constructed as follows. BAC plasmid C3X [15] was used as template for PCR using primer pair M105_EcoRV_for (5′-atatgatatcgccctggttcgtcacgttcc-3′) and M105_SphI_rev (5′-atatgcatgccagaccgatgccctgactcttg-3′). The 4056-bp product was subcloned into pDrive by means of UA-based ligation (Qiagen, Hilden Germany) leading to the intermediate construct pdrive-M105. This construct was cleaved with DraI and SphI, and the resulting 4090-bp fragment was inserted into the SmaI-cleaved shuttle vector pst76-KSR [16].
Generation of mCMV Δ4IDE-rev
Revertant virus Δ4IDE-rev was generated starting from the parental BAC plasmid C3X-IE1Ala + m164Ala + m145Ala + M105Ala [13]. Markerless BAC mutagenesis [17] was performed to first revert the single aa-replacement at the C-terminal MHC-I anchor residue of the antigenic peptide m145 451CYYASRTKL459 A459L as described [13]. For nested PCR, the following primer pairs were used: m145-A-L_for_long (5′-gcgcgagccttttcgatgtcgagttctcccgagtattttgtgcggctcgcgtaatagaggatgacgacgataagtaggg-3′)/pEPKAN-S_rev (5′-aggatgacgacgataagtaggg-3′) and m145-A-L_long_rev (5′-atggtattttgttattattgctattacgcgagccgcacaaaataccgttcgggagaactccaaccaattaaccaattctgattag-3′)/m145-A-L_short_fwd (5′-gcgcgagccttttcgatgtc-3′). The PCR fragment was inserted into C3X-IE1Ala + m164Ala + m145Ala + M105Ala by RED recombination, and subsequently, the KanR cassette was removed resulting in a markerless insertion of codon ‘tac’ at positions 204,120–204,118 [18]. The resulting BAC plasmid C3X-IE1Ala + m164Ala + m105Ala was subcloned into E. coli strain DH10b and used as a template for the subsequent reversions of the single aa-replacements in the mutated peptide sequences m164 257AGPPRYSRA265, IE1 168YPHFMPTNA176, and M105 207TYWPVVSDA215 that resulted in A265I, A176L, and A215I, respectively. For all replacements, two-step BAC mutagenesis [19] with shuttle plasmids pST76K-m164Ile [20], pST76KIE1Leu [21], and pST76K-M105 (see above) was performed, resulting in the BAC plasmid C3X-Δ4IDE-rev. Correctness of codon replacements was verified by restriction pattern analysis and subsequent sequencing (GATC, Freiburg, Germany). For reconstitution of recombinant virus mCMV Δ4IDE-rev, BAC plasmid C3X-Δ4IDErev DNA was transfected into primary mouse embryo fibroblasts (MEF), followed by several rounds of propagation in MEF until the elimination of BAC vector sequences could be verified by PCR as described in detail previously [22]. A verified BAC vector-free virus clone was used to prepare high-titer stocks of sucrose gradient-purified revertant virus Δ4IDE-rev [22].
Infection of mice
Adult, 8-to-10-week-old female BALB/cJ mice (haplotype H-2d) were infected with cell culture-purified, BAC-derived mCMV MW97.01 [15], here referred to as mCMV-wild-type (WT), or with mCMV mutant Δ4IDE-mut or revertant Δ4IDE-rev (see above). Infection was performed either intraperitoneally (i.p.) with 105 PFU/500 µl PBS or subcutaneously with 105 PFU/25 µl PBS at the left hind footpad (intrafootpad, i.f.; intraplantar infection) as specified in the figure legends. Mice were bred and housed under specified pathogen-free conditions in the Central Laboratory Animal Facility (CLAF) at the University Medical Center of the Johannes Gutenberg-University, Mainz. Animal experiments were approved according to German federal law, permission numbers 177-07/G09-1-004 and 177-07/G 14-1-015.
Synthetic peptides
All peptides used in this work were synthesized by JPT Peptide Technologies (Berlin, Germany), and purified peptides (see below) were provided with a purity of >80 %.
Purified peptides
Synthetic peptides m18346–354 (SGPSRGRII), M5483–90 (RGPYSDEL), M5483–91 (RGPYSDELR), M5483–92 (RGPYSDELRF), M105207–215 (TYWPVVSDI), IE1168–176 (YPHFMPTNL), m145451–459 (CYYASRTKL), and m164257–265 (AGPPRYSRI) were used for exogenous loading of P815 mastocytoma cells as target cells in the ELISpot assay, or of P815 cells and L-cell transfectants in the cytotoxicity assay (see below). Peptide loading concentrations are specified in the figure legends.
M54-peptide library
The PepTrackTM, Fast Track microscale peptide library, covering the complete aa-sequence of the mCMV protein M54, consisted of 549 unpurified 10-mer peptides, each with an amine at the N-terminus and an individual aa-residue at the C-terminus. Peptides were delivered freeze-dried in 96-well round-bottom plates (50–100 nmol each). Lyophilizates were resolved in 5 µl DMSO 100 % (v/v) per well and diluted with 95 µl PBS resulting in an approximate concentration of 5 × 10−4 M of each peptide. A concentration of 1 × 10−5 M for usage in the ELISpot assay (see below) was achieved by further dilution with PBS in polypropylene tubes. For the ELISpot assay with duplicate assay cultures, P815 target cells were added to 20 µl of 1 × 10−5 M of each peptide, resulting in a final loading concentration of 1 × 10−6 M.
Alanine (Ala)-peptide library
The PepTrackTM, Fast Track microscale Ala-peptide library, comprised of peptides with single Ala replacements at all aa-positions, was synthesized for the antigenic 12-mer M5481–92 and all possible 11-, 10- and 9-mers derived thereof, of ten candidate peptides altogether (Fig. 3b). Conditions of synthesis and delivery were the same as for the M54-peptide library with a total peptide amount of ca. 20 nmol per peptide. Lyophilizates were resolved and diluted as described for the M54-peptide library. In contrast to the M54-peptide library, the loading concentration of P815 target cells for usage in the ELISpot assay was reduced to 1 × 10−8 M, as higher Ala-peptide concentrations obliterated the differences in antigenicity.
Generation of CTLL and in vitro cytotoxicity assay
A polyclonal CTLL specific for peptide M5483–90 RGPYSDEL was generated by repetitive stimulation of spleen cells with the purified synthetic peptide as described [22]. Specifically, memory spleen cells from Δ4IDE-mut-infected mice were repeatedly stimulated with peptide M5483–90 at a concentration of 10−10 M with P815/B7 as feeder cells. Cytolytic activity was measured in a standard 4-h 51Cr-release assay with 1000 51Cr-labeled target cells at an effector-to-target cell ratio (E:T) of 20:1. Target cells were P815 (haplotype H-2d) coexpressing the major histocompatibility complex (MHC) class I (MHC-I) molecules Ld, Dd, and Kd, or the L-cell transfectants L-Ld, L-Dd, and L-Kd expressing the respective MHC-I molecules selectively. The parental fibroblast cell line Ltk− (ATCC CCL-1.3™, haplotype H-2k), negative for H-2d MHC-I molecules, served as a specificity control. Target cells were exogenously loaded with peptide M5483–90 in the graded molar concentrations indicated, and cytolytic activity was measured in three replicate cultures.
ELISpot assay
Frequencies and functional avidities of peptide-specific CD8 T cells were determined by an IFN-γ ELISpot assay essentially as described previously ([23, 24] and references therein). In brief, graded numbers of immunomagnetically purified CD8 T cells from the spleens were sensitized in duplicate or triplicate assay cultures by incubation with P815 cells as stimulator cells that were exogenously loaded with synthetic antigenic peptides at the molar concentrations indicated. A list of all currently known mCMV-specific MHC class I-restricted antigenic peptides in haplotype H-2d can be found in review articles [8, 9]. The frequencies of responding cells were calculated by intercept-free linear regression analysis. Spots were counted automatically based on standardized criteria using ImmunoSpot S4 Pro Analyzer (CTL Shaker Heights, OH, USA) and CTL-ImmunoSpot software V5.1.36.
Viral genome-wide ORF library screening
An mCMV-ORF library of expression plasmids [25] comprising all ORFs defined by Rawlinson et al. [18] was employed for ORF-specific stimulation of pulmonary mononuclear leukocytes [9] that were isolated as described [14, 26]. For this, SV40-transformed BALB/c fibroblasts were transfected with the individual mCMV-ORF expression plasmids, and sensitized, IFN-γ-expressing CD8 T cells were quantitated by cytofluorometric analysis (method described in greater detail in [22]). A list allocating library numbers to annotated ORFs can be found as supplemental material in Ref. [20].
Bioinformatic algorithms for prediction of CD8 T cell epitopes
For the prediction of CD8 T cell epitopes within mCMV protein M54, we used the following computational programs: MHC-Pathway.net (http://www.mhc-pathway.net), SYFPEITHI (http://www.syfpeithi.de), RANKPEP (http://imed.med.ucm.es/Tools/rankpep.html), and the IEDB analysis resource Consensus tool [27] that combines predictions from ANN aka NetMHC (3.4) [28, 29], SMM [30], and Comblib [31] (http://tools.immuneepitope.org/main).
Results and discussion
ORF-M54 codes for an antigenic CD8 T cell peptide
In search of a novel class of CMV antigens that (1) elicit T cell-mediated protection, that (2) are highly conserved among different CMV species, and that (3) are essential for virus replication, the group of D. H. Spector identified M54 DNA of mCMV to induce protective immunity against systemic challenge when used as an expression plasmid for DNA vaccination [11]. Yet, the corresponding T cell epitope remained unknown.
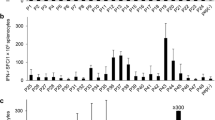
Genome-wide antigenicity testings of all mCMV-ORFs performed by us in the past (reviewed in [9]) did not give us any hint of an M54-derived peptide that stimulates CD8 T cells from mCMV-WT-infected mice, neither in the acute phase of infection [9, 13, 32, 33] nor in the memory phase [32]. Even after experimental hematopoietic cell transplantation (HCT) in the mouse model (reviewed in [9]), conditions under which no alternative immune control mechanisms besides CD8 T cells can mediate protection, thus rendering survival completely dependent on the reconstitution of CD8 T cells [34, 35], no reactivity against an ORF-M54-encoded peptide was detectable (Fig. 1a). However, by analyzing the mCMV-specific CD8 T cell repertoire in the absence of the 4 IDEs in the HCT model, an M54 reactivity became visible and was actually found to dominate the antiviral CD8 T cell response (Fig. 1b). This reactivity could be attributed to at least one CD8 T cell specificity, but stimulation by more than one M54-peptide could not be excluded at this stage.

mCMV-specific CD8 T cell immunome in the presence (a) or absence (b) of the immunodominant CD8 T cell epitopes (IDEs). ORF library screening was performed to analyze the antiviral specificity repertoire at 4 weeks after syngeneic HCT and i.f. infection, either with mCMV-WT (a) or with mutant virus mCMV-Δ4IDE (b). Stimulation of CD8 T cells within pulmonary mononuclear leukocytes by ORF-transfected cells was monitored by cytofluorometric analysis of intracellular IFN-γ. Arrows point to the positions of those ORFs that code for the 4 IDEs deleted in the corresponding mutant virus Δ4IDE. The library screening shown in (a) is taken from an own previous publication [9] with permission by Caister Academic Press, Norfolk, UK
These data strongly corroborated the DNA vaccination studies, performed previously by D. H. Spector’s group, having predicted the existence of at least one CD8 T cell epitope within the mCMV protein M54 [11]. Furthermore, our observation that in the presence of the immunodominant CD8 T cell specificities no M54 reactivity was detectable regardless of the time after infection and of the particular infection model used, is in line with the results obtained by Morello et al. [11] who reported frequencies of M54-specific CD8 T cells close to background levels after primary mCMV infection. Nonetheless, for the M54-based DNA vaccine to be protective, M54 epitope(s) were apparently presented by professional antigen-presenting cells in the absence of the IDEs [13] as well as in the absence of the viral immune evasion molecules [36, 37] in order to prime and expand the corresponding CD8 T cells. For an understanding, it is important to call to mind that priming by M54-plasmid vaccination and priming by infection with the IDE deletion mutant have in common that immunodominance hierarchies, and thus immunodomination, are avoided. On the other hand, upon challenge infection in the vaccination study mentioned above, the M54 epitope(s) must have been efficiently presented by infected tissue cells even in the presence of both, IDEs [13] as well as immune evasion molecules [38]. This implies that IDE-mediated immunodomination operates during CD8 T cell priming and/or subsequent clonal expansion, but does not affect antigen presentation by infected cells in the antiviral effector phase.
M54-peptide library screening resulted in two candidate peptides
In a first attempt to identify the M54-encoded CD8 T cell epitope(s) indicated by the ORF library screening assay, we applied bioinformatic algorithms searching for top-scoring MHC-I-binding peptides [39], an approach we have already utilized successfully for the identification of 8 CD8 T cell epitopes of mCMV in haplotype H-2d (reviewed in [9]). The predicted epitopes were synthesized as peptides but failed in stimulating ex vivo-isolated CD8 T cells for IFN-γ production and in generating CTLL (data not shown). Therefore, we decided to utilize a library of overlapping peptides covering the complete aa-sequence of the M54 protein (Fig. 2a). The advantage of this approach is that no preselection of peptides is performed on the basis of prediction algorithms. Such an unbiased library reduces the risk of missing an antigenic peptide, in particular when it does not fit to the consensus motif of a defined MHC molecule or to predictions of epitope length, proteasomal cleavage sites, or efficiency of transporter associated with antigen processing (TAP) transport from the cytosol into the ER.

M54-peptide library screening. a A peptide library of mCMV protein M54 was synthesized, consisting of 549 10-mers with an offset of two amino acids (aa). b One week after i.p. infection of BALB/c mice with mutant virus Δ4IDE, CD8 T cells from the spleens were immunomagnetically enriched and stimulated with P815 mastocytoma cells exogenously loaded with every single M54-peptide in duplicates in a final concentration of 10−6 M in the ELISpot assay. Peptides stimulating a significant number of effector CD8 T cells for IFN-γ production are marked with the corresponding peptide sequence. These peptides are highlighted in (a) by larger font and in bold face (and shown in red in the online version). Note that the order of peptides in the library is based on the schema of peptide synthesis and does not reflect their consecutive order within the protein. Part of the data was included in a technical note [45]
The M54-peptide library consisting of 549 10-mers with an overlap of 8 aa was used to stimulate CD8 T cells derived from Δ4IDE-infected mice. This screening resulted in three candidate peptides activating a significant number of CD8 T cells (Fig. 2b): M5481–90, M5483–92, and M54375–384. Two of them were consecutive peptides with an overlap of 8 aa, making it highly probable that an antigenic peptide covered by the corresponding region of 12 aa, namely M5481–92, was the actually stimulating peptide. Using computational programs for CD8 T cell epitope prediction, we chose three top-scoring 9-mers contained in the 12-mer M5481–92, and two top-scoring 9-mers contained in the 10-mer M54375–384. CD8 T cells specific for the M54375–384-derived peptides could not be detected in ex vivo ELISpot assays, whereas antigenicity from within the 12-mer M5481–92 was confirmed (data not shown).
The 10-mer M5483–92 represents a strong CD8 T cell epitope
To confine the optimal M54 epitope, as defined by stimulating the highest number of CD8 T cells primed in vivo, the antigenic 12-mer M5481–92 and all possible 9-, 10- and 11-mers derived thereof, 10 peptides altogether, were synthesized. Stimulating CD8 T cells with these peptides indicated the 10-mer M5483–92 RGPYSDELRF to be the peptide with the highest antigenicity (Fig. 3a). Notably, in accordance with the initial observation made with the ORF library screening assays (recall Fig. 1), mutant virus Δ4IDE stimulated a higher number of M54-specific CD8 T cells in vivo compared to the wild-type virus (Fig. 3a, compare right and left panels).

M54 Ala-peptide library screening. One week after i.p. infection with mCMV-WT (a, left diagram) or mutant virus Δ4IDE (a, right diagram, b, c), CD8 T cells from the spleens were immunomagnetically enriched and stimulated separately with the 12-mer M5481–92 and all possible 11-, 10-, and 9-mers derived thereof (a), or were stimulated with the Ala-peptide library (b). For this, P815 target cells in duplicate assay cultures were exogenously loaded with each peptide at a final concentration of 10−8 M. Frequencies of responding effector CD8 T cells, shown as bars, were determined in the IFN-γ-based ELISpot assay. The table (c) summarizes the significance of every single aa in the respective peptide as determined by the Ala replacement shown in (b). The arrows point to the data derived from stimulation with the 10-mer RGPYSDELRF (a) or from stimulation with the Ala peptides derived thereof (b + c). Part of the data was included in a technical note [45]
An ‘Ala-walk’ performed for all ten candidate peptides disclosed the functional significance of every single aa in the respective peptide (Fig. 3b). Replacement of a single aa—provided it is essential for binding to the MHC molecule or for recognition by the T cell receptor—with Ala results in a reduction or abrogation of antigenicity of the respective peptide. This approach was successfully applied previously to analyze the role of individual aa of the IE1-peptide 168YPHFMPTNL176 of mCMV, identifying aa-residues as essential MHC or TCR contact residues or as spacer residues non-essential for either interaction [40]. Here, abrogation of antigenicity by Ala replacement (Fig. 3b) was consistently observed for aa83 [R], aa84 [G], aa85 [P], aa86 [Y], aa88 [D], aa89 [E], and aa90 [L] (Fig. 3c). Ala replacements at position aa81 [W] or aa82 [C] at the N-terminus, as well as at position aa91 [R] or aa92 [F] at the C-terminus, did not negatively affect the antigenicity, but either had no influence on antigenicity or even improved it (Fig. 3c).
In summary, out of the peptides considered so far, the 10-mer M5483–92 proved to be the peptide with the highest antigenicity, and the sequence spanning aa83–aa90 appeared to be the antigenic core.
The 10-mer M5483–92 does not represent the optimal epitope
In order to confirm M5483–92 being the optimal epitope, 10-mer M5483–92, 9-mer M5483–91, and 8-mer M5483–90 were tested as purified synthetic peptides. Truncation of the 10-mer was performed only at the C-terminus, as the ‘Ala-walk’ had already identified aa 83–86 as being essential, whereas Ala replacement of aa91 or aa92 even improved the antigenicity (Fig. 3c). Stimulation of CD8 T cells derived from Δ4IDE-mutant and Δ4IDE-revertant virus-infected mice with graded concentrations of the three peptides clearly revealed the 8-mer M5483–90 RGPYSDEL to be the peptide with the highest stimulatory potential (Fig. 4a). Even at the low exogenous loading concentration of 10−10 M, a significant number of CD8 T cells were activated for IFN-γ secretion, whereas ~100-fold and ~1000-fold concentrations were required with the 10- and the 9-mers, respectively. The impressive increase in CD8 T cell frequency in the absence of the 4 IDEs paralleled—and thus confirmed—our findings with the ORF library and the peptide libraries (recall Figs. 1 and 3, respectively). Furthermore, this gain in frequency coincided with a strong increase in the number of high-avidity M5483–90 RGPYSDEL peptide-specific CD8 T cells (Fig. 4a) as defined by detection at the relatively low peptide concentration of 10−9 M. As we have shown previously that higher avidity implies higher protective potential [7, 41], we conclude from the avidity distribution that the protective potential of the population of 8-mer epitope-specific CD8 T cells is higher when they are primed in the absence of IDE-mediated immunodomination, not only because of a higher total cell number but also, in particular, because of an increased number of cells with high avidity. As a DNA plasmid vaccine can avoid immunodomination, enhanced CD8 T cell avidity further argues in favor of an experimental DNA plasmid vaccine based on M54 for mCMV [11] or, in translation, on UL54 for hCMV.

Identification and MHC class I restriction of the optimal M54-peptide. a For verification of the optimal CD8 T cell epitope, the 10-mer M5483–92 and the corresponding C-terminally truncated 9-mer M5483–91 and 8-mer M5483–90 were exogenously loaded in triplicates as purified synthetic peptides on P815 target cells in the graded molar concentrations indicated. Stimulation of CD8 T cells, immunomagnetically enriched from the spleens of Δ4IDE-mut and Δ4IDE-rev infected mice at 1 week after i.p. infection, was monitored in the IFN-γ-based ELISpot assay. Bars represent most probable numbers determined by intercept-free linear regression analysis. Error bars indicate 95 % confidence intervals. b For analysis of the presenting MHC-I molecule, an M5483–90-specific CTLL was stimulated with the corresponding synthetic peptide exogenously loaded on radioactively labeled P815 target cells or on L-cell transfectants L-Ld, L-Dd, or L-Kd in the graded molar concentrations indicated. Data of the cytotoxicity assay represent the mean percentage of specific lysis from three replicate cultures
The optimal epitope M5483–90 is presented by the MHC-I molecule H-2 Dd
Generation of an M5483–90-specific CTLL using a peptide concentration of 10−10 M for repetitive restimulation resulted in a sensitive CTLL with a detection limit of approximately 10−11 M measured in a cytotoxicity assay (Fig. 4b). M5483–90-peptide loading on L-cell transfectants, selectively expressing the MHC-I molecules Ld, Dd, or Kd, identified Dd as the peptide-presenting molecule. This is unusual, as Dd-presented peptides are supposed to be 9-mers with a strict requirement of the rather infrequently occurring binding motif xGPxxxxxI,L,F [39, 42]. Although the 8-mer peptide M5483–90 RGPYSDEL does not perfectly fit to the Dd motif, it fulfills the condition at its N-terminal part xGP and position eight at least shares a hydrophobic residue with position 9 of the more conventional Dd-presented peptides. In general, Dd-presented 8-mers were rarely found [39, 42], so that M5483–90 is an atypical CD8 T cell epitope, which explains why such an effort was needed to find it.
Concluding remarks
Our data unraveled the suggested CD8 T cell epitope encoded by mCMV-ORF-M54. The hCMV counterpart UL54 codes for the essential DNA polymerase, and its sequence is highly conserved between different CMV species, thus qualifying it as a promising CD8 T cell target for inclusion in a vaccine [11].
The remarkable expansion of M54-peptide-specific CD8 T cells in the absence of the IDEs strongly supports the assumption that the low frequency detectable after mCMV-WT infection is the result of a highly efficient suppression by the IDE-specific CD8 T cells. Such an ‘immunodomination’ of subdominant specificities by dominant specificities, as defined by response quantity rather than quality, is an established factor known to influence the immunodominance hierarchy [43, 44]. With respect to the development of an efficient vaccine, the fact that an otherwise subdominant CD8 T cell specificity significantly expands in the absence of the dominant specificities, which coincides with a significant increase in the absolute numbers of high-avidity cells, indeed makes the corresponding epitope an interesting candidate for a T cell-based vaccine. The success of a DNA plasmid vaccine [11] can thus be explained, at least in part, by the absence of competing immunodominating epitopes that, when present, could stimulate high numbers of CD8 T cells, however primarily CD8 T cells of low-avidity and hence of reduced protective value [7, 20, 41].
References
Ho M (2008) The history of cytomegalovirus and its diseases. Med Microbiol Immunol 197:65–73
Seo S, Boeckh M (2013) Clinical cytomegalovirus research: hematopoietic cell transplantation. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol II, chap 16. Caister Academic Press, Norfolk, pp 337–353
Adler SP, Nigro G (2013) Clinical cytomegalovirus research: congenital infection. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol I, chap 3. Caister Academic Press, Norfolk, pp 55–73
Cannon MJ, Grosse SD, Fowler KB (2013) The epidemiology and public health impact of congenital cytomegalovirus infection. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol I, chap 2. Caister Academic Press, Norfolk, pp 26–48
Plotkin SA, Plachter B (2013) Cytomegalovirus vaccine: on the way to the future? In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol II, chap 20. Caister Academic Press, Norfolk, pp 424–449
Reddehase MJ, Weiland F, Münch K, Jonjic S, Lüske A, Koszinowski UH (1985) Interstitial murine cytomegalovirus pneumonia after irradiation: characterization of cells that limit viral replication during established infection of the lungs. J Virol 55:264–273
Ebert S, Podlech J, Gillert-Marien D, Gergely KM, Büttner JK, Fink A, Freitag K, Thomas D, Reddehase MJ, Holtappels R (2012) Parameters determining the efficacy of adoptive CD8 T-cell therapy of cytomegalovirus infection. Med Microbiol Immunol 201:527–539
Holtappels R, Böhm V, Podlech J, Reddehase MJ (2008) CD8 T-cell-based immunotherapy of cytomegalovirus infection: “proof of concept” provided by the murine model. Med Microbiol Immunol 197:125–134
Holtappels R, Ebert S, Podlech J, Fink A, Böhm V, Lemmermann NA, Freitag K, Renzaho A, Thomas D, Reddehase MJ (2013) Murine model for cytoimmuntherapy of CMV disease after hematopoietic cell transplantation. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol II, chap 17. Caister Academic Press, Norfolk, pp 354–381
Holtappels R, Podlech J, Pahl-Seibert MF, Jülch M, Thomas D, Simon CO, Wagner M, Reddehase MJ (2004) Cytomegalovirus misleads its host by priming of CD8 T cells specific for an epitope not presented in infected tissues. J Exp Med 199:131–136
Morello CS, Kelley LA, Munks MW, Hill AB, Spector DH (2007) DNA immunization using highly conserved murine cytomegalovirus genes encoding homologs of human cytomegalovirus UL54 (DNA polymerase) and UL105 (helicase) elicits strong CD8 T-cell responses and is protective against systemic challenge. J Virol 81:7766–7775
Reddehase MJ, Rothbard JB, Koszinowski UH (1989) A pentapeptide as minimal antigenic determinant for MHC class I-restricted T lymphocytes. Nature 337:651–653
Ebert S, Lemmermann NA, Thomas D, Renzaho A, Reddehase MJ, Holtappels R (2012) Immune control in the absence of immunodominant epitopes: implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med Microbiol Immunol 201:541–550
Podlech J, Holtappels R, Grzimek NKA, Reddehase MJ (2002) Animal models: murine cytomegalovirus. In: Kaufmann SHE, Kabelitz D (eds) Methods in microbiology, vol 32. Immunology of infection. Academic Press, London, pp 493–525
Wagner M, Jonjic S, Koszinowski UH, Messerle M (1999) Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J Virol 73:7056–7060
Borst EM, Pósfai G, Pogoda F, Messerle M (2004) Mutagenesis of herpesvirus BACs by allele replacement. Methods Mol Biol 256:269–279
Tischer BK, von Einem J, Kaufer B, Osterrieder N (2006) Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197
Rawlinson WD, Farrell HE, Barrell BG (1996) Analysis of the complete DNA sequence of murine cytomegalovirus. J Virol 70:8833–8849
Messerle M, Crnkovic I, Hammerschmidt W, Ziegler H, Koszinowski UH (1997) Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci USA 94:14759–14763
Holtappels R, Simon CO, Munks MW, Thomas D, Deegen P, Kühnapfel B, Däubner T, Emde SF, Podlech J, Grzimek NK, Oehrlein-Karpi SA, Hill AB, Reddehase MJ (2008) Subdominant CD8 T-cell epitopes account for protection against cytomegalovirus independent of immunodomination. J Virol 82:5781–5796
Simon CO, Holtappels R, Tervo HM, Böhm V, Däubner T, Oehrlein-Karpi SA, Kühnapfel B, Renzaho A, Strand D, Podlech J, Reddehase MJ, Grzimek NK (2006) CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J Virol 80:10436–10456
Lemmermann NA, Podlech J, Seckert CK, Kropp KA, Grzimek NK, Reddehase MJ, Holtappels R (2010) CD8 T-cell immunotherapy of cytomegalovirus disease in the murine model. In: Kabelitz D, Kaufmann SHE (eds) Methods in microbiology, vol 37. Immunology of infection. Academic Press, London, pp 369–420
Pahl-Seibert MF, Jülch M, Podlech J, Thomas D, Deegen P, Reddehase MJ, Holtappels R (2005) Highly protective in vivo function of cytomegalovirus IE1 epitope-specific memory CD8 T cells purified by T-cell receptor-based cell sorting. J Virol 79:5400–5413
Böhm V, Simon CO, Podlech J, Seckert CK, Gendig D, Deegen P, Gillert-Marien D, Lemmermann NA, Holtappels R, Reddehase MJ (2008) The immune evasion paradox: immunoevasins of murine cytomegalovirus enhance priming of CD8 T cells by preventing negative feedback regulation. J Virol 82:11637–11650
Munks MW, Gold MC, Zajac AL, Doom CM, Morello CS, Spector DH, Hill AB (2006) Genome-wide analysis reveals a highly diverse CD8 T cell response to murine cytomegalovirus. J Immunol 176:3760–3766
Ebert S, Becker M, Lemmermann NA, Büttner JK, Michel A, Taube C, Podlech J, Böhm V, Freitag K, Thomas D, Holtappels R, Reddehase MJ, Stassen M (2014) Mast cells expedite control of pulmonary murine cytomegalovirus infection by enhancing the recruitment of protective CD8 T cells to the lungs. PLoS Pathog 10:e1004100
Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, Lundegaard C, Sette A, Lund O, Bourne PE, Nielsen M, Peters B (2012) Immune epitope database analysis resource. Nucleic Acids Res 40:W525–W530
Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, Brunak S, Lund O (2003) Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 12:1007–1017
Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M (2008) NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res 36:W509–W512
Peters B, Sette A (2005) Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 6:132
Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, Sette A, Peters B (2008) Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res 4:2
Seckert CK, Schader SI, Ebert S, Thomas D, Freitag K, Renzaho A, Podlech J, Reddehase MJ, Holtappels R (2011) Antigen-presenting cells of haematopoietic origin prime cytomegalovirus-specific CD8 T-cells but are not sufficient for driving memory inflation during viral latency. J Gen Virol 92:1994–2005
Lemmermann NA, Böhm V, Holtappels R, Reddehase MJ (2011) In vivo impact of cytomegalovirus evasion of CD8 T-cell immunity: facts and thoughts based on murine models. Virus Res 157:161–174
Podlech J, Holtappels R, Wirtz N, Steffens HP, Reddehase MJ (1998) Reconstitution of CD8 T cells is essential for the prevention of multiple-organ cytomegalovirus histopathology after bone marrow transplantation. J Gen Virol 79:2099–2104
Podlech J, Holtappels R, Pahl-Seibert MF, Steffens HP, Reddehase MJ (2000) Murine model of interstitial cytomegalovirus pneumonia in syngeneic bone marrow transplantation: persistence of protective pulmonary CD8-T-cell infiltrates after clearance of acute infection. J Virol 74:7496–7507
Reddehase MJ (2002) Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat Rev Immunol 2:831–844
Lemmermann NA, Fink A, Podlech J, Ebert S, Wilhelmi V, Böhm V, Holtappels R, Reddehase MJ (2012) Murine cytomegalovirus immune evasion proteins operative in the MHC class I pathway of antigen processing and presentation: state of knowledge, revisions, and questions. Med Microbiol Immunol 201:497–512
Fink A, Lemmermann NA, Gillert-Marien D, Thomas D, Freitag K, Böhm V, Wilhelmi V, Reifenberg K, Reddehase MJ, Holtappels R (2012) Antigen presentation under the influence of ‘immune evasion’ proteins and its modulation by interferon-gamma: implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med Microbiol Immunol 201:513–525
Rammensee HG, Bachmann J, Stevanovic S (1997) MHC ligands and peptide motifs. Molecular Biology Intelligence Unit, Landes Bioscience, Austin
Reddehase MJ, Koszinowski UH (1991) Redistribution of critical major histocompatibility complex and T cell receptor-binding functions of residues in an antigenic sequence after biterminal substitution. Eur J Immunol 21:1697–1701
Nauerth M, Weißbrich B, Knall R, Franz T, Dössinger G, Bet J, Paszkiewicz PJ, Pfeifer L, Bunse M, Uckert W, Holtappels R, Gillert-Marien D, Neuenhahn M, Krackhardt A, Reddehase MJ, Riddell SR, Busch DH (2013) TCR-ligand koff rate correlates with the protective capacity of antigen-specific CD8+ T cells for adoptive transfer. Sci Transl Med 5:192ra87
Corr M, Boyd LF, Padlan EA, Margulies DH (1993) H-2Dd exploits a four residue peptide binding motif. J Exp Med 178:1877–1892
Chen W, McCluskey J (2006) Immunodominance and immunodomination: critical factors in developing effective CD8+ T-cell-based cancer vaccines. Adv Cancer Res 95:203–247
Yewdell JW, Bennink JR (1999) Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu Rev Immunol 17:51–88
Holtappels R (2014) Strategy for identification of CD8 T-cell epitopes in a viral protein. Application note Immunology, PepTrack™ Peptide Libraries. JPT Peptide Technologies, Berlin
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft, SFB 490, individual projects E3 ‘Persistence of murine cytomegalovirus after modulation of the CD8 T cell immunome’ (R.H., and D.T.) and E2 ‘Immunological control of latent cytomegalovirus infection’ (A.R., and M.J.R.). N.A.W.L. and M.J.R. were also supported by the Deutsche Forschungsgemeinschaft, Clinical Research Group KFO 183, individual project TP8 ‘Establisment of challenge models for optimizing the immunotherapy of cytomegalovirus disease’, and N.A.W.L. received intramural funding in the young investigator program MAIFOR of the University Medical Center of the Johannes Gutenberg-University, Mainz.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Special Issue on Cytomegalovirus.
Rights and permissions
About this article

Cite this article
Holtappels, R., Lemmermann, N.A.W., Thomas, D. et al. Identification of an atypical CD8 T cell epitope encoded by murine cytomegalovirus ORF-M54 gaining dominance after deletion of the immunodominant antiviral CD8 T cell specificities. Med Microbiol Immunol 204, 317–326 (2015). https://doi.org/10.1007/s00430-015-0404-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-015-0404-3