
Abstract
Human cytomegalovirus (HCMV) is a ubiquitous pathogen that causes morbidity risk in immunologically suppressed and immunodeficient patients including congenital infections. Approaches to curb the consequences of HCMV infections are restricted by a lack of complete understanding of viral pathogenesis. The infection of mice with murine cytomegalovirus (MCMV) as a model of HCMV infection has been particularly useful in elucidating the role of innate and adaptive immune response mechanisms. A large number of cytomegalovirus genes modulate the innate and the adaptive host immune response. The products of several MCMV genes are involved in subverting the natural killer (NK) cell response by down-modulating cellular ligands for the NKG2D receptor expressed on NK cells and CD8+ T cells. Mutant viruses lacking these immunoevasion genes are attenuated with respect to virus growth in vivo. Given the importance of the NKG2D receptor in controlling both NK- and T cell-mediated immunity, it is of tremendous importance to understand the molecular mechanisms and consequences of viral regulation of the NKG2D ligands.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cytomegaloviruses (CMVs) are ubiquitous pathogens which cause morbidity risk in immunosuppressed and immunodeficient hosts [1]. As members of the herpesvirus family, CMVs share large double-stranded DNA genomes of about 230 kbp in size, are widely distributed in nature, but exhibit a highly restricted host range, each one allowing the infection of a single mammalian species. Thus, in vivo models of human cytomegalovirus (HCMV) pathogenesis and immunosurveillance rely on the infection of animals with their respective natural CMV. The infection of mice with murine cytomegalovirus (MCMV) represents the most frequently used model to study immunobiology and pathogenesis of CMV infection. The immune response provides only partial protection against CMVs, and viral genomes remain life-long in their animal hosts, thus periodic reactivation and virus shedding may occur [2]. The nonproductive state of CMV infection is governed by a hierarchical immune control with the CD8+ T cell subset playing a dominant role, whereas the contribution of antiviral antibodies is limited to the prevention of virus spread after reactivation from latency [3–5]. Furthermore, the immune response is unable to fully prevent re-infection as was demonstrated for congenital CMV infections in infants of mothers with pre-conceptional immunity [6, 7].
It is still not clear how CMVs manage to persist in their animal hosts in spite of a fully primed immune response. CMVs encode numerous proteins able to regulate immunosurveillance mechanisms of their hosts [8]. In order to prevent infected cells from lysis by CD8+ T lymphocytes, HCMV encodes several glycoproteins (US2, US3, US6, US11) which can compromise antigen presentation by MHC class I molecules [9]. The same is achieved by three MCMV proteins encoded by the m04, m06 and m152 genes [9]. We have previously shown that the deletion of MCMV gene m152 sensitizes the virus to the control by CD8+ T cells in vivo [10]. However, as proposed by the missing self-theory, the lack of engagement of inhibitory natural killer (NK) cell receptors by MHC class I molecules should make the infected cells more prone to NK cell control [11]. To avoid this situation, CMVs have adopted several mechanisms enabling them to subvert not only CD8+ T cells but also NK cell mediated control.
NK cells and CMV control
It is now well established that NK cells play a crucial role in the control of CMV replication during the time preceding the induction of the adaptive immune response, whereas the role of NK cells in the control of chronic CMV infection and prevention of virus recurrence remains poorly defined. NK cells are activated through soluble mediators as well as direct cell-to-cell contact; upon activation, they control the infection by cytolysis and secretion of proinflammatory cytokines. Interactions of NK cells and dendritic cells (DCs) proved to be important for the regulation of adaptive immunity [12, 13]. While T and B lymphocytes express clonally distributed antigen-specific receptors, NK cells express numerous inhibitory and activating receptors and their activity is regulated via the integration of signals gathered through them [14]. Thus, NK cell activation occurs either due to the lack of inhibitory signals or because of the prevalence of activating ones. The engagement of inhibitory NK receptors with their ligands, such as MHC class I molecules, prevents healthy cells from NK cell mediated lysis.
NK cell receptors deployed for the surveillance of MHC class I cell surface expression include the members of the killer cell Ig-like receptor (KIR) family and leukocyte Ig-like receptors (LIR) in humans, the Ly49 family in mice and CD94/NKG2 heterodimers in humans and mice [15–17]. They all possess an immunoreceptor tyrosine-based inhibitory motif (ITIM) in their cytoplasmic tail. The phosphorylation of tyrosine within this motif results in the recruitment of phosphatases to the receptor complex which interferes with the signal transduction pathway that activates NK cells. These NK cell receptor classes also include the activating members. Activating NK cell receptors utilize adaptor proteins (DAP12, DAP10, FcεRIγ, CD3ζ) that harbor an immunoreceptor tyrosine-based activating motif (ITAM) to transduce signals. The engagement of some activating NK cell receptors can override inhibition and destroy target cells even if they express surface MHC class I molecules [18, 19]. Several other NK cell receptors encoded by single genes and gene families have been described, such as NKG2D, natural cytotoxicity receptors (NCRs), NKR-P1 gene family and others [16, 20].
Taking the above into consideration, a straightforward approach for the virus to subvert NK cell activation would be to increase the quantity of ligands for inhibitory receptors or to disable the activating ones. Indeed, CMVs encode viral homologues for the major inhibitory ligands, MHC class I molecules [21]. The HCMV UL18 gene product binds inhibitory LIR-1 receptor [22], while the receptor binding the MCMV class I homologue, m144, is still not identified. However, the role of UL18 in immune subversion must further be investigated since it may also lead to NK cell activation [23–25]. The differential regulation of MHC class I molecules could be an additional mechanism to avoid both CD8 and NK cell responses simultaneously. It is well established that HCMV down-modulates most of MHC class Ia while preserving MHC class Ib to serve as ligands for inhibitory NK cell receptors [26]. A peptide derived from the HCMV encoded UL40, as a substitute for the leader sequence of classical MHC class I molecules, binds to and supports the surface expression of an MHC class Ib molecules, HLA-E [27, 28]. Another mode of CMV-mediated NK cell inhibition is the down-modulation of cellular ligands for activating NK cell receptors as discussed below.
MCMV specific NK cell receptors
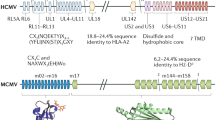
In contrast to most of mouse strains including wild mice, which fail to mount an efficient NK cell response to MCMV, certain mouse strains are able to control the infection by mounting a robust NK cell response immediately after infection. It has been shown that the resistance of C57BL/6 mice to MCMV is determined by a single dominant locus, Cmv1r, located in the natural killer gene complex (NKC) on the mouse chromosome 6 [29, 30] that encodes the stimulatory Ly49H NK cell receptor [31–33]. Unlike most other Ly49 receptors which use cellular MHC class I molecules as their ligands, Ly49H binds to an MCMV-encoded protein, the product of the m157 gene (Fig. 1 a) [34, 35]. Similarly to several other MCMV m145 gene family members, the m157 protein has some structural but not sequence homology to MHC-class I molecules [35]. The crystal structure of the m157 suggested that its interaction with Ly49 receptors could be significantly stronger than that of classical inhibitory Ly49-MHC interactions [36]. Under selective pressure by Ly49H, the m157 is subject of mutations and deletions which eventually lead to mutant viruses resistant to Ly49H-mediated control, similarly to recombinant viruses in which m157 was deliberately deleted [37, 38].

Schematic representation of the role of MCMV immunoevasins on NK cell activity. a MCMV resistant mice control MCMV infection by recognizing viral protein m157 through the activating Ly49H receptor (C57BL/6 mice) or by the recognition of so far undefined viral protein in the context of MHC class I through the activating Ly49P receptor (MA/My mice). Consequently, the deletion of viral gene encoding the ligand for the activating NK cell receptor abolishes the virus sensitivity to NK cells. b NKG2D is an NK cell activating receptor which recognizes different ligands: MULT-1, H60 and five RAE-1 isoforms in mouse. These proteins are poorly expressed on healthy cells but can be induced by cellular stress. MCMV encodes immunoevasins able to down-modulate the surface expression of NKG2D ligands and prevent NK cell activation. c MCMV genes (m138, m145, m152 and m155) encode proteins that specifically target NKG2D ligands for down-modulation. Viral mutants lacking either of these genes are no longer able to regulate NKG2D ligands and are therefore sensitive to NK cell control in vivo
Additional NKC loci associated with specific recognition of infected cells have been recently discovered [39, 40]. The genetic resistance in MA/My mice maps to the Cmv3 locus encoding Ly49P NK cell receptor that recognizes a so far unknown viral protein associated with the H-2Dk MHC class I molecule (Fig. 1a) [39]. It has been suggested that the Cmv4 locus encodes an NK cell activating receptor and mediates resistance in PWK mice [40], while the loci outside the NKC complex regulate NK-resistance in other mouse strains [41]. Although it is unexpected that viruses acquire genes that encode ligands for the activating NK cell receptors, it is of note that m157 also serves as a ligand for the inhibitory NK cell receptor Ly49I [34]. Therefore, it is likely that m157 and other viral ligands for activating NK receptors had been originally selected to serve as ligands for inhibitory NK cell receptors. According to this scenario, the generation of activating Ly49 receptors specific for infected cells could be a host response to viral immunosubversion.
CMV avoidance of NK cell activation via NKG2D
The lack of activating Ly49H and other NK cell receptors that specifically recognize infected cells fails to explain the inability of most of the mouse strains to mount an efficient NK cell response against MCMV. Namely, these mouse strains can mediate an NK cell response against other viral infections and tumors. Therefore, we proposed that MCMV must be able to actively compromise NK cell activation via activating receptors such as NKG2D. NKG2D is a potent activating receptor recognizing cellular ligands that are inducible by cellular stress, such as infection, and thus represents a prominent candidate for viral immunoevasion [20, 42]. It is a highly conserved C-lectin type molecule expressed at the cell surface as a homodimer. The key feature of this receptor is its ability to bind various ligands, all being distant homologues to the MHC class I molecules, but poorly expressed on healthy cells. NKG2D is also expressed on T lymphocytes where it mediates co-stimulation rather than an independent activating signal. It is expressed on human and mouse NK cells, but with a different pattern. All human CD8+ T lymphocytes constitutively express NKG2D, while in mice it is expressed only on activated CD8+ T lymphocytes. Moreover, there are two distinct isoforms of NKG2D in mice, the longer and the shorter one; the short one associates with DAP10 and DAP12 while the long isoform binds only to DAP10 [43, 44]. In humans, there are only the longer NKG2D isoform. Both HCMV and MCMV encode proteins able to target NKG2D ligands for the down-modulation in infected cells. Negative regulation of NKG2D ligands in humans has first been established for the HCMV protein UL16, able to affect ULBP-1, ULBP-2 and MICB [45, 46]. More recently it was demonstrated that the UL142 down-regulates the full length MICA protein [47]. In turn, MCMV regulates the NKG2D ligands MULT-1, RAE-1 and H60 through the m145, m152 and m155 proteins, respectively (Fig. 1b) [48–52]. Furthermore, the deletion of either of MCMV genes encoding inhibitors of NKG2D ligands resulted in rescuing the expression of their target ligand and an enhanced sensitivity of mutant viruses to NK cell control in vivo (Fig. 1c). We have recently shown that the expression of MULT-1 and H60 is also inhibited by m138/fcr-1 [53]. The m138/fcr-1 was originally described as an MCMV receptor for the Fc portion of immunoglobulin G (IgG) and was designated fcr-1 [54]. Since the deletion of this gene results in attenuation in vivo, it was assumed that the latter is caused by enhanced susceptibility of the virus to antibody dependent cellular cytotoxicity. However, the m138 deletion mutant virus was also attenuated in B cell-deficient mice, suggesting an additional function of this protein [55]. We have shown that this mutant virus was attenuated as early as three days post infection [53]. Since this attenuation could be reversed by the depletion of NK cells, or even by blocking the NKG2D receptor, the observed phenotype is most likely due to the expression of NKG2D ligands on the surface of infected cells. Another, recently discovered mode of escaping NKG2D signaling includes the regulation of NKG2D ligands by viral miRNA [56]. The specific miRNA of HCMV, hcmv-miR-UL112, binds to and inhibits the translation of the transcript encoding NKG2D ligand MICB. Unlike CMVs, some other viruses inhibit NK cell activation via NKG2D by encoding soluble proteins which bind to both human and rodent NKG2D and block the interaction between the NKG2D receptor and its host-encoded ligands [57].
The existence of four different MCMV immunoregulatory genes aimed at regulating the NKG2D ligands implies a strong selective pressure for the virus to oppose the NKG2D receptor mediated immune response. This finding is not unexpected if we take into consideration the nature of the NKG2D receptor. Namely, as in the case of m157-Ly49H interaction [36], ligand engagement by NKG2D can override the inhibitory signals gathered through the engagement of the inhibitory NK cell receptors and their ligands [19]. The magnitude of the NKG2D signal can also be explained by the fact that there are four, rather than two, DAP10 chains in an NKG2D-DAP10 complex and thus the binding of a single ligand can result in the phosphorylation and the subsequent signaling of four DAP10 chains [58]. The true potential of NKG2D signaling lies in the NKG2D ligands. The NKG2D receptor shows a remarkable feature of recognizing at least seven different ligands in humans (ULBP 1–4, RAET-1G, MICA, MICB) and seven in mouse (H60, MULT-1, RAE-1α-ε) [42, 59]. These ligands must be tightly controlled in order to avoid an autoimmune response. Furthermore, the infection itself induces the expression of most of the NKG2D ligands and therefore the viruses must have the means to cope with this situation.
Molecular mechanisms of MCMV regulation of NKG2D ligands
In contrast to numerous CMV inhibitors of NKG2D ligands characterized so far, the molecular mechanisms by which they disrupt the expression of their target proteins are not sufficiently understood. Among the MCMV inhibitors of NKG2D, m152 was the first to be described [49]. In addition to CD8-dependent attenuation, the deletion of m152 gene strongly attenuates the mutant virus in NK cell-dependent manner. Namely, the m152 deletion mutant is attenuated 3 days post infection, before the maturation of the T cell response. Since the deletion of m152 gene rescued the expression of NKG2D ligands on the surface of infected cells, the NK cell-dependent phenotype of the mutant virus was expected. A subsequent study demonstrated that m152 down-regulates RAE-1 proteins [51]. As mentioned above, m152 was originally characterized for its ability to arrest the maturation of MHC class I molecules [60], while its mode of down-modulation of RAE-1 proteins remains to be defined. RAE-1 genes are inducible by retinoic acid [61] but also after MCMV infection [51]. In spite of this, the virus is able to down-modulate the surface expression of RAE-1 and prevent recognition via NKG2D. However, our unpublished data demonstrates the differential regulation of RAE-1 isoforms by m152 (J. Arapovic, unpublished). In addition, we have now evidence that a particular motif inside the RAE-1 sequence determines the ligand sensitivity to m152-mediated down-regulation. Of note is that allele-specific preferences is also characteristic of the down-modulation of MHC class I molecules by m152 [62].
The mutant virus lacking the m155 gene was also sensitive to NK cell control in vivo, due to its inability to down-regulate the NKG2D ligand H60 [50]. We have demonstrated that m155 is able to affect H60 in isolated conditions, without the involvement of other MCMV proteins. Namely, the co-infection of CV-1 cells with vaccinia viruses (VV), H60-VV and m155-VV, resulted in a lack of H60 surface expression, while the co-infection with H60-VV and WT VV resulted in a strong H60 surface staining. The same system was used to explore the possible effect of m155 on H60 maturation. H60 passed through its immature 46 kDa form and reached the Endo H resistance as a mature 70 kDa protein in equal amount and time period independently of the presence of m155. Also, m155-VV did not cause a change in the half-life of the H60 glycoprotein. Thus, m155 must mediate its effect on H60 only after this protein has reached the ERGIC/cis-Golgi compartment [50]. There was a slight shift in the migration of the glycosylated H60 protein form in the presence of m155, which could suggest an altered posttranslational modification of H60. Using transfectant cells expressing the m155 gene, others have shown that proteasomal inhibitors can prevent H60 down-regulation [52]. In contrast, we have found increased intracellular accumulation of H60 when drugs that specifically prevent the lysosomal degradation were used (T. Lenac, unpublished).
Unlike H60 [51], the level of MULT-1 mRNA as well as the protein synthesis is up-regulated in the MCMV-infected cells [48]. However, in spite of up-regulation of its expression, MCMV successfully prevents the surface expression of MULT-1 by two inhibitors. The ability of m145 regulator to down-modulate MULT-1 in the absence of other MCMV proteins was tested by co-infection of CV-1 cells with recombinant VV harboring genes encoding MULT-1 and m145. Analogous to the m155 effect on H60, the m145 successfully prevented MULT-1 surface expression. Our results suggest that m145 must also regulate its target after ERGIC/cis-Golgi compartment, since the rate of MULT-1 intracellular transport to EndoH-resistant forms as well as its half-life was unaffected in the presence of m145. This is in agreement with our confocal studies of WT MCMV infected cells showing co-localization of MULT-1 with the trans-Golgi marker (Fig. 2a).

MCMV mechanism for down-regulation of NKG2D ligand MULT-1. a Intracellular distribution of MULT-1 in uninfected and MCMV-infected cells. Confocal analysis was performed on MULT-1-3T3 cells, either mock infected or infected with indicated viruses. MULT-1 was stained with rat anti-MULT-1 mAb followed by the TRITC labeled secondary antibodies (red). In addition, Rab6, a marker of medial- and trans-Golgi compartment is stained in green. In spite of the presence of viral inhibitors, m145 or m138, MULT-1 co-localizes with the trans-Golgi compartment. b Down-modulation of surface resident MULT-1 in MCMV-infected cells. MULT-1-3T3 transfectants were mAb-tagged for surface MULT-1 and incubated for the indicated periods of time, when the MULT-1-mAb complexes were examined with the fluorescence labeled secondary antibodies. The mAb-tagged surface portion of protein is very stable on uninfected cells and completely down-regulated in cells infected with WT MCMV. Deletion of m138 abolished the down-modulation almost completely, whereas in cells infected with the virus lacking m145 the surface portion of MULT-1 was only partially down-modulated indicating that the m145 is essential for the function of m138 in down-modulation of MULT-1. c Degradation of surface resident MULT-1 in MCMV-infected cells. MULT-1-3T3 cells, either mock infected or infected with indicated viruses, were mAb-tagged for surface MULT-1. After incubation for 10 h the fluorescence labeled secondary antibodies were added to visualize MULT-1-mAb complexes. In uninfected cells surface resident MULT-1 is recycling through discrete vesicles whereas in WT MCMV infected cells it is subject to endocytosis (b) and degradation. In contrast, in cells infected with the virus lacking m138 the surface resident MULT-1 is not only preserved on the cell surface but also accumulated intracellularly. Cells infected with virus lacking m145 showed only partial down-modulation of surface resident protein (b) and in addition a significant portion of the protein is localized in the intracellular vesicles, similar to those observed in uninfected cells. Altogether, for the efficient down-modulation and degradation of surface resident MULT-1 both MCMV inhibitors are needed. Parts of data shown in b are reproduced from The Journal of Experimental Medicine, 2006, 203:1843–1850. Copyright 2006 The Rockefeller University Press (53)
Down-modulation of surface resident MULT-1: a joint effort of two viral immunoevasins
A better understanding of the molecular mechanisms by which MCMV proteins m145 and m155 interfere with the expression of MULT-1 and H60 is further complicated by the fact that both proteins are also targeted by m138/fcr-1 [53]. The study showed that m138/fcr-1 targets MULT-1 and H60 for down-regulation and thus contributes to viral resistance to NK cell-control in vivo. Since we knew that MULT-1 is unaffected by MCMV before it reaches the trans-Golgi network (TGN), we have further analyzed the surface resident MULT-1 molecules. The surface portion of MULT-1 was tagged with specific mAb and its destiny was followed during infection by flow cytometry and confocal studies (Fig. 2b, c). Note that in normal cells newly synthesized MULT-1 protein reaches the plasma membrane and recycles via the AP2-positive clathrin vesicles [53]. The surface resident MULT-1 appears to be very stable with the half-life of approximately 10 h. However, in WT MCMV-infected cells the surface resident MULT-1 was completely down-modulated from the cell membrane in no more than 6 h (Fig. 2b). Cells infected with virus lacking m138 preserved surface MULT-1 similar to uninfected cells indicating that m138/fcr-1 is essential for the down-modulation of the surface portion of this protein. Interestingly, in spite of the presence of m138, cells infected with the virus lacking m145 showed only marginal down-modulation of the surface protein, suggesting the requirement of m145 for the function of m138. We showed that inhibitors of clathrin-dependent endocytosis could prevent the effect of m138/fcr-1 on the surface resident MULT-1, whereas inhibitors of the lysosomal proteases prevented the degradation but could not abolish the down-modulation of surface MULT-1 [53]. It is worth mentioning that the m138/fcr-1 protein sequence includes a clathrin box motif (LFAFD), which is in accordance with our finding that inhibitors of clathrin endocytosis can block the down-modulation of surface resident MULT-1. This motif was found on cargo adaptor proteins (APs) and interacts with the N-terminus of clathrin heavy chain (the beta propeller structure) [63]. It was identified that the Ig1 domain of the fcr-1 ectodomain is necessary and sufficient to down-regulate MULT-1 [53]. In contrast to MULT-1, the surface portion of H60 seems not to be recycling (not shown), while its down-regulation required the preserved composition of the complete fcr-1 ectodomain [53]. Furthermore, our recent unpublished data indicate that m138/fcr-1 might also target some RAE-1 family members for down-modulation (J. Arapovic, unpublished). In addition to NKG2D ligands, m138/fcr-1 is able to down-regulate the costimulatory molecule B7-1 (CD80) [64] and thus has a strong impact on the T cell response since it impairs the ability of DCs to activate CD8+ T cells. This might be an explanation for the much broader in vivo attenuation of the MCMV mutant lacking m138/fcr-1 as compared to the mutant viruses lacking other NKG2D ligand regulators.
What could be the reason for the functional redundancy of MCMV inhibition of NKG2D ligands? The multiple inhibitors may be related to the possible tissue specific role of these viral proteins. Indeed, we have observed that the level of attenuation of MCMV mutants lacking inhibitors for the NKG2D ligands varies between different organs, suggesting either differential expression of viral genes, differential expression of cellular ligands or both. Therefore, in order to achieve a complete down-modulation of NKG2D ligands, the co-operation between two regulators might be needed. For instance, some of the inhibitors may change the microlocalization of the ligands in the membrane making them more accessible to other regulators. We have observed differential localization of recycling MULT-1 between uninfected cells and cells infected with mutant virus lacking m138. While in normal cells the surface portion of MULT-1 is localized in discrete recycling vesicles, in cells infected with the Δm138 MCMV, MULT-1 accumulates intracellularly and partially co-localizes with TGN (Fig. 2a and data not shown). Since this was observed in cells infected with the virus lacking m138, differential distribution must be caused by m145. Moreover, in cells infected with the m145 deletion mutant, surface resident MULT-1 was only partially down-modulated, but also preserved intracellularly in vesicles comparable to those in uninfected cells. Therefore, it can be speculated that m145 dislocates MULT-1 proteins or slows down their recycling, making them more accessible to m138/fcr-1. Once it approaches MULT-1, m138/fcr-1 could act as a cargo adaptor protein for clathrin-mediated endocytosis (Fig. 3). Altogether, our results suggested the co-operative effect of m138 and m145 in down-modulation of MULT-1. However, more specific studies on the interaction between NKG2D ligands and their regulators are required to understand the details of molecular mechanisms involved. It seems worth mentioning that the cytoplasmic tail of m145 contains the YKTI motif, whereas MULT-1 possesses a dileucin motif. One can speculate that m145 affects MULT-1 by competing for the same adaptor proteins. On the other hand, m138/fcr-1 has a short cytoplasmic tail with no tyrosine or dileucin motifs. It might be that m138/fcr-1 could benefit from the motifs present in the m145 protein.

Model of the m138/fcr-1- and m145-mediated MULT-1 down-regulation. MULT-1 is regulated by two MCMV proteins, m138/fcr-1 and m145. Both regulators must affect MULT-1 dominantly after trans-Golgi compartment, since confocal studies of WT MCMV infected cells show co-localization of MULT-1 with the trans-Golgi marker (Fig. 2 a). After acquiring its mature form, MULT-1 reaches the plasma membrane and recycles via the AP2-positive clathrin vesicles. m138/fcr-1 specifically targets membrane resident MULT-1 and facilitates its endocytosis and lysosomal degradation. Since the cells infected with the m138 deletion mutant rescued the membrane expression of MULT-1 and showed its intracellular accumulation, it is suggested that m145 dislocates MULT-1 proteins making them more accessible to m138/fcr-1
Future perspectives
Because of the importance of the NKG2D receptor in controlling both NK- and T cell-mediated immunity, it is of tremendous significance to understand how and why NKG2D ligands are regulated upon viral infection. The studies performed by our lab and others have identified several MCMV proteins involved in the regulation of NKG2D ligands. Further studies are needed to understand the regulation of the expression of MCMV immunoevasion proteins that target NKG2D ligands, as well as the molecular mechanisms by which these viral proteins disrupt NKG2D ligand expression. We also need to identify the cellular proteins that interact with viral immunoevasins and evaluate how viral regulation of NKG2D ligands impacts the efficiency of NK, NKT and memory T cell responses in controlling latency and in resolving recurrent virus infection. We believe that these research approaches will advance our understanding of CMVs lifetime persistence in the infected host. Eventually, this may lead to the development of novel probes to characterize the cellular molecules and systems that control the expression of NK receptor ligands.
References
Roizman B, Pellett PE (2001) The family Herpesviridae: a brief introduction. In: Knipe DM, Howley PM (eds) Virology. Lippincott Williams and Wilkins, Philadelphia, pp 2381–2397
Reddehase MJ, Podlech J, Grzimek NK (2002) Mouse models of cytomegalovirus latency: overview. J Clin Virol 25(Suppl 2):S23–36
Jonjic S, Pavic I, Polic B, Crnkovic I, Lucin P, Koszinowski UH (1994) Antibodies are not essential for the resolution of primary cytomegalovirus infection but limit dissemination of recurrent virus. J Exp Med 179(5):1713–7
Polic B, Hengel H, Krmpotic A, Trgovcich J, Pavic I, Luccaronin P, Jonjic S, Koszinowski UH (1998) Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J Exp Med 188(6):1047–54
Simon CO, Seckert CK, Grzimek NKA, Reddehase MJ (2006) Murine model of cytomegalovirus latency and reactivation: the silencing/desilencing and immune sensing hypothesis. In: Reddehase MJ (ed) Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Wymondham, pp 483–500
Boppana SB, Rivera LB, Fowler KB, Mach M, Britt WJ (2001) Intrauterine transmission of cytomegalovirus to infants of women with preconceptional immunity. N Engl J Med 344(18):1366–71
Fowler KB, Stagno S, Pass RF (2003) Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA 289(8):1008–11
Jonjic S, Bubic I, Krmpotic A (2006) Innate immunity to cytomegloviruses. In: Reddehase MJ (ed) Cytomegaloviruses molecular biology and immunology. Caister Academic Press, Norwich, pp 285–320
Reddehase MJ (2002) Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat Rev Immunol 2(11):831–44
Krmpotic A, Messerle M, Crnkovic-Mertens I, Polic B, Jonjic S, Koszinowski UH (1999) The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J Exp Med 190(9):1285–96
Karre K, Ljunggren HG, Piontek G, Kiessling R (1986) Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319(6055):675–8
Degli-Esposti MA, Smyth MJ (2005) Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol 5(2):112–24
Robbins SH, Bessou G, Cornillon A, Zucchini N, Rupp B, Ruzsics Z, Sacher T, Tomasello E, Vivier E, Koszinowski UH, Dalod M (2007) Natural killer cells promote early CD8 T cell responses against cytomegalovirus. PLoS Pathog 3(8):e123
Kirwan SE, Burshtyn DN (2007) Regulation of natural killer cell activity. Curr Opin Immunol 19(1):46–54
Dimasi N, Moretta L, Biassoni R (2004) Structure of the Ly49 family of natural killer (NK) cell receptors and their interaction with MHC class I molecules. Immunol Res 30(1):95–104
Lanier LL (2005) NK cell recognition. Annu Rev Immunol 23:225–74
Yokoyama WM, Plougastel BF (2003) Immune functions encoded by the natural killer gene complex. Nat Rev Immunol 3(4):304–16
Diefenbach A, Jensen ER, Jamieson AM, Raulet DH (2001) Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 413(6852):165–71
Cerwenka A, Baron JL, Lanier LL (2001) Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci USA 98(20):11521–6
Raulet DH (2003) Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 3(10):781–90
Farrell HE, Davis-Poynter NJ, Andrews DM, Degli-Esposti MA (2002) Function of CMV-encoded MHC class I homologues. Curr Top Microbiol Immunol 269:131–51
Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, Hsu ML (1997) A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity 7(2):273–82
Leong CC, Chapman TL, Bjorkman PJ, Formankova D, Mocarski ES, Phillips JH, Lanier LL (1998) Modulation of natural killer cell cytotoxicity in human cytomegalovirus infection: the role of endogenous class I major histocompatibility complex and a viral class I homolog. J Exp Med 187(10):1681–7
Prod’homme V, Griffin C, Aicheler RJ, Wang EC, McSharry BP, Rickards CR, Stanton RJ, Borysiewicz LK, Lopez-Botet M, Wilkinson GW, Tomasec P (2007) The human cytomegalovirus MHC class I homolog UL18 inhibits LIR-1+ but activates LIR-1- NK cells. J Immunol 178(7):4473–81
Wagner CS, Riise GC, Bergstrom T, Karre K, Carbone E, Berg L (2007) Increased expression of leukocyte Ig-like receptor-1 and activating role of UL18 in the response to cytomegalovirus infection. J Immunol 178(6):3536–43
Lopez-Botet M, Llano M, Ortega M (2001) Human cytomegalovirus and natural killer-mediated surveillance of HLA class I expression: a paradigm of host-pathogen adaptation. Immunol Rev 181:193–202
Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S, Cerundolo V, Borysiewicz LK, McMichael AJ, Wilkinson GW (2000) Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 287(5455):1031
Ulbrecht M, Martinozzi S, Grzeschik M, Hengel H, Ellwart JW, Pla M, Weiss EH (2000) Cutting edge: the human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J Immunol 164(10):5019–22
Scalzo AA, Fitzgerald NA, Simmons A, La Vista AB, Shellam GR (1990) Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J Exp Med 171(5):1469–83
Scalzo AA, Fitzgerald NA, Wallace CR, Gibbons AE, Smart YC, Burton RC, Shellam GR (1992) The effect of the Cmv-1 resistance gene, which is linked to the natural killer cell gene complex, is mediated by natural killer cells. J Immunol 149(2):581–9
Brown MG, Dokun AO, Heusel JW, Smith HR, Beckman DL, Blattenberger EA, Dubbelde CE, Stone LR, Scalzo AA, Yokoyama WM (2001) Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 292(5518):934–7
Daniels KA, Devora G, Lai WC, O’Donnell CL, Bennett M, Welsh RM (2001) Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J Exp Med 194(1):29–44
Lee SH, Girard S, Macina D, Busa M, Zafer A, Belouchi A, Gros P, Vidal SM (2001) Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat Genet 28(1):42–5
Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL (2002) Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296(5571):1323–6
Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, Scalzo AA, Fremont DH, Yokoyama WM (2002) Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci USA 99(13):8826–31
Adams EJ, Juo ZS, Venook RT, Boulanger MJ, Arase H, Lanier LL, Garcia KC (2007) Structural elucidation of the m157 mouse cytomegalovirus ligand for Ly49 natural killer cell receptors. Proc Natl Acad Sci USA 104(24):10128–33
Voigt V, Forbes CA, Tonkin JN, Degli-Esposti MA, Smith HR, Yokoyama WM, Scalzo AA (2003) Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc Natl Acad Sci USA 100(23):13483–8
French AR, Pingel JT, Wagner M, Bubic I, Yang L, Kim S, Koszinowski U, Jonjic S, Yokoyama WM (2004) Escape of mutant double-stranded DNA virus from innate immune control. Immunity 20(6):747–56
Desrosiers MP, Kielczewska A, Loredo-Osti JC, Adam SG, Makrigiannis AP, Lemieux S, Pham T, Lodoen MB, Morgan K, Lanier LL, Vidal SM (2005) Epistasis between mouse Klra and major histocompatibility complex class I loci is associated with a new mechanism of natural killer cell-mediated innate resistance to cytomegalovirus infection. Nat Genet 37(6):593–9
Adam SG, Caraux A, Fodil-Cornu N, Loredo-Osti JC, Lesjean-Pottier S, Jaubert J, Bubic I, Jonjic S, Guenet JL, Vidal SM, Colucci F (2006) Cmv4, a new locus linked to the NK cell gene complex, controls innate resistance to cytomegalovirus in wild-derived mice. J Immunol 176(9):5478–85
Rodriguez M, Sabastian P, Clark P, Brown MG (2004) Cmv1-independent antiviral role of NK cells revealed in murine cytomegalovirus-infected New Zealand White mice. J Immunol 173(10):6312–8
Cerwenka A, Lanier LL (2003) NKG2D ligands: unconventional MHC class I-like molecules exploited by viruses and cancer. Tissue Antigens 61(5):335–43
Diefenbach A, Tomasello E, Lucas M, Jamieson AM, Hsia JK, Vivier E, Raulet DH (2002) Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol 3(12):1142–9
Gilfillan S, Ho EL, Cella M, Yokoyama WM, Colonna M (2002) NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat Immunol 3(12):1150–5
Kubin M, Cassiano L, Chalupny J, Chin W, Cosman D, Fanslow W, Mullberg J, Rousseau AM, Ulrich D, Armitage R (2001) ULBP1, 2, 3: novel MHC class I-related molecules that bind to human cytomegalovirus glycoprotein UL16, activate NK cells. Eur J Immunol 31(5):1428–37
Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ (2001) ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 14(2):123–33
Chalupny NJ, Rein-Weston A, Dosch S, Cosman D (2006) Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem Biophys Res Commun 346(1):175–81
Krmpotic A, Hasan M, Loewendorf A, Saulig T, Halenius A, Lenac T, Polic B, Bubic I, Kriegeskorte A, Pernjak-Pugel E, Messerle M, Hengel H, Busch DH, Koszinowski UH, Jonjic S (2005) NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J Exp Med 201(2):211–20
Krmpotic A, Busch DH, Bubic I, Gebhardt F, Hengel H, Hasan M, Scalzo AA, Koszinowski UH, Jonjic S (2002) MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat Immunol 3(6):529–35
Hasan M, Krmpotic A, Ruzsics Z, Bubic I, Lenac T, Halenius A, Loewendorf A, Messerle M, Hengel H, Jonjic S, Koszinowski UH (2005) Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J Virol 79(5):2920–30
Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, Lanier LL (2003) NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med 197(10):1245–53
Lodoen MB, Abenes G, Umamoto S, Houchins JP, Liu F, Lanier LL (2004) The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J Exp Med 200(8):1075–81
Lenac T, Budt M, Arapovic J, Hasan M, Zimmermann A, Simic H, Krmpotic A, Messerle M, Ruzsics Z, Koszinowski UH, Hengel H, Jonjic S (2006) The herpesviral Fc receptor fcr-1 down-regulates the NKG2D ligands MULT-1 and H60. J Exp Med 203(8):1843–50
Thale R, Lucin P, Schneider K, Eggers M, Koszinowski UH (1994) Identification and expression of a murine cytomegalovirus early gene coding for an Fc receptor. J Virol 68(12):7757–65
Crnkovic-Mertens I, Messerle M, Milotic I, Szepan U, Kucic N, Krmpotic A, Jonjic S, Koszinowski UH (1998) Virus attenuation after deletion of the cytomegalovirus Fc receptor gene is not due to antibody control. J Virol 72(2):1377–82
Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M, Horwitz E, Prokocimer Z, Prichard M, Hahn G, Goldman-Wohl D, Greenfield C, Yagel S, Hengel H, Altuvia Y, Margalit H, Mandelboim O (2007) Host immune system gene targeting by a viral miRNA. Science 317(5836):376–81
Campbell JA, Trossman DS, Yokoyama WM, Carayannopoulos LN (2007) Zoonotic orthopoxviruses encode a high-affinity antagonist of NKG2D. J Exp Med 204(6):1311–7
Garrity D, Call ME, Feng J, Wucherpfennig KW (2005) The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc Natl Acad Sci USA 102(21):7641–6
Eagle RA, Trowsdale J (2007) Promiscuity and the single receptor: NKG2D. Nat Rev Immunol 7(9):737–44
Ziegler H, Thale R, Lucin P, Muranyi W, Flohr T, Hengel H, Farrell H, Rawlinson W, Koszinowski UH (1997) A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 6(1):57–66
Zou Z, Nomura M, Takihara Y, Yasunaga T, Shimada K (1996) Isolation and characterization of retinoic acid-inducible cDNA clones in F9 cells: a novel cDNA family encodes cell surface proteins sharing partial homology with MHC class I molecules. J Biochem (Tokyo) 119(2):319–28
Wagner M, Gutermann A, Podlech J, Reddehase MJ, Koszinowski UH (2002) Major histocompatibility complex class I allele-specific co-operative and competitive interactions between immune evasion proteins of cytomegalovirus. J Exp Med 196(6):805–16
Dell’Angelica EC (2001) Clathrin-binding proteins: got a motif? Join the network! Trends Cell Biol 11(8):315–8
Mintern JD, Klemm EJ, Wagner M, Paquet ME, Napier MD, Kim YM, Koszinowski UH, Ploegh HL (2006) Viral interference with B7-1 costimulation: a new role for murine cytomegalovirus fc receptor-1. J Immunol 177(12):8422–31
Acknowledgments
Our work is supported by Croatian Ministry of Science, Education and Sport grants: 0621261-1263 (SJ) and 0621261-1268 (AK). AK is supported by the Howard Hughes Medical Institute Scholars grant.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lenac, T., Arapović, J., Traven, L. et al. Murine cytomegalovirus regulation of NKG2D ligands. Med Microbiol Immunol 197, 159–166 (2008). https://doi.org/10.1007/s00430-008-0080-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-008-0080-7