
Abstract
Serotonin 1A receptors are known to play an important role in many psychiatric and neurodegenerative disorders. Currently, all available 5-HT1A receptor PET radiopharmaceuticals that are radiolabeled with fluorine-18 are antagonists. As agonists bind preferentially to the high-affinity state of receptors, it would be of great interest to develop agonist radioligands which could provide a measure of the functional 5-HT1A receptors in pathophysiological processes. The 5-HT1A receptor agonist candidates we recently proposed had promising in vitro properties but were not optimal in terms of PET imaging. F13640, a.k.a befiradol or NLX-112, is a 5-HT1A receptor agonist with a high affinity (Ki = 1 nM) and a high selectivity that would be suitable for a potential PET radiopharmaceutical. With propose here the first preclinical evaluation of 18F-F13640. 18F-F13640’s nitro-precursor was synthesized and radiolabeled via a fluoro-nucleophilic substitution. Its radiopharmacological characterization included autoradiographic studies, metabolic studies, and in vivo PET scans in rat, cat and non-human primate. Some of the results were compared with the radiotracer 18F-MPPF, a 5-HT1A receptor antagonist. The radiochemical purity of 18F-F13640 was > 98%. In vitro binding pattern was consistent with the 5-HT1A receptor distribution. Metabolic studies revealed that the radiotracer rapidly entered the brain and led to few brain radiometabolites. Although 18F-F13640 in vivo binding was blocked by the 5-HT1A antagonist WAY-100635 and the 5-HT1A agonist 8-OH-DPAT, the distribution pattern was markedly different from antagonist radiotracers in the three species, suggesting it provides novel information on 5-HT1A receptors. Preliminary studies also suggest a high sensitivity of 18F-F13640 to endogenous serotonin release. 18F-F13640 has suitable characteristics for probing in vitro and in vivo the 5-HT1A receptors in high-affinity state. Quantification analyses with kinetic modeling are in progress to prepare the first-in-man study of 18F-F13640.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Serotonin 1A (5-HT1A) receptors are involved in a wide range of physiological processes such as cognition, behavior, movement and pain modulation, and have been proposed as attractive therapeutic targets in various pathologies in psychiatry including mood disorders, schizophrenia and, more recently in neurological pathologies, e.g., Parkinson’s disease, Alzheimer’s disease. Positron emission tomography (PET) imaging of cerebral 5-HT1A receptors has been used to provide further insight into their role and potential as a therapeutic target or a biomarker for the serotonergic system.
Currently, several selective 5-HT1A antagonist PET ligands are available for mapping and quantifying these receptors in human brain, such as 11C-WAY-100,635 and 18F-MPPF (Zimmer and Le Bars 2013). Interestingly, it has been shown several decades ago that 5-HT1A receptors can exist in a high or a low affinity state, depending on if they are coupled or not to their G proteins (Mongeau et al. 1992). This property implies that whereas 5-HT1A receptor antagonists bind to the total pool of receptors, 5-HT1A receptor agonists preferentially bind to a subpopulation of receptors in their high affinity state (Gozlan et al. 1995). However, and until recently, this pharmacological property was not exploited in molecular neuroimaging (Zimmer 2016).
We proposed in 2006 that the use of a 5-HT1A receptor agonist tracer in PET imaging may provide a more functional representation of this receptor by specifically targeting the pool of receptors in their active state (Aznavour et al. 2006). Such a strategy would enable in vivo investigation of the proportion of 5-HT1A receptors that are actually G-protein-coupled and which is likely to be altered in pathological conditions such as psychiatric disorders or neurodegenerative diseases (Avissar and Schreiber 2006; Becker et al. 2014; Vidal et al. 2016).
In addition, use of agonist PET probes would allow the determination of receptor occupancy by 5-HT1A receptor therapeutic drugs, most of which show agonist activity at this site. Examples include the anxiolytic, buspirone, the antidepressants, vortioxetine and vilazodone, and the antipsychotics, aripiprazole, cariprazine and clozapine. Previous attempts to quantify agonist receptor occupancy using antagonist PET radiopharmaceuticals have been largely unsuccessful (Bantick et al. 2004a, b).
Moreover and according to these pharmacological properties, a 5-HT1A agonist tracer may be more sensitive to extracellular changes in endogenous serotonin (5-HT), because they both compete for receptors in the same high-affinity state (Paterson et al. 2010). Indirect monitoring of endogenous 5-HT release by use of an agonist PET radiotracer would be helpful to improve understanding of the serotonergic system.
Overall, compelling arguments support the development of a 5-HT1A radiotracer with agonistic properties, but until now all attempts remained unsuccessful. Most of the agonist radiotracer-candidates previously evaluated turned out to be unsuitable or non-optimal for PET imaging of functional 5-HT1A receptors in vivo. Among of these, 11C-CUMI-101 has provided promising results in animals and humans (Kumar et al. 2007; Milak et al. 2010) but some recent studies showed that it is not a full agonist of 5-HT1A receptors and that it binds significantly to alpha-1 receptors (Hendry et al. 2011; Shrestha et al. 2014, 2016), although another study showed no displacement of 3H-CUMI-101 by prazosin in baboon and human brain sections (Kumar et al. 2013). Moreover, 11C-CUMI-101 may not be sensitive enough to 5-HT release to explore physiological conditions (Pinborg et al. 2012). To date, there is no radiofluorinated 5-HT1A agonist available for PET imaging in humans. Our team previously reported the preclinical evaluations of two full agonists, 18F-F15599 and 18F-F13714 (Lemoine et al. 2010, 2012). Although results were encouraging, these candidates were not optimal for in vivo imaging, because of a too low target-to-background ratio (for 18F-F15599) or an irreversible binding (for 18F-F13714).
In this context, we chose to evaluate another selective agonist from the same series, F13640 (a.k.a. befiradol or NLX-112), whose chemical structure is close to the previous candidates but with an intermediate affinity of 1 nM for 5-HT1A receptors, more likely to be suitable for in vivo imaging. F13640, also known as befiradol or NLX-112, is undergoing clinical development as a treatment for CNS disorders and is a well characterized and highly selective full agonist of 5-HT1A receptors (Maurel et al. 2007; Colpaert et al. 2002; Heusler et al. 2010). In this article, we present the first characterization of 18F-F13640 from rodent to rhesus monkey, as a candidate for future use in PET studies in human.
Materials and methods
Synthesis of 18F-F13640 nitro-precursor
Synthesis of 2
To a stirred solution of 1 (5.45 g, 27.04 mmol, 1.00 eq) in SOCl2 (100.00 ml) was added dimethylformamide (DMF), then the mixture was heated to 65 °C for 2 h. The mixture was concentrated under vacuum to give crude 2, which was directly used in next step. Liquid chromatography-mass spectrometry (LC-MS) was conducted after quenched a sample by MeOH. As a result, MS indicated the molecule of methyl ester. [M + H]+ = 216.0.
Synthesis of 4
To a stirred solution of 3 (4.5 g, 16.9 mmol) and Triethylamine (6.16 g, 60.84 mmol) in CH2Cl2 (250 ml) was added 2 (7.5 g, 31.77 mmol) at 0 °C, then the mixture was stirred at 25 °C for 1 h. The reaction mixture was concentrated under vacuum to give crude product. The residue was purified by column chromatography silica gel (Petroluem Ether/Ethyl Acetate = 2:1) to afford 4 (6.56 g, 14.58 mmol) as solid.
1H nuclear magnetic resonance (NMR) (CDCl3; 400 MHz): δ 1.64-2.0 (m, 4H), 3.30–3.50 (m, 4H), 4.48 (br, 1H), 5.10 (s, 3H), 7.28–7.43 (m, 6H), 7.58 (s, 1H), 7.90 (d, J = 8.16 Hz, 1H). LCMS: [M + H]+ = 450.1.
Synthesis of 5
4 (5.5 g, 12.23 mmol) was dissolved in trifluoroacetic acid (TFA, 80 ml), and mixture was stirred at 70 °C for 2 h. Then the mixture was concentrated under vacuum to afford crude 5 (6.27 g, 14.59 mmol), which was directly used in next step. 1H NMR (MeOD-d4; 400 MHz): δ 1.70–2.18 (m, 4H), 3.25 (m, 3H), 3.41–3.68 (m, 2H), 4.57 (br, 1H), 7.60 (dd, J 1 = 8.40 Hz, J 2 = 1.6 Hz, 1H), 7.79 (d, J = 1.20 Hz, 1H), 8.05 (d, J = 8.40 Hz, 1H). LCMS: [M + H]+ = 316.1.
Synthesis of 7
To a stirred suspension of 6 (1.69 g, 13.96 mmol), NaOAc (1.2 g, 14.66 mmol) and MgSO4 (840.28 mg, 6.98 mmol) in EtOH (140 ml) was added 5 (6 g, 13.96 mmol) at 25 °C. After addition, mixture was stirred for 1 h at 25 °C. Then the mixture was cooled to 0 °C and NaBH3CN (877.35 mg, 13.96 mmol) was added, then the mixture was stirred at 25 °C for 12 h. The mixture was filtered and concentrated under vacuum to give crude product. The residue was purified by Neutral Pre-HPLC to afford 7 (1.2 g, 2.85 mmol, 20.42%).1H NMR (CDCl3; 400 MHz): δ 1.68 (br, 2H), 1.96 (br, 1H), 2.00-2.20 (m, 2H), 2.34 (s, 3H), 2.73–2.89 (m, 2H), 3.20 (br, 1H), 3.44–3.44 (m, 2H), 3.91 (s, 2H), 4.46–4.64 (m, 1H), 7.20 (d, J = 8.0 Hz, 1H), 7.42–7.48 (m, 2H), 7.60 (d, J = 1.2 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H), 8.40 (s, 1H). LCMS: [M + H]+ = 421.2.
18F-F13640 radiosynthesis and quality controls
After initial fluoride preparation (18O(p,n)18F cyclotron reaction, collection, drying, and Kryptofix activation), 5 mg of F13640 nitro precursor were introduced with 3 mL of dimethyl sulfoxide, and the reaction mixture was heated at 150 °C for 10 min. After dilution with water, the reaction mixture was passed through an activated C18 cartridge for purification, and the crude product was eluted from the cartridge with methanol. Pure 18F-F13640 was obtained after separation on a preparative HPLC column (SymmetryPrep C18, 7 µm, 7.80 × 300 mm, Waters) eluted with H3PO4 (20 mM)/tetrahydrofuran/trifluoroacetic acid 85%/15%/0.1% at 3 ml min−1, with a retention time of 40 min (λ =254 nm). The radiotracer was formulated via solid-phase extraction techniques using a Sep-Pak Light C18 cartridge (Waters); the final product eluted with 1 mL of ethanol was diluted with isotonic saline and sterilized by filtration (sterile filter Millex-GS, 0.22 µm; Millipore). The radiochemical purity and specific activity of the injectable solution were measured by analytical HPLC assay (λ =220 nm and radioactive detection; C18 Nucleodur 5 µm, 4.6 × 250 mm column [Macherey–Nagel]; elution with HCO2H 10 mM/CH3CN 67%/33% at 0.9 ml min−1, with a retention time of 7 min).
In some experiments, the 5-HT1A antagonist 18F-MPPF was also used for comparison. 18F-MPPF was synthetized in an automated synthesizer, using the chemical pathway previously described (Le Bars et al. 1998).
Animals
Adult male Sprague–Dawley rats (Charles River Laboratories; 300 ± 50 g) and three European male cats (5.2 ± 0.3 kg) were used in Lyon. Two female rhesus macaques (Macaca mulatta), (11.1 ± 2.3 kg) housed at the Yale University School of Medicine (New Haven, CT, USA) were used in New Haven.
Determination of brain unmetabolized radiotracer fraction
Rats were anesthetized by i.p. injection of urethane (1.25 g/kg) and a catheter was placed into their caudal vein. The rats were killed by decapitation 10, 20, 40, or 50 min after a bolus injection of 18F-F13640 (58 ± 6 MBq). The brains were carefully removed; each hemisphere was homogenized in 400 µL of perchloric acid at 0.4 mol/L and centrifuged at 1,000 g for 10 min. The supernatant was neutralized by 120 µL of 4M potassium acetate and filtered (0.45 µm) before HPLC. The HPLC system consisted of a C-18 reversed phase column (C18 Nucleodur 5 µm, 4.6 × 250 mm column; elution with H2O/Acetonitrile/TFA 60%/40%/0.1%) at a flow rate of 0.9 ml min−1. During elution, 2-min fractions were collected and counted for radioactivity with an automated gamma-counter.
In vitro autoradiography studies
After a short inhalation of isoflurane, rats were decapitated and brains were carefully removed and immediately frozen in 2-methylbutane cooled with dry ice (− 29 °C). Coronal sections (30 µm thick) were cut using a − 20 °C cryostat, thaw-mounted on glass slides, and allowed to air dry before storage at − 80 °C until used. The day of 18F-F13640 synthesis, the slides were incubated for 20 min in Tris phosphate-buffered saline buffer, pH adjusted to 7.5, containing 37 kBq/ml of 18F-F13640. For competition experiments, the slides were placed in the same buffer supplemented with the selective 5-HT1A antagonist WAY-100635 (1, 10 and 100 nM), or with 5-HT (10 nM, 100 nM, and 1 µM). For uncoupling experiments, the buffer was supplemented with 10 µM of Gpp(NH)p, a nonhydrolysable analog of guanosine 5′-triphosphate, and for comparison some slices were incubated with 37 kBq/ml of 18F-MPPF instead of 18F-F13640. After incubation, the slides were dipped in distilled cold water (4 °C) and then dried and juxtaposed to a phosphor imaging plate for 60 min (BAS-1800 II; Fujifilm). Regions of interest (ROIs) were drawn manually using Multigauge software (Fujifilm). The results were expressed in optical densities (PSL/mm2) or in percentage of control.
Ex vivo autoradiography studies
Rats were anesthetized with 4–5% isoflurane for 5 min (induction phase), then lowered to 2% isoflurane. A catheter was inserted into the caudal vein. Rats were pre-injected with saline or fenfluramine, a serotonin releaser (0.25, 0.5, 1, 2 or 5 mg/kg ; three rats per dose), 20 min before the radiotracer injection. 18F-F13640 (or 18F-MPPF) was injected at a dose of 111 ± 11 KBq/g. Twenty minutes after injection, rats were decapitated and brains were carefully removed and immediately frozen in 2-methylbutane cooled with dry ice (− 29 °C). Coronal sections (30 µm thick) were immediately cut using a − 20 °C cryostat, thaw-mounted on glass slides, dried and juxtaposed to a phosphor imaging plate for 60 min. ROIs were drawn manually using Multigauge software (Fujifilm) according to a stereotaxic atlas of the rat brain (Paxinos et Watson, 1986). For each radiotracer, the results were expressed in percentage of optical density compared to control (saline injection).
MicroPET studies in rat
Rats were anesthetized with 4–5% isoflurane for 5 min (induction phase), and a catheter was placed into their caudal vein. Anesthesia was then lowered to 2% isoflurane during the acquisition on the PET/CT Inveon (Siemens), with monitoring of the respiratory rate using a pressure sensor. The acquisition started with a CT image acquisition for 10 min, followed by the intravenous injection of 18F-F13640 (36 ± 5 KBq/g). The total duration of the scan was 60 min. The images were reconstructed in three dimensions in a series of 24 sequential frames of increasing duration from 10 s to 5 min. ROIs were automatically delineated using a multi-atlas dataset (Lancelot et al. 2014), after manual positioning of the PET images on an anatomical MRI template using the software Inveon Research Workplace (IRW, Siemens). The time-activity curves were expressed in Bq/cm3 normalized to the injected dose corrected for weight, to obtain standardized uptake values (SUVs). 5 rats were used for baseline experiments. For competition experiments, 2 rats received 2 mg/kg of the 5-HT1A antagonist WAY-100635 in co-injection with the tracer. Two other rats received an intravenous injection of the 5-HT1A agonist 8-OH-DPAT at 2 mg/kg, 20 min before acquisition.
PET studies in cat
The PET images were acquired on three European male cats which underwent three experiments separated by at least 7 days. The cats were anesthetized with isoflurane set at 5% for 5 min for induction, then lowered to 2.5% for maintenance. The cat was placed in an acrylic stereotactic apparatus with ear bars, and a catheter was inserted into the forearm branch of the brachiocephalic vein continuously perfused with NaCl 0.9%. Heart rate and oxygen saturation were continuously monitored during the experiment. The PET scan was acquired on a PET/CT Biograph mCT (Siemens) used in 3-dimensional mode. The transmission scan was performed during the first minutes of acquisition with the X-ray source of the machine, followed by an intravenous injection of 18F-F13640 (99 ± 10 MBq). The total duration of the scan was 90 min. The images obtained were reconstructed in a series of 31 sequential frames of increasing duration from 30 s to 5 min. ROIs were manually drawn in IRW according to a stereotaxic atlas of the cat brain (Lancelot et al. 2010), after manual registration on a MRI template. Radioactivity in all ROIs was calculated as the average of the values in the left and right sides. The time-activity curves were expressed in Bq/cm3, normalized to the injected dose corrected for weight to obtain SUVs. One cat underwent two baseline experiments (test–retest) and a blockade experiment with the 5-HT1A antagonist WAY-100635 (1 mg/kg i.v., 30 min before the scan). Two other cats underwent a baseline experiment, a blockade experiment with WAY-100635 (2 mg/kg i.v., 5 min before the scan) and a blockade experiment with the 5-HT1A agonist 8-OH-DPAT (1 mg/kg i.v., 20 min before the scan).
PET studies in rhesus monkey
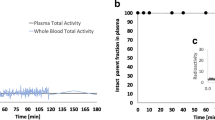
The PET images were acquired on two female rhesus monkeys which underwent two experiments separated by at least 14 days. Each macaque was imaged two times (one baseline and one blockade experiments) following identical procedures with a Focus 220 PET scanner (Siemens). The animals were induced into anesthesia with ketamine (10 mg/kg) and glycopyrrolate (0.01 mg/kg i.m.) 2 h before radiotracer injection, positioned in the Focus 220 PET camera, and immediately intubated with an endotracheal tube for continued anesthesia with 2% isoflurane. An intravenous line was placed and used for injection of the radiotracer or the antagonist WAY-100635 in the blockade study (2 mg/kg of the 5-HT1A antagonist WAY-100635, 5 min prior to the injection of 18F-F13640). Body temperature was kept at 37–38 °C using a heated water blanket. Vital signs, including heart rate, blood pressure, respiration rate, oxygen saturation and body temperature, were monitored every 10 to 15 min during the study. Following the i.v. injection of 18F-F13640 (172 ± 11 MBq), dynamic 3D PET data were acquired continuously in list-mode from 2 to 3 h and rebinned into a series of 45 frames as follows: 6 × 0.5 min, 3 × 1 min, 2 × 2 min, 34 × 5 min. The dynamic series were subsequently reconstructed using a filtered back projection algorithm with standard corrections for random, scatter, and attenuation. Reconstructed dynamic PET images were transferred and analyzed using the image processing PMOD software package (PMOD Technologies). The first baseline image of each primate was normalized to an INIA19 MR Rhesus brain template (Rohlfing et al. 2012). Blockade condition images were co-registered to the corresponding baseline image thus transforming each image to a standard space. An atlas including relevant brain regions was applied. Average activity concentration (kBq/cc) within each ROI was determined and time activity curves (TAC) representing the regional brain activity concentration over time were generated. Time activity curves were also expressed in SUVs by normalizing by the weight of the animal and the injected dose.
Statistical analysis
All statistical tests were performed using a one-way ANOVA followed by Dunnett’s post-hoc tests using GraphPad Prism 6 software. The statistical significance was set at p < 0.05.
Results
Radiosynthesis
Labeling of 18F-F13640 was obtained from the nitro precursor with a radiochemical yield of 6% corrected for decay and 90-min radiosynthesis time. No radioactive by-products were observed, and the HPLC conditions chosen ensured good separation of 18F-F13640 from its precursor, as confirmed by quality control (Fig. 1). Radiochemical purity was greater than 98%, and specific activity ranged between 30 and 122 GBq µmol−1, corrected at EOS.18F-MPPF was obtained from the nitro precursor with a radiochemical yield of 25% corrected for decay. Radiochemical purity was higher than 98% and specific activity ranged between 37 and 111 GBq µmol−1, corrected at EOS.
Unmetabolized radiotracer in rat brain
The amount of total radioactivity was found to correspond mainly to unmetabolized 18F-F13640 (Fig. 2). Even at the latest time point of 50 min after injection, more than 87% of the radioactivity was attributed to 18F-F13640 (experiments performed in duplicate). Overall, during the first hour after injection, the amount of radiometabolites was largely a minority compared to the unmetabolized radiotracer.
In vitro distribution studies
In vitro distribution of 18F-F13640 was evaluated by autoradiography in rat brains (Fig. 1a). The binding pattern matched the known distribution of 5-HT1A receptors, with a strong binding in the cingulate and entorhinal cortex, hippocampus, lateral septum, raphe nucleus, and, to a lesser extent, brainstem. Almost no binding was observed in the cerebellum. Specific binding of 18F-F13640 (ratio of the region of interest to cerebellum) was 9.4 in hippocampus, 4.4 in cingulate cortex, 4.6 in dorsal raphe nucleus and 1.6 in thalamus.

a Synthesis of nitro-precursor 7 and radiosynthesis of 18F-F13640. b Quality chromatogram of final 18F-F13640 (UV ultraviolet absorbance and radio, radioactivity) shows radiochemical purity. DCM dichloromethane, DMF dimethylformamide, DMSO dimethylsulfoxide, TFA trifluoroacetic acid
In vitro competition studies
In 5-HT1A receptor-rich areas, 18F-F13640 binding was reduced in a concentration-dependent manner by WAY-100,635 (− 29, − 64 and − 83% in the hippocampus, and 32, 49 and 67% of decrease in the cingulate cortex, with 1, 10 and 100 nM of WAY-100635, respectively), whereas it was unchanged in the cerebellum (Fig. 1b). 18F-F13640 binding was also inhibited by serotonin (7, 21, 54 and 75%) of decrease in the hippocampus with 1, 10, 100 nM and 1 µM of 5-HT, respectively (Fig. 1c).
In vitro uncoupling studies
Incubation with 10 µM of the uncoupling agent Gpp(NH)p reduced 18F-F13640 binding by 54% in the cingulate cortex and 47% in the hippocampus and the dorsal raphe nucleus, with no modification in the cerebellum (Fig. 4a). Conversely, 18F-MPPF binding was not statistically different when Gpp(NH)p was added, although there was an increase in some experiments (as shown on Fig. 4a).
Ex vivo competition studies
Sensitivity of 18F-F13640 to endogenous 5-HT release was evaluated in rats by ex vivo autoradiography (Fig. 4b). 18F-F13640 binding was dose-dependently inhibited by fenfluramine in various regions. In hippocampus, cingulate cortex and dorsal raphe nucleus, the competition was significant at the dose of 0.5 mg/kg (28, 26 and 35%, respectively) and highest at 5 mg/kg (72, 74 and 77%, respectively). As a comparison, in similar experiments performed using 18F-MPPF, fenfluramine did not induce any significant decrease of radioactivity, although a tendency was observed at 5 mg/kg (Fig. 4b).
In vivo distribution in rat brain
18F-F13640 showed a high and fast uptake in rat brain, and a slow wash-out during the hour following injection (Fig. 2b). The distribution pattern was different from in vitro observations, with lower differences across regions and highest signal in the brainstem, the thalamus, the dorsal raphe and cortical areas (Fig. 2a). For each ROI, standard uptake value ratios (SUVRs) were calculated with time curves integrated from 10 to 60 min, and cerebellum taken as the non-target region. The highest SUVRs were found in the dorsal raphe nucleus (1.55), the brainstem (1.44) and the thalamus (1.26). The binding was lower but significant in the hippocampus, the cingulate cortex and the striatum (SUVRs about 1.2).

Radiochromatogram of brain radioactivity from 10 to 50 min after intravenous injection of 18F-F13640 in the rat. The main amount of radioactivity (about 90%) was eluted at 6 min, corresponding to the unmetabolized radiotracer (n = 2)
In vivo blockade of 18F-F13640 in rat brain
The intravenous co-injection of the antagonist WAY-100635 with 18F-F13640 induced a significant decrease of tracer uptake in the brain (Fig. 2b). The blockade varied between 30 and 45%. The uptake was also lower in the cerebellum, resulting in unchanged SUVRs compared to baseline scans. 18F-F13640 brain uptake was also strongly reduced by intravenous injection of the agonist 8-OH-DPAT, 20 min prior to the PET scan (Fig. 2b). The blockade varied between 83 and 89% and also changed the slope of the time-activity curves compared to baseline scans. However, the signal was also reduced in the cerebellum, resulting in few differences in SUVRs compared to baseline scans.
In vivo distribution in cat brain
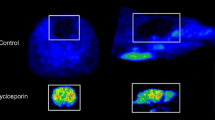
18F-F13640 showed a high and fast uptake in the whole cat brain, and a very slow wash-out during the 90 min of acquisition (Figs. 3, 7). Inter-regional differences were greater than in rat brain (see Table 1). 18F-F13640 uptake was high in cortical areas (especially anterior cingulate cortex, somatosensory cortex and entorhinal cortex), in insula, amygdala, thalamus, dorsal raphe nucleus and pontine nuclei. The cerebellum exhibited the lowest signal, but binding was heterogeneous in this region (moderate in the cerebellar lobes and low in the nuclei, see Fig. 3a). For comparison, standard uptake value ratios (SUVRs) were calculated with time curves integrated from 30 to 90 min, and cerebellar nuclei taken as the non-target region. The highest SUVRs were found in the thalamus (2.00), the anterior cingulate cortex (1.99), the amygdala (1.98), the dorsal raphe nucleus (1.96) and the pontine nuclei (1.80). The binding was moderate in the hippocampus (1.4) and the lateral septum (1.38) and lower in the striatum (1.12). The test–retest scans in cat#1 showed a good reproducibility of results: all regions included, the mean difference between the scans was about 15% in SUVs and 2% in SUVRs (with cerebellar nuclei as the non-target region).

a In vitro autoradiograms of brain sections of rat incubated with 18F-F13640 and corresponding anatomic slices. b In vitro competition in rat brain between 18F-F13640 and 5-HT1A antagonist WAY-100635 at increasing concentrations (n = 3), with corresponding autoradiograms. c In vitro competition in rat brain between 18F-F13640 and serotonin at increasing concentrations (n = 3), with corresponding autoradiograms. Results expressed in percentages of optical densities compared to control ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001, versus control; One-way ANOVA followed by a Dunnett’s post-hoc test). Cereb cerebellum, Cing Cx cingulate cortex, Hip hippocampus
In vivo blockade of 18F-F13640 in cat brain
The intravenous pre-injection of WAY-100635, either at 1 mg/kg 30 min before, or 2 mg/kg 5 min before, decreased 18F-F13640 uptake in the three cats (see Figs. 3b, 7). At 2 mg/kg, the blockade varied between 8 and 35% depending on the regions (Table 1). The strongest blockade was found in the cingulate cortex, dorsal raphe nucleus, thalamus and amygdala. In cat #2, the uptake was also lower in the cerebellar nuclei, resulting in unchanged SUVRs compared to the baseline scan (Fig. 7b). 18F-F13640 brain uptake was also strongly reduced by intravenous injection of the agonist 8-OH-DPAT at 1 mg/kg, 20 min prior to the PET scan (Figs. 3b, 7b, c). The blockade varied between 52 and 71% and also changed the slope of the time-activity curves compared to baseline scans (Table 1). The blockade was strong in cingulate cortex, dorsal raphe nucleus, thalamus and amygdala, but also in brainstem and pontine nuclei. Globally, the level of radioactivity in all ROIs was close to the cerebellar nuclei (Fig. 7b, c).
In vivo distribution in Rhesus monkey brain
18F-F13640 showed a high and fast uptake in the whole brain, and a slow wash-out over the 120–180 min of acquisition (Figs. 4, 5). 18F-F13640 binding was high in amygdala, insula, hippocampus, anterior cingulate cortex and raphe nuclei. In particular, the posterior parahippocampal gyrus (PPHG) displayed a very strong signal compared to the other regions. The lowest uptake was observed in occipital cortex and cerebellum. 18F-F13640 binding was heterogeneous in cerebellum, with more uptake in the lobes than in the nuclei (Fig. 4a). For comparison, standard uptake value ratios (SUVRs) were calculated with time curves integrated from 30 to 90 min, and cerebellar nuclei taken as the non-target region. The highest SUVRs were found in the PPHG (2.93), the anterior cingulate cortex (2.22), the insula (2.20), the dorsal raphe nucleus (1.91), the amygdala (1.91) and the hippocampus (1.89). The binding was moderate in the caudate-putamen (1.75), the thalamus (1.68) and the brainstem (1.46).

a Comparison of effects of Gpp(NH)p, decoupling receptors from G proteins, on 5-HT1A agonist 18F-F13640 and antagonist 18F-MPPF in vitro binding in rat brain. b Comparison of effects of fenfluramine administration at different doses on 18F-F13640 and 18F-MPPF ex vivo binding in rat brain (n = 3). Results expressed in percentages of optical densities compared to control ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001, versus control; One-way ANOVA followed by a Dunnett’s post-hoc test). Cing Cx cingulate cortex, Hip hippocampus

a Coronal microPET images summed for 60 min on corresponding MRI images, showing 18F-F13640 distribution in rat brain. Scale in SUVs. b Transverse microPET images summed for 60 min in three different experiments; from left to right: control experiment/co-injection of WAY-100635 (2 mg/kg)/pre-injection of 8-OH-DPAT (2 mg/kg). Scale in SUVs. c Time–activity curves of 18F-F13640 at baseline (n = 4) and in co-injection with WAY-100635 (2 mg/kg; n = 2) or with pre-injection of 5-HT1A agonist 8-OH-DPAT (2 mg/kg, 20 min before radiotracer injection; n = 2). Results expressed in mean SUVs ± SEM. Amy Amygdala, Brainst Brainstem, Cereb cerebellum, Col colliculus, DRN dorsal raphe nucleus, Ent Ctx entorhinal cortex, Hip hippocampus, Ins insula, Sept lateral septum, SS Ctx somatosensory cortex, Striat striatum, Sub subiculum, Thal thalamus
In vivo blockade of 18F-F13640 in Rhesus monkey brain
The intravenous injection of WAY-100635 at 2 mg/kg, 5 min prior to the PET scan, led to a decrease of 18F-F13640 binding in the two monkeys (Figs. 4b, 5). The blockade was homogeneous and varied between 38 and 49% (Table 2). In monkey #2, the uptake was also lowered in the cerebellar nuclei, resulting in unchanged SUVRs in certain regions compared to the baseline scan (Fig. 5b).
Discussion
Despite numerous attempts, there is currently no specific and full agonist of 5-HT1A receptors available for clinical PET imaging. In this context, our team focused on a series of highly selective and fully active 5-HT1A agonists (Maurel et al. 2007). We previously radiolabeled two chemical analogs from this series, F15599 and F13714 (Lemoine et al. 2010, 2012; Yokoyama et al. 2016). Even though both molecules allowed specific in vivo imaging of functional 5-HT1A receptors, the signal/noise ratio of 18F-F15599 was insufficient for development as a radiopharmaceutical (Lemoine et al. 2010). On the contrary, the signal/noise ratio of 18F-F13714 was better but binding was shown to be irreversible in non-human primates, complicating the quantification of binding parameters (Lemoine et al. 2012; Yokoyama et al. 2016; Tavares et al. 2013). Having taken into account these limitations, we identified another analog from this series, F13640 and present here its radiopharmacological evaluations in rats and rhesus monkey.
Since F13640 affinity for 5-HT1A receptors is intermediate compared to the previous molecules, F15599 and F13714 – with a Ki of 1 vs. 2.24 nM and 0.1 nM, respectively (Maurel et al. 2007); it may be better adapted to achieving an improved signal/noise ratio, but sufficiently modest to avoid eliciting irreversible binding. Interestingly, F13640, a drug that is currently undergoing clinical development as is also known as befiradol or NLX-112 (Iderberg et al. 2015), is a well-characterized 5-HT1A agonist which presents several advantages. First, it exhibits an outstanding selectivity for 5-HT1A receptors, a property that is rarely obtained for a brain PET radiopharmaceutical: its affinity for 46 other receptors, enzymes, and transporters is at least 1,000-fold lower (Colpaert et al. 2002). Second, F13640 exhibits a particularly efficacious agonist activity at 5-HT1A receptors in all essays in which it has been characterized, both in vitro and in vivo (Colpaert et al. 2002; Heusler et al. 2010; Pauwels and Colpaert 2003; Wurch et al. 2003; Newman-Tancredi et al. 2017). Third, its chemical structure presents a fluorine atom enabling a straightforward PET radiofluorination. All these properties make F13640 a good candidate as a 5-HT1A agonist PET radiotracer and encouraged its exploration.
Interestingly, analysis of radiometabolites in rat brain showed that a large majority of brain radioactivity was due to unchanged 18F-F13640, from 10 to 50 min after injection, thus ruling out any significant contribution from radiometabolites to brain uptake.
In vitro distribution of 18F-F13640 in rat brain sections matched the reported distribution of 5-HT1A receptors, with a strong binding in dorsal raphe nucleus, hippocampus, lateral septum and cortical areas, and almost no binding in striatum and cerebellum (Lanfumey and Hamon 2000). Competition studies confirmed the specificity of 18F-F13640 for 5-HT1A receptors as binding in 5-HT1A-rich regions was reduced by increasing concentrations of serotonin and WAY-100635. 18F-F13640 binding was also markedly reduced by Gpp(NH)p, an agent that switches receptors into an uncoupled state, demonstrating that it binds preferentially to G-protein coupled 5-HT1A receptors in vitro, a property that is consistent with its agonist activity. In contrast, uncoupling tends to increase the binding of the antagonist 18F-MPPF, an observation that was previously reported (Lemoine et al. 2010; Vidal et al. 2016). These results highlight that 18F-F13640 and 18F-MPPF, both 5-HT1A receptor radioligands, bind to different pools of receptors depending on their coupling state.
This difference of binding pattern between an antagonist and an agonist radiotracer was confirmed in vivo. Interestingly, 18F-F13640 distribution pattern in rat brain was markedly different between in vitro and in vivo studies. In vitro observations were consistent with the known distribution of 5-HT1A receptors, suggesting a classical correlation between coupled and total receptors densities. On the contrary, in vivo binding in PET studies (which may be presumed to reflect the percentage of coupled receptors) was disconnected from the known density of the whole population of 5-HT1A receptors. 18F-F13640 binding was particularly high in brainstem and thalamus, which are regions usually described as containing a moderate to low density of receptors. 18F-F13640 uptake was relatively moderate in hippocampus and high in the dorsal raphe nucleus, both regions particularly rich in 5-HT1A receptors. This last observation is interesting because raphe nuclei exert pivotal control of the serotonergic system (Andrade et al. 2015) and is usually too small area to be clearly detected in microPET by other 5-HT1A PET radiotracers. In rhesus monkey, 18F-F13640 in vivo binding was high in cingulate cortex, insula, amygdala, dorsal raphe nucleus, and also in hippocampus contrary to the rat. Remarkably, 18F-F13640 uptake in the parahippocampal gyrus, a small region next to the hippocampus, was at least two times higher than in any other region, suggesting that this region contains a very high density of G-coupled 5-HT1A receptors in rhesus monkey.
Taken together, the results showed that in vivo distribution pattern of 18F-F13640, a specific 5-HT1A agonist radiotracer, are very different from those of an antagonist radiotracer. Similar results have been recently reported in our study with marmosets, showing binding pattern differences between 18F-MPPF and the previous agonist radiotracer, 18F-F13714, both in conscious and anesthetized animals (Yokoyama et al. 2016). We also observed striking differences between in vitro and in vivo results in rat brain. The differences in binding patterns between radiotracers and in vitro/in vivo conditions are unlikely to be explained by non-specific binding, as 18F-F13640 binding was strongly inhibited by the antagonist WAY-100635 even in regions with unexpectedly high signal such as thalamus (Figs. 2–5). In the rat and the cat, although WAY-100635 blockade was incomplete, 18F-F13640 binding was almost completely blocked by the agonist 8-OH-DPAT. This may be due to a more effective competition between agonists as they both compete for receptors in the high-affinity state only. In addition, as indicated above, the properties of F13640 are well documented both in vitro and in vivo. All these data describe F13640 as a “super-agonist” highly specific and with a high affinity for 5-HT1A receptors, tested on many other brain targets, including other 5-HT receptors (Colpaert et al. 2002; Heusler et al. 2010) and rendering very unlikely a potential interaction with an unwanted off-target. A probable explanation for the distinct binding patterns between 5-HT1A agonist and antagonist radiotracers could be the ratio of coupled versus uncoupled receptors which might be much lower in vivo compared to in vitro findings, an hypothesis that has already been suggested to explain the lack of sensitivity of 5-HT1A antagonists radiotracers to competition with 5-HT1A agonists and endogenous serotonin release (Bantick et al. 2004a, b; Udo de Haes et al. 2006). As it has been shown that cell surface dynamics of 5-HT1A receptors are influenced by G-protein coupling, it might also be possible that agonist and antagonist binding sites (i.e. G-protein-coupled and uncoupled receptors) are actually differently located on the membrane in vivo (Pucadyil and Chattopadhyay 2007) (Fig. 6).

a Coronal PET images summed for 90 min on corresponding MRI images, showing 18F-F13640 distribution in cat brain. Scale in SUVs. b Transverse PET images summed from 30 to 90 min in three different experiments; from left to right: control experiment/pre-injection of WAY-100635 (2 mg/kg)/pre-injection of 8-OH-DPAT (1 mg/kg). Scale in SUVs. All images belong to the same animal (cat #2). Ant Cing anterior cingulate cortex, Caud caudate, Cereb GM cerebellar grey matter, Cereb Nucl cerebellar nuclei, Cort WM cortical white matter, PN pontine nuclei, Post Cing posterior cingulate cortex

a Time–activity curves of 18F-F13640 in cat #1; in black; mean SUVs of test-retest (± SEM); in white, pre-injection of WAY-100635 at 1 mg/kg, 30 min before the scan. b Time–activity curves of 18F-F13640 in cat #2; in black; baseline scan; in white, pre-injection of WAY-100635 at 2 mg/kg, 5 min before the scan; in grey, pre-injection of 8-OH-DPAT at 1 mg/kg, 20 min before the scan. c Time–activity curves of 18F-F13640 in cat #3; in black; baseline scan; in white, pre-injection of WAY-100635 at 2 mg/kg, 5 min before the scan; in grey, pre-injection of 8-OH-DPAT at 1 mg/kg, 20 minutes before the scan
To go further, some remarks should also be added concerning the biased properties of F13640. The recent concept of biased agonism states that some agonists are able to target specifically certain subpopulations of receptors depending on their coupling with different intracellular effectors, such as G proteins (Newman-Tancredi, 2011). F13640 was recently shown to be a biased 5-HT1A agonist, preferentially activating Gαo proteins (Newman-Tancredi et al. 2017), which is likely to influence the central distribution of the radiotracer as it was shown that 5-HT1A receptors can couple to different G protein subtypes depending on their localization in the brain (Mannoury La Cour et al. 2006). For now, these different hypotheses remain speculative and new studies are needed to understand the molecular basis of the distinctive binding profile of 18F-F13640 compared to classical 5-HT1A radiotracers.
In any case, increasing evidence including our own results suggest that the use of 5-HT1A agonists in PET imaging could generate new information that was not apparent with antagonist tracers or accessible by the use of in vitro techniques. For instance, it has been reported recently that the strong effects of F13640 on opiate-induced respiratory depression in rats involve sites or mechanisms in the brainstem that are not functional in vitro (Ren et al. 2015). Consistent with these findings, in rat brain we observed a very strong labeling in the brainstem in vivo but not in vitro, thus reinforcing the idea that 18F-F13640 binding has a strong functional meaning. Similarly, although the striatum is usually described as devoid of 5-HT1A receptors, significant specific binding was observed in our PET experiments in the three species. Supporting our findings, it has been demonstrated thanks to microinjection experiments that the antidyskinetic properties of 5-HT1A agonists are conveyed in part via a small population of functional receptors within this region (Bishop et al. 2009; Meadows et al. 2017). In this context, it should be mentioned that F13640 is currently in clinical development for Parkinson’s disease patients who suffer from L-DOPA-induced dyskinesia. Our results also suggest that there are significant species differences in the distribution of G-coupled 5-HT1A receptors—a piece of information which would not have been accessible from experiments using antagonist tracers that bind the total pool of receptors, irrespective of their affinity state (Figs. 8, 9).

a Coronal PET images summed between 60 to 180 minutes on corresponding MRI images, showing 18F-F13640 distribution in rhesus monkey brain. Scale in SUVs. b Transverse and coronal PET images summed from 60 to 180 min in two different experiments; on the left, baseline experiment; on the right, pre-injection of WAY-100635 (2 mg/kg). Scale in SUVs. All images belong to the same animal (monkey #1). PPHG posterior parahippocampal gyrus

a Time–activity curves of 18F-F13640 in monkey #1; in black, baseline scan; in white, pre-injection of WAY-100635 at 2 mg/kg, 5 min before the scan. b Time-activity curves of 18F-F13640 in monkey #2; in black, baseline scan; in white, pre-injection of WAY-100635 at 2 mg/kg, 5 min before the scan.
In preliminary ex vivo experiments, we also evaluated 18F-F13640 sensitivity to endogenous 5-HT release by pharmacological challenge with increasing doses of fenfluramine in rats. Our results suggest that 18F-F13640 binding is dose-dependently inhibited by fenfluramine, with a significant effect starting at a low dose of 0.5 mg/kg. The competition occurred in various regions, including hippocampus, dorsal raphe nucleus, thalamus and cortical areas. In contrast, using 18F-MPPF a decreasing tendency was observed at the high dose of 5 mg/kg only, which is not surprising considering previous studies that showed 5-HT1A antagonists radiotracers are probably not sensitive enough to detect physiological changes in 5-HT release (Paterson et al. 2010; Jagoda et al. 2006). These results support the hypothesis that agonist radiotracers are more sensitive than antagonists to extracellular endogenous agonist fluctuations, but even more than expected, as in our findings 18F-F13640 uptake was decreased at doses of fenfluramine at least ten times lower than 18F-MPPF. Nevertheless, additional studies are necessary to confirm that 18F-F13640 is really sensitive to 5-HT release and not to other pharmacological effects of fenfluramine.
Finally at this stage, some methodological and radiopharmacological issues need to be exercised. First, caution is advisable with the choice of a reference region for SUVRs with this tracer. For simple comparisons between regions, we here chose the cerebellar nuclei, because it was the region displaying the lowest signal in cat and rhesus monkey. The whole cerebellum could not be taken as a non-target region because the lobes and vermis included significant uptake. However, in one cat and one rhesus monkey, the TACs in the cerebellar nuclei were also slightly decreased in the blockade conditions, which could be due to partial volume effects considering the small size of this region, or by the presence of a non-negligible amount of 5-HT1A receptors. Although these results were unexpected, as the cerebellum is often described as a region devoid of 5-HT1A receptors, some data already reported significant binding of 11C-WAY-100635 in the cerebellar lobes and vermis and suggested that cerebellar white matter may be a more suitable reference region (Parsey et al. 2005; Hirvonen et al. 2007; Ganz et al. 2017). Moreover, it is noteworthy that an iontophoresis study actually reported the presence of functional 5-HT1A receptors located in the cerebellar gray matter in cats (Kerr et Bishop, 1992). In any case, further kinetic modeling studies will be needed to fully understand and quantify 18F-F13640, and given our results it can be expected that the choice of a reference region will have to be carefully validated for each species of interest by comparisons with arterial input function. Second, in certain animals, the time-activity curves showed almost no decline during the scan, meaning that dissociation of 18F-F13640 from 5-HT1A receptors was very slow. This is comparable to the previously evaluated structural analog, 18F-F13714, although 18F-F13640 affinity for 5-HT1A receptors is ten times lower. The slow kinetics is probably inherent to the high affinity and efficacy of these agonists for 5-HT1A receptors, and has already been reported in vitro compared to other 5-HT1A ligands (Heusler et al. 2010). However, unlike 18F-F13714, the wash-out was faster in certain animals (namely, cat #3 and monkey #1), which constitutes an interesting point that deserve to be explored, and preliminary findings using fenfluramine in cats show that 18F-F13640 binding is reversible despite a slow wash-out (unshowed data) which is more advantageous for quantification and constitutes a favorable point for a future use in humans.
Conclusion
We report here the first preclinical evaluation of the 5-HT1A receptor agonist radiotracer 18F-F13640. 18F-F13640 is highly selective for 5-HT1A receptors and sensitive to G-protein uncoupling in vitro. In anesthetized rats, the radioligand rapidly entered the brain without significant presence of radiometabolites.18F-F13640 binding in vivo in anesthetized rats, cat and non-human primate was specific as demonstrated by blocking experiments, but with striking differences in distribution patterns compared to in vitro results and classical observations with antagonist radiotracers. 18F-F13640 uptake was dose-dependently inhibited by fenfluramine in various regions in rat brain, suggesting it is sensitive to 5-HT endogenous release. 18F-F13640 is, therefore, a promising PET tracer for in vivo imaging and quantification of functional 5-HT1A receptors in the human brain. This justifies that a first-in-man study with 18F-F13640 is now scheduled.
Conflict of interest
Dr. Newman-Tancredi is an employee and stockholder of Neurolixis. The other authors report no conflict of interest and have nothing to disclose.
Ethical approval
All experiments in Lyon were performed in accordance with European guidelines for care of laboratory animals (2010/63/EU) and were approved by the ethics animal committee of the Université de Lyon. All imaging studies in New Haven were carried out under institutional animal care protocols complying with US federal regulations and according to the Yale University Institutional Animal Care and Use Committee.
References
Andrade R, Huereca D, Lyons JG, Andrade EM, McGregor KM (2015) 5-HT1A receptor-mediated autoinhibition and the control of serotonergic cell firing. ACS Chem Neurosci 6(7):1110–1115. https://doi.org/10.1021/acschemneuro.5b00034
Avissar S, Schreiber G (2006) The involvement of G proteins and regulators of receptor-G protein coupling in the pathophysiology, diagnosis and treatment of mood disorders. Clin Chim Acta (1–2):37–47. https://doi.org/10.1016/j.cca.2005.11.003
Aznavour N, Rbah L, Leger L, Buda C, Sastre JP, Imhof A, Charnay Y, Zimmer L (2006) A comparison of in vivo and in vitro neuroimaging of 5-HT 1A receptor binding sites in the cat brain. J Chem Neuroanat 31(3):226–232. https://doi.org/10.1016/j.jchemneu.2006.01.006
Bantick RA, Montgomery AJ, Bench CJ, Choudhry T, Malek N, McKenna PJ, Quested DJ, Deakin JF, Grasby PM (2004a) A positron emission tomography study of the 5-HT1A receptor in schizophrenia and during clozapine treatment. J Psychopharmacol (3):346–354. https://doi.org/10.1177/026988110401800304
Bantick RA, Rabiner EA, Hirani E, de Vries MH, Hume SP, Grasby PM (2004b) Occupancy of agonist drugs at the 5-HT1A receptor. Neuropsychopharmacology 29(5):847–859. https://doi.org/10.1038/sj.npp.1300390
Becker G, Streichenberger N, Billard T, Newman-Tancredi A, Zimmer L (2014) A postmortem study to compare agonist and antagonist 5-HT1A receptor-binding sites in Alzheimer’s disease. CNS Neurosci Ther 20(10):930–934. https://doi.org/10.1111/cns.12306
Bishop C, Krolewski DM, Eskow KL, Barnum CJ, Dupre KB, Deak T, Walker PD (2009) Contribution of the striatum to the effects of 5-HT1A receptor stimulation in L-DOPA-treated hemiparkinsonian rats. J Neurosci Res 87(7):1645–1658. https://doi.org/10.1002/jnr.21978
Colpaert FC, Tarayre JP, Koek W, Pauwels PJ, Bardin L, Xu XJ, Wiesenfeld-Hallin Z, Cosi C, Carilla-Durand E, Assie MB, Vacher B (2002) Large-amplitude 5-HT1A receptor activation: a new mechanism of profound, central analgesia. Neuropharmacology 43(6):945–958
Ganz M, Feng L, Hansen HD, Beliveau V, Svarer C, Knudsen GM, Greve DN (2017) Cerebellar heterogeneity and its impact on PET data quantification of 5-HT receptor radioligands. J Cereb Blood Flow Metab 37(9):3243–3252. https://doi.org/10.1177/0271678X16686092
Gozlan H, Thibault S, Laporte AM, Lima L, Hamon M (1995) The selective 5-HT1A antagonist radioligand [3H]WAY 100635 labels both G-protein-coupled and free 5-HT1A receptors in rat brain membranes. Eur J Pharmacol 288(2):173–186
Hendry N, Christie I, Rabiner EA, Laruelle M, Watson J (2011) In vitro assessment of the agonist properties of the novel 5-HT1A receptor ligand, CUMI-101 (MMP), in rat brain tissue. Nucl Med Biol 38(2):273–277. https://doi.org/10.1016/j.nucmedbio.2010.08.003
Heusler P, Palmier C, Tardif S, Bernois S, Colpaert FC, Cussac D (2010) [(3)H]-F13640, a novel, selective and high-efficacy serotonin 5-HT(1A) receptor agonist radioligand. Naunyn Schmiedebergs Arch Pharmacol 382(4):321–330. https://doi.org/10.1007/s00210-010-0551-4
Hirvonen J, Kajander J, Allonen T, Oikonen V, Nagren K, Hietala J (2007) Measurement of serotonin 5-HT1A receptor binding using positron emission tomography and [carbonyl-(11)C]WAY-100635-considerations on the validity of cerebellum as a reference region. J Cereb Blood Flow Metab 27(1):185–195. https://doi.org/10.1038/sj.jcbfm.9600326
Iderberg H, McCreary AC, Varney MA, Kleven MS, Koek W, Bardin L, Depoortere R, Cenci MA, Newman-Tancredi A (2015) NLX-112, a novel 5-HT1A receptor agonist for the treatment of L-DOPA-induced dyskinesia: Behavioral and neurochemical profile in rat. Exp Neurol 271:335–350. https://doi.org/10.1016/j.expneurol.2015.05.021
Jagoda EM, Lang L, Tokugawa J, Simmons A, Ma Y, Contoreggi C, Kiesewetter D, Eckelman WC (2006) Development of 5-HT1A receptor radioligands to determine receptor density and changes in endogenous 5-HT. Synapse 59(6):330–341. https://doi.org/10.1002/syn.20246
Kumar JS, Prabhakaran J, Majo VJ, Milak MS, Hsiung SC, Tamir H, Simpson NR, Van Heertum RL, Mann JJ, Parsey RV (2007) Synthesis and in vivo evaluation of a novel 5-HT1A receptor agonist radioligand [O-methyl- 11C]2-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)-4-methyl-1,2,4-triazine-3,5(2H,4H)dione in nonhuman primates. Eur J Nucl Med Mol Imaging 34(7):1050–1060. https://doi.org/10.1007/s00259-006-0324-y
Kumar JS, Parsey RV, Kassir SA, Majo VJ, Milak MS, Prabhakaran J, Simpson NR, Underwood MD, Mann JJ, Arango V (2013) Autoradiographic evaluation of [3H]CUMI-101, a novel, selective 5-HT1AR ligand in human and baboon brain. Brain Res 1507:11–18. https://doi.org/10.1016/j.brainres.2013.02.035
Lancelot S, Roche R, Slimen A, Bouillot C, Levigoureux E, Langlois JB, Zimmer L, Costes N (2014) A multi-atlas based method for automated anatomical rat brain MRI segmentation and extraction of PET activity. PloS one 9(10):e109113. https://doi.org/10.1371/journal.pone.0109113
Lanfumey L, Hamon M (2000) Central 5-HT(1A) receptors: regional distribution and functional characteristics. Nucl Med Biol 27(5):429–435
Le Bars D, Lemaire C, Ginovart N, Plenevaux A, Aerts J, Brihaye C, Hassoun W, Leviel V, Mekhsian P, Weissmann D, Pujol JF, Luxen A, Comar D (1998) High-yield radiosynthesis and preliminary in vivo evaluation of p-[18F]MPPF, a fluoro analog of WAY-100635. Nucl Med Biol 25(4):343–350
Lemoine L, Verdurand M, Vacher B, Blanc E, Le Bars D, Newman-Tancredi A, Zimmer L (2010) [18F]F15599, a novel 5-HT1A receptor agonist, as a radioligand for PET neuroimaging. Eur J Nucl Med Mol Imaging 37(3):594–605. https://doi.org/10.1007/s00259-009-1274-y
Lemoine L, Becker G, Vacher B, Billard T, Lancelot S, Newman-Tancredi A, Zimmer L (2012) Radiosynthesis and preclinical evaluation of 18F-F13714 as a fluorinated 5-HT1A receptor agonist radioligand for PET neuroimaging. J Nucl Med 53(6):969–976. https://doi.org/10.2967/jnumed.111.101212
Mannoury la Cour C, El Mestikawy S, Hanoun N, Hamon M, Lanfumey L (2006) Regional differences in the coupling of 5-hydroxytryptamine-1A receptors to G proteins in the rat brain. Mol Pharmacol 70(3):1013–1021. https://doi.org/10.1124/mol.106.022756
Maurel JL, Autin JM, Funes P, Newman-Tancredi A, Colpaert F, Vacher B (2007) High-efficacy 5-HT1A agonists for antidepressant treatment: a renewed opportunity. J Med Chem (20):5024–5033. https://doi.org/10.1021/jm070714l
Meadows SM, Chambers NE, Conti MM, Bossert SC, Tasber C, Sheena E, Varney M, Newman-Tancredi A, Bishop C (2017) Characterizing the differential roles of striatal 5-HT1A auto- and hetero-receptors in the reduction of l-DOPA-induced dyskinesia. Exp Neurol 292:168–178. https://doi.org/10.1016/j.expneurol.2017.03.013
Milak MS, DeLorenzo C, Zanderigo F, Prabhakaran J, Kumar JS, Majo VJ, Mann JJ, Parsey RV (2010) In vivo quantification of human serotonin 1A receptor using 11C-CUMI-101, an agonist PET radiotracer. J Nucl Med 51(12):1892–1900. https://doi.org/10.2967/jnumed.110.076257
Mongeau R, Welner SA, Quirion R, Suranyi-Cadotte BE (1992) Further evidence for differential affinity states of the serotonin1A receptor in rat hippocampus. Brain Res 590(1–2):229–238
Newman-Tancredi A, Martel JC, Cosi C, Heusler P, Lestienne F, Varney MA, Cussac D (2017) Distinctive in vitro signal transduction profile of NLX-112, a potent and efficacious serotonin 5-HT1A receptor agonist. J Pharm Pharmacol 69(9):1178–1190. https://doi.org/10.1111/jphp.12762
Parsey RV, Arango V, Olvet DM, Oquendo MA, Van Heertum RL, John Mann J (2005) Regional heterogeneity of 5-HT1A receptors in human cerebellum as assessed by positron emission tomography. J Cereb Blood Flow Metab 25(7):785–793. https://doi.org/10.1038/sj.jcbfm.9600072
Paterson LM, Tyacke RJ, Nutt DJ, Knudsen GM (2010) Measuring endogenous 5-HT release by emission tomography: promises and pitfalls. J Cereb Blood Flow Metab 30(10):1682–1706. https://doi.org/10.1038/jcbfm.2010.104
Pauwels PJ, Colpaert FC (2003) Ca2 + responses in Chinese hamster ovary-K1 cells demonstrate an atypical pattern of ligand-induced 5-HT1A receptor activation. J Pharmacol Exp Ther 307(2):608–614. https://doi.org/10.1124/jpet.103.055871
Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates. Academic Press, New York
Pinborg LH, Feng L, Haahr ME, Gillings N, Dyssegaard A, Madsen J, Svarer C, Yndgaard S, Kjaer TW, Parsey RV, Hansen HD, Ettrup A, Paulson OB, Knudsen GM (2012) No change in [(1)(1)C]CUMI-101 binding to 5-HT(1A) receptors after intravenous citalopram in human. Synapse 66(10):880–884. https://doi.org/10.1002/syn.21579
Pucadyil TJ, Chattopadhyay A (2007) The human serotonin1A receptor exhibits G-protein-dependent cell surface dynamics. Glycoconj J 24(1):25–31. https://doi.org/10.1007/s10719-006-9008-x
Ren J, Ding X, Greer JJ (2015) 5-HT1A receptor agonist Befiradol reduces fentanyl-induced respiratory depression, analgesia, and sedation in rats. Anesthesiology 122(2):424–434. https://doi.org/10.1097/ALN.0000000000000490
Rohlfing T, Kroenke CD, Sullivan EV, Dubach MF, Bowden DM, Grant KA, Pfefferbaum A (2012) The INIA19 Template and NeuroMaps Atlas for Primate Brain Image Parcellation and Spatial Normalization. Front Neuroinf 6:27. https://doi.org/10.3389/fninf.2012.00027
Shrestha SS, Liow JS, Lu S, Jenko K, Gladding RL, Svenningsson P, Morse CL, Zoghbi SS, Pike VW, Innis RB (2014) (11)C-CUMI-101, a PET radioligand, behaves as a serotonin 1A receptor antagonist and also binds to alpha(1) adrenoceptors in brain. J Nucl Med 55(1):141–146. https://doi.org/10.2967/jnumed.113.125831
Shrestha SS, Liow JS, Jenko K, Ikawa M, Zoghbi SS, Innis RB (2016) The 5-HT1A Receptor PET Radioligand 11C-CUMI-101 Has Significant Binding to alpha1-Adrenoceptors in Human Cerebellum, Limiting Its Use as a Reference Region. J Nucl Med 57(12):1945–1948. https://doi.org/10.2967/jnumed.116.174151
Tavares A, Becker G, Barret O, Morley T, Alagille D, Vacher B, Newman-Tancredi A, Tamagnan G, Zimmer L (2013) Initial evaluation of [18F]F13714, a novel 5-HT1A receptor agonist in non-human primates. Annual Congress of EANM
Udo de Haes JI, Harada N, Elsinga PH, Maguire RP, Tsukada H (2006) Effect of fenfluramine-induced increases in serotonin release on [18F]MPPF binding: a continuous infusion PET study in conscious monkeys. Synapse 59(1):18–26. https://doi.org/10.1002/syn.20209
Vidal B, Sebti J, Verdurand M, Fieux S, Billard T, Streichenberger N, Troakes C, Newman-Tancredi A, Zimmer L (2016) Agonist and antagonist bind differently to 5-HT1A receptors during Alzheimer’s disease: a post-mortem study with PET radiopharmaceuticals. Neuropharmacology 109:88–95. https://doi.org/10.1016/j.neuropharm.2016.05.009
Wurch T, Colpaert FC, Pauwels PJ (2003) Mutation in a protein kinase C phosphorylation site of the 5-HT1A receptor preferentially attenuates Ca2 + responses to partial as opposed to higher-efficacy 5-HT1A agonists. Neuropharmacology 44(7):873–881
Yokoyama C, Mawatari A, Kawasaki A, Takeda C, Onoe K, Doi H, Newman-Tancredi A, Zimmer L, Onoe H (2016) Marmoset serotonin 5-HT1A receptor mapping with a biased agonist PET probe 18F-F13714: comparison with an antagonist tracer 18F-MPPF in awake and anesthetized states. Int J Neuropsychopharmacol 19 (12). https://doi.org/10.1093/ijnp/pyw079
Zimmer L (2016) Pharmacological agonists for more-targeted CNS radio-pharmaceuticals. Oncotarget 7(49):80111–80112. https://doi.org/10.18632/oncotarget.13418
Zimmer L, Le Bars D (2013) Current status of positron emission tomography radiotracers for serotonin receptors in humans. J Labelled Comp Radiopharm 56(3–4):105–113. https://doi.org/10.1002/jlcr.3001
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vidal, B., Fieux, S., Colom, M. et al. 18F-F13640 preclinical evaluation in rodent, cat and primate as a 5-HT1A receptor agonist for PET neuroimaging. Brain Struct Funct 223, 2973–2988 (2018). https://doi.org/10.1007/s00429-018-1672-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-018-1672-7