
Abstract
The basal forebrain (BF) is an important regulator of hippocampal and cortical activity. In Alzheimer’s disease (AD), there is a significant loss and dysfunction of cholinergic neurons within the BF, and also a hypertrophy of fibers containing the neuropeptide galanin. Understanding how galanin interacts with BF circuitry is critical in determining what role galanin overexpression plays in the progression of AD. Here, we examined the location and function of galanin in the medial septum/diagonal band (MS/DBB) region of the BF. We show that galanin fibers are located throughout the MS/DBB and intermingled with both cholinergic and GABAergic neurons. Whole-cell patch clamp recordings from MS/DBB neurons in acute slices reveal that galanin decreases tetrodotoxin-sensitive spontaneous GABA release and dampens muscarinic receptor-mediated increases in GABA release in the MS/DBB. These effects are not blocked by pre-exposure to β-amyloid peptide (Aβ1–42). Optogenetic activation of cholinergic neurons in the MS/DBB increases GABA release back onto cholinergic neurons, forming a functional circuit within the MS/DBB. Galanin disrupts this cholinergic-GABAergic circuit by blocking the cholinergic-induced increase in GABA release. These data suggest that galanin works in the BF to reduce inhibitory input onto cholinergic neurons and to prevent cholinergic-induced increase in inhibitory tone. This disinhibition of cholinergic neurons could serve as a compensatory mechanism to counteract the loss of cholinergic signaling that occurs during the progression of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cholinergic neurons in the basal forebrain (BF) send projections to the hippocampus and cortex (Mesulam et al. 1983; Dutar et al. 1995), where they regulate synaptic plasticity and cognition (Huerta and Lisman 1993; Ovsepian et al. 2004; Hasselmo 2006; Gu and Yakel 2011; Hasselmo and Sarter 2011). A decrease in cholinergic signaling in the brain due to the loss or dysfunction of cholinergic neurons in the BF is one of the hallmark features of Alzheimer’s disease (AD) (Davies and Maloney 1976; Bartus et al. 1982; Whitehouse et al. 1982; Mufson et al. 2008; Schliebs and Arendt 2011). Changes in cholinergic transmission have been shown to coincide with the onset of β-amyloid (Aβ) accumulation, and are detectable even during the preclinical stages of AD (Beach et al. 2000; Lim et al. 2015). Altered input onto cholinergic cells has the potential to alleviate or exacerbate this diminished cholinergic neurotransmission by disinhibiting or inhibiting surviving cholinergic cells. Cholinergic neurons in the BF receive input from local GABAergic and glutamatergic neurons, as well as afferent projections from other brain regions (Zaborszky et al. 1999; Colom 2006). The relative strength of these inputs has been shown to change with age (Jasek and Griffith 1998; Griffith et al. 2014), and may also be affected during AD, resulting in an altered ratio of excitatory and inhibitory transmission (Nava-Mesa et al. 2014). However, the extent to which synaptic transmission onto cholinergic neurons in the BF is altered in AD is still unknown. Determining how BF circuitry is altered during AD will be fundamental to understanding disease progression and developing novel therapeutic strategies to alleviate the symptoms of cholinergic dysfunction.
One known alteration that happens to BF circuitry during AD is the hyperinnervation of the BF by fibers containing galanin (Chan-Palay 1988; Mufson et al. 1993; Bowser et al. 1997), a 29–30 amino acid peptide that is expressed throughout the central nervous system (Tatemoto et al. 1983; Perez et al. 2001; Lang et al. 2015). Concurrent with the increase in galanin fiber density, there is an increase in galanin receptor expression (Mufson et al. 2000). The elevated levels of galanin in the BF of AD patients could indicate an important role for this peptide in the etiology of the disease, either as a compensatory mechanism or as an exacerbating factor during disease progression. Several studies have examined the potential role of galanin in AD, with some showing that galanin overexpression impairs learning and memory, and decreases choline acetyltransferase (ChAT) levels in the BF (Steiner et al. 2001; Crawley 2008). Conversely, other studies have shown that hyperinnervation of the BF can increase ChAT levels (Counts et al. 2008) and preserve the expression of neuroprotective genes (Counts et al. 2009) in AD brains, and that galanin can protect against Aβ-induced toxicity and learning deficits (Ding et al. 2006; Elliott-Hunt et al. 2011; Li et al. 2013).
The key to discerning the role of galanin in disease progression may lie in examining the interaction between cholinergic and galaninergic signaling within the BF. Galanin infused into the medial septum can increase acetylcholine release in the ventral hippocampus and improve learning and memory (Elvander et al. 2004; Elvander and Ogren 2005). This enhancement may be partly due to a direct excitation of cholinergic neurons by galanin binding to the galanin receptor subtype 2 (GAL2) (Jhamandas et al. 2002). However, galanin receptors are found throughout the BF, including non-cholinergic cells (Miller et al. 1997; O’Donnell et al. 1999; Mennicken et al. 2002; He et al. 2005), indicating that galanin may have additional roles in regulating synaptic transmission within the BF. Here, we sought to ascertain the role of galaninergic signaling in the MS/DBB region of the BF, which provides the primary cholinergic input to the hippocampus (Dutar et al. 1995). To do this, we examined the location of galaninergic fibers within the MS/DBB and determined whether galanin can modulate synaptic transmission in the MS/DBB or alter the function of endogenous cholinergic signaling. Our results show that galanin is involved in regulating inhibitory input onto cholinergic neurons and can modulate a local cholinergic-GABAergic circuit by blunting acetylcholine-induced increases in GABAergic signaling.
Methods
Animals
Wild-type C57BL/6J mice were obtained from Charles River and ChAT-IRES-cre (Rossi et al. 2011) (ChAT-Cre) mice were initially obtained from Jackson labs and subsequently bred in-house. Mice were maintained on a 12 h light/dark cycle under constant temperature control with free access to food and water. All procedures were approved and performed in compliance with the NIEHS/NIH Humane Care and Use of Animals in Research protocols.
Immunohistochemistry
Eight-week-old C57BL/6J mice, n = 4–6 of each sex, were anesthetized with phenobarbital and transcardially perfused with 0.1 M phosphate-buffered saline (PBS) followed by 4 % paraformaldehyde (PFA) in PBS. Brains were removed from the skull and postfixed overnight in 4 % PFA at 4 °C. Following rinse in PBS, brains were cryoprotected in 30 % sucrose in PBS and embedded in tissue-freezing medium (Triangle Biomedical Sciences). 40 μm free-floating coronal cryosections were collected in PBS and every fourth section was processed for immunolabeling. Following a 3 min antigen retrieval in dH2O at 100 °C, sections were rinsed in PBS, PBS with 0.3 % Triton X-100 (PBST), and then blocked in either 5 % normal goat serum or 5 % normal donkey serum in PBST at room temperature for 1 h. Sections were then incubated overnight at 4 °C with antibodies against Galanin (rabbit anti-Galanin 1:2000; T-4334 Peninsula Labs) and the cholinergic marker choline acetyltransferase (Goat anti-ChAT 1:1000; AB144P Millipore), the GABAergic marker parvalbumin (goat anti-Parvalbumin 1:2000; ab32895 Abcam), or the GABAergic marker somatostatin (mouse anti-Somatostatin [SOM-018] 1:500; GTX71935 GeneTex). Following rinse in PBST, tissue was incubated with Alexa Fluor secondary antibodies at a 1:1,000 dilution (488 goat anti-rabbit; A11034, 488 donkey anti-rabbit; A21206, 568 goat anti-mouse; A11031, and 568 donkey anti-goat; A11057) for two hours at room temperature. Sections were rinsed with PBS, mounted onto slides, and coverslipped with vectashield hard set mounting medium (Vector Laboratories, Inc.; H-1400). Images were collected on Zeiss LSM 780 and 880 inverted confocal microscopes.
Electrophysiology
At 6–10 weeks of age, mice were decapitated under isoflurane anesthesia. Their brains were removed and immediately place in an ice-cold cutting solution-containing (in mM) 230 sucrose, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 5 glucose, 0.5 CaCl2, 7 MgCl2, 1 ascorbate, and 3 sodium pyruvate. Coronal slices (300 μm) were taken though the MS/DBB using a Leica VT1200 s vibratome (Leica Biosystems Inc.). Slices were then transferred to an artificial cerebrospinal fluid (ACSF) solution-containing (in mM) 126 NaCl, 3.5 KCl, 1.2 NaH2PO4, 25 NaHCO3, 11 glucose, 2 CaCl2, and 1.3 MgCl2 that was continuously oxygenated with 95 % O2/5 % CO2. Slices were warmed to 35º C for 30 min, and then cooled to room temperature for 1 h prior to recording.
Following a 1 h recovery period, slices were transferred to a recording chamber that was continuously perfused with oxygenated ACSF at a rate of 2 ml/min. Whole-cell patch clamp recordings were taken from MS/DBB neurons in slices using patch pipettes that were pulled with a P-97 horizontal puller (Sutter Instrument Co.) to a resistance of 3–7 MΩ. For voltage clamp recordings, cells were held at -60 mV. Cells were only used for analysis if access resistance was below 40 MΩ and varied by less than 20 % throughout the experiment. Inhibitory postsynaptic current (IPSC) recordings were performed using an internal pipette solution that contained (in mM) 135 CsCl, 10 EGTA, 2 MgCl, 10 HEPES, 4 MgATP, and 0.3 NaGTP, with pH 7.2–7.3 and osmolarity 280–290 mOSm. IPSCs were isolated by bath applying 10 μM DNQX and 40 μM AP5 to block AMPA and NMDARs, respectively. Excitatory postsynaptic current (EPSC) and current clamp recordings were performed using an internal pipette solution that contained (in mM) 130 K+-gluconate, 10 KCl, 10 HEPES, 1 EGTA, 2 MgCl2, 3 MgATP, and 0.3 NaGTP with pH 7.2–7.3 and osmolarity 280–290 mOSm. EPSCs were isolated by bath applying 10 μM gabazine to block GABAARs. In a subset of experiments, 0.5 μM tetrodotoxin (TTX) was added to the bath solution to isolate action potential-independent postsynaptic currents. Signals were digitized using an Axon Digidata 1550 digitizer (Molecular Devices) and collected using pClamp 10 software (Molecular Devices) with an Axopatch 200B amplifier (Molecular Devices). Data were filtered at 2 kHz and sampled at 10 kHz.
Peptides were allowed to wash-on for at least 5 min. Recordings of spontaneous postsynaptic currents were analyzed for 3–4 min prior to and after drug application. Carbachol often resulted in transient changes in spontaneous activity. Therefore, responses to carbachol were recorded throughout a 6 min wash-on and the peak 30 s of activity was used for statistical analysis. In cells where there was a decrease or no change in frequency, data were analyzed 90–120 s after the start of carbachol wash-on, at which point complete exchange of the bath solution would have occurred.
Stereotaxic injections
Expression of mCherry and ChR2 into cholinergic neurons solely in the MS/DBB was achieved by stereotaxic injection of virus directly into the MS/DBB of ChAT-Cre mice. pAAV-EF1a-double-floxed-hChR2(H134R)-mCherry-WPRE-HGHpA (Addgene plasmid #20297 from Karl Deisseroth) and was packaged with AAV stereotype 9 by the viral core facility at the NIEHS. For stereotaxic injections, 5–7-week-old ChAT-Cre mice were anesthetized with an intraperitoneal (IP) injection of 100 mg/kg ketamine and 7 mg/kg xylazine. Once fully anesthetized, Marcaine (50–75 ul of 0.5 % solution of bupivacaine) was delivered subcutaneously to the scalp as a local analgesic. Mice were secured in a stereotaxic instrument (Kopf Instruments) using ear bars. A Hamilton syringe with a 30 gauge needle (Hamilton Co.) was slowly lowered into position and 1 μl of virus was infused at a rate of 0.1 μl/min for 10 min. Stereotaxic coordinates used were 0.05 mm ML, 0.8 mm AP, and -4.4 mm DV. Following surgery, the mouse was given 0.1 mg/kg buprenorphine subcutaneously (SC), 0.5 cc of sterile saline SC, and 0.1 cc of Antisedan (0.2 mg/ml) IP to reverse the effects of the ketamine/xylazine. The mouse was allowed to recover on a heating pad and monitored for any signs of distress. 2–3 weeks following surgery, mice were decapitated under isoflurane anesthesia and their brains were used for electrophysiology as described above.
Optogenetic stimulation
Activation of ChR2 was achieved by delivering square pulses of a 470 nm blue LED light through a 400 μM diameter, and 0.39 numerical aperture fiber optic cannula (ThorLabs, Inc.) positioned directly over the slice. Output power of the LED was 12.5 mW, as measured by an X-Cite XR2100 power meter (EXFO, Inc.).
Drugs
All drugs were purchased from Tocris Bioscience unless otherwise noted. Working solutions of all drugs were made fresh from frozen stock solutions each morning. Human Aβ1–42 (Anaspec, Inc.) was prepared according to package instructions to prevent fibril formation; lyophilized powder was reconstituted in 1 % NH4OH and then diluted in ACSF to a stock concentration of 200 μM. Stock solutions of Aβ1–42 were aliquoted and stored at −20 °C and used within 10 days.
Data Analysis
Electrophysiology data were analyzed using MiniAnalysis 6.03 (Synaptosoft, Inc.). SigmaPlot 13 (Systat Software, Inc.) was used for statistical analysis and graphical illustrations. Data were tested for normality using a Shapiro–Wilk test and then analyzed using the appropriate statistical test: a paired t test or Wilcoxon signed-rank test for single-paired comparisons and one-way repeated measures ANOVA with Holm–Sidak post-hoc analysis or Freidman test with Tukey’s post-hoc analysis for multiple comparisons. Significance was defined as a p value of <0.05. Data are presented ± SEM.
Results
Galanin-containing fibers are found throughout the medial septum and diagonal band of Broca
To examine what role galanin may play in regulating BF function, we first sought to determine where galanin fibers are located in relation to two main cell types in the MS/DBB: cholinergic and GABAergic neurons. Galanin expression in the MS/DBB has been studied previously, but there is still some debate as to the precise location of galanin this region (Melander et al. 1985; Miller et al. 1998; Perez et al. 2001; Keimpema et al. 2014). We performed double immunofluorescence labeling to examine the expression of galanin-containing fibers relative to cholinergic and GABAergic neurons in the MS/DBB of adult C57BL/6J mice. We targeted choline acetyltransferase (ChAT) as a marker for cholinergic neurons, somatostatin (SST) as a marker for GABAergic interneurons, and parvalbumin (PV) as a marker for GABAergic projection neurons (Zaborszky and Duque 2000; Xu et al. 2015). We found that galaninergic fibers are located throughout the MS/DBB, and are intermingled with cholinergic neurons as well as both SST+ and PV+ GABAergic neurons (Fig. 1). We did not observe any co-expression of galanin with either cholinergic or GABAergic neurons. These data show that galanin is expressed in fibers throughout the MS/DBB, and may, therefore, be capable of altering synaptic transmission.

Galanin+ fibers are present throughout the MS/DBB. Fluorescent images show expression of galanin and ChAT in the MS/DBB. Galanin+ fibers (green) were observed in the vicinity of ChAT+ neurons (red) in the medial septum (MS) (a), vertical limb of the diagonal band (VDB) (b), and horizontal limb of the diagonal band (HDB) (c). Fluorescent images show expression of galanin and SST in the MS/DBB. Galanin+ fibers (green) were observed in the vicinity of SST+ neurons (red) in the MS (d), VDB (e) and HDB (f). Fluorescent images show expression of galanin and PV in the MS/DBB. Galanin+ fibers (green) were observed in the vicinity of PV+ neurons (red) in the MS (g), VDB (h), and HDB (i). Scale bars 50 μm
Galanin reduces inhibitory input to basal forebrain neurons
We performed whole-cell patch clamp recordings from basal forebrain neurons of unknown neurochemical identity in acute slices taken from 6- to 10-week-old C57BL/6J mice to determine how galanin affects synaptic transmission within the basal forebrain. To examine the impact of galanin on GABAergic transmission, we recorded spontaneous IPSCs (sIPSCs) from MS/DBB neurons. We found that 500 nM galanin (1–29 rat, mouse) significantly decreased the frequency of sIPSCs without altering their amplitude (Fig. 2a). This effect was consistently seen in recorded neurons, with all cells responding to galanin with at least a 5 % decrease in sIPSC frequency, and 10 of 11 cells showing at least a 15 % decrease in sIPSC frequency. The in vivo galanin receptor antagonist M40 (500 nM) also caused a significant decrease in sIPSC frequency (n = 5, data not shown). These results are consistent with the previous studies showing that in vivo galanin receptor antagonists often act as partial agonists in vitro (Papas and Bourque 1997; Kinney et al. 1998). To determine if the galanin-induced decrease in GABA release was action potential-dependent, we recorded miniature IPSCs (mIPSCs) in the presence of 0.5 μM TTX to block voltage-gated Na+ channels. Galanin had no effect on mIPSC frequency or amplitude (Fig. 2b), indicating that galanin decreases GABA release in a TTX-dependent manner.

Galanin decreases TTX-dependent GABA release in the MS/DBB. (a) Galanin (500 nM) significantly decreased the frequency, but not amplitude, of spontaneous inhibitory postsynaptic currents (sIPSCs) recorded from MS/DBB neurons in slices. This effect was reversible following a 10 min wash-out, n = 11. (b) Galanin had no effect on frequency or amplitude of miniature (m)IPSCs, n = 10. (c) Galanin had no effect on the frequency or amplitude of spontaneous excitatory postsynaptic currents (sEPSCs), n = 10. (d) Galanin significantly reduced the frequency, but not amplitude, of mEPSCs, n = 10. Data presented ± SEM. Asterisk indicates p < 0.05
To determine if galanin could alter spontaneous glutamate release, we recorded sEPSCs from MS/DBB neurons. Galanin had no effect on sEPSC frequency or amplitude (Fig. 2c). There was a small, but significant, decrease in mEPSC frequency, but not amplitude, induced by galanin (Fig. 2d). These data suggest that galanin may work directly at glutamatergic presynaptic terminals to decrease TTX-independent glutamate release without altering TTX-dependent glutamate release.
The most pronounced effect of galanin on synaptic transmission that we observed was a decrease in TTX-dependent GABA release. Galanin binds to three known receptors, GAL1-3 (Habert-Ortoli et al. 1994; Howard et al. 1997; Wang et al. 1997), so we next wanted to determine which galanin receptor subtypes were responsible for decreasing TTX-dependent GABA release in the MS/DBB. To test this, we recorded sIPSCs from MS/DBB neurons prior to and following bath application of the GAL1-specific agonist M617, and the GAL2/3-specific agonist galanin (2–11) (Gal 2–11, also known as AR-M 1896). M617 (500 nM) significantly reduced the frequency, but not amplitude, of sIPSCs (Fig. 3a), whereas Gal 2–11 (500 nM) had no effect on sIPSC frequency or amplitude (Fig. 3b). These results suggest that galanin reduces GABA release in the MS/DBB via activation of the GAL1 receptor.

Activation of GAL1 decreases GABA release in the MS/DBB. a The GAL1 agonist M617 (500 nM) significantly decreased the frequency, but not amplitude, of sIPSCs recorded from MS/DBB neurons, n = 8. b The GAL2/3 agonist Gal 2–11 (500 nM) had no effect on the frequency or amplitude of sIPSCs, n = 5. Data presented ± SEM. Asterisk indicates p < 0.05
Galanin blocks muscarinic receptor-induced GABA release
Previous studies have shown that the muscarinic agonist carbachol can excite GABAergic neurons and increase GABA release in the BF (Alreja et al. 2000; Wu et al. 2000; Yang et al. 2014). Because these effects are opposed to those of galanin, we wanted to determine if galanin was capable of interfering with the actions of carbachol. We found that bath application of 50 μM carbachol significantly increased sIPSC frequency, but not amplitude, in MS/DBB neurons (Fig. 4a–c). The effects of carbachol were blocked by the muscarinic acetylcholine receptor (mAChR) antagonist atropine (5 μM) (Fig. 4d). To determine if galanin could alter carbachol-induced GABA release, we bath applied galanin prior to and during the wash-on of carbachol. In the presence of galanin, carbachol had no significant effect on sIPSC frequency or amplitude (Fig. 4e–g). Atropine did not block the inhibitory effect of galanin on GABA release, indicating that galanin’s blockade of carbachol-induced GABA release is not due to a direct antagonism of muscarinic antagonists (Fig. 4h).

Galanin blunts carbachol-induced GABA release in the MS/DBB. a Sample traces from a MS/DBB neuron showing sIPSCs before and after bath application of carbachol (50 µM). b Mean data and individual cell data showing the effects of carbachol on sIPSC frequency, n = 15. c Mean data and individual cell data showing the effects of carbachol on sIPSC amplitude, n = 15. d Atropine (5 µM) blocked the effects of carbachol, n = 8. e Sample traces showing sIPSCs recorded in the presence of galanin (500 nM) before and after bath application of carbachol. f Mean data and individual cell data showing the effects of carbachol on sIPSC frequency in the presence of galanin, n = 11. g Mean data and individual cell data showing the effects of carbachol on sIPSC amplitude in the presence of galanin, n=11. h Atropine did not block the inhibitory effect of galanin on sIPSC frequency, n = 9. Data presented ± SEM. Asterisk indicates p < 0.05
β-amyloid peptide decreases glutamate release and does not block the disinhibition caused by galanin
Galanin expression is increased in the basal forebrain of AD patients (Chan-Palay 1988; Mufson et al. 1993; Bowser et al. 1997), at the same time in which Aβ levels in the brain will also be high (McLean et al. 1999; Wang et al. 1999). In addition, there is some evidence that galanin and Aβ can be co-released from dense core vesicles (Toneff et al. 2013). Because these peptides will be highly expressed at the same time, we sought to classify the effects of Aβ on synaptic transmission within the BF, and determine if there is any interaction between the actions of galanin and Aβ on synaptic transmission. We found that bath application of 100 nM Aβ1–42 significantly decreased sEPSC frequency, with no change in amplitude (Fig. 5a). Aβ1–42 did not significantly alter sIPSC frequency or amplitude, and did not inhibit the effects of galanin or carbachol on sIPSC frequency (Fig. 5b, c). Thus, galanin and Aβ1–42 have distinct and opposing actions on synaptic transmission in the MS/DBB; i.e., galanin decreases inhibitory synaptic transmission, while Aβ1–42 decreases excitatory synaptic transmission.

β-Amyloid 1–42 peptide (Aβ1–42) decreases glutamate, but not GABA, release in the MS/DBB. a Aβ1–42 (100 nM) significantly decreased the frequency, but not amplitude, of sEPSCs recorded from MS/DBB neurons in slices, n = 13. b Aβ1–42 had no effect of sIPSC frequency or amplitude, and did not block the inhibitory effects of galanin on sIPSC frequency, n = 13. c Aβ1–42 did not block the excitatory effects of carbachol on sIPSC frequency, n = 4. Data presented ± SEM. Asterisk indicates p < 0.05
Galanin reduces GABA release onto cholinergic neurons
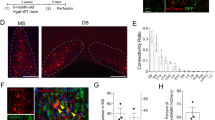
The galanin-induced decrease in sIPSC frequency shown above was recorded from neurons that likely represented a mixture of cell types. We next sought to confirm that galanin could selectively decrease GABA release specifically onto cholinergic neurons in the MS/DBB by utilizing knock-in mice that expressed Cre recombinase under the control of the ChAT promoter (Rossi et al. 2011). At 5–7 weeks of age, ChAT-Cre mice were stereotaxically injected with a Cre-inducible AAV-containing double-floxed ChR2 (Tsai et al. 2009; Witten et al. 2010) fused with mCherry into the MS/DBB, resulting in selective expression of mCherry and ChR2 in cholinergic neurons (Gu and Yakel 2011). Two to three weeks following injection, whole-cell patch clamp recordings were made from mCherry-expressing neurons in MS/DBB slices (Fig. 6a), which had the electrophysiological characteristics of BF cholinergic neurons, including a low spontaneous firing rate (1.17 ± 0.28 Hz, n = 7), inward rectification, and lack of depolarizing sag (Fig. 6b) (Griffith and Matthews 1986; Unal et al. 2012; McKenna et al. 2013). Bath application of galanin significantly reduced the frequency, but not amplitude, of sIPSCs onto identified cholinergic neurons (Fig. 6c). These results confirm that galanin is reducing GABAergic input directly onto cholinergic neurons in the MS/DBB.

Galanin decreases GABA release directly onto cholinergic neurons in the MS/DBB. a Example of an mCherry-expressing cholinergic neuron in the MS/DBB 2–3 weeks following stereotaxic injection of Cre-inducible AAV-containing double-floxed ChR2(H134R) and mCherry into the MS/DBB. b mCherry-expressing neurons had electrophysiological properties characteristic of cholinergic neurons in the basal forebrain. c Galanin significantly decreased the frequency of sIPSCs onto mCherry-expressing cholinergic neurons, n = 11. Data presented ± SEM. Asterisk indicates p < 0.05
Galanin disrupts a local cholinergic-GABAergic circuit in the MS/DBB
Data presented here and elsewhere (Alreja et al. 2000; Wu et al. 2000; Yang et al. 2014) show that cholinergic activation of muscarinic receptors can activate GABAergic neurons in the BF. However, it has not been shown that activation of cholinergic neurons within the MS/DBB can increase GABA release back onto cholinergic neurons. To examine the potential existence of a cholinergic-GABAergic circuit within the MS/DBB, we utilized optogenetics to selectively stimulate MS/DBB cholinergic neurons. In ChAT-Cre mice that had undergone stereotaxic injections as described above, cholinergic neurons-expressing mCherry responded to 10 Hz 470 nm blue light stimulation by firing action potentials, confirming the presence and activation of ChR2 in these neurons (Fig. 7a). To test the impact of cholinergic activation on GABA release onto cholinergic neurons, we recorded sIPSCs from identified cholinergic neurons for 45 s prior to and 30 s after a light stimulation protocol that consisted of 4 bursts of 25 ms pulses at 10 Hz that lasted for 1 s and were separated by 3 s inter-burst intervals (Fig. 7b). We found that this light stimulation protocol resulted in a significant increase in sIPSC frequency, but not amplitude, in cholinergic neurons (Fig. 7c). Individual data revealed that 8/17 cells (47 %) had at least a 20 % increase in frequency, and 5/17 cells (29 %) had at least a 50 % increase in frequency (Fig. 7d). In the presence of gabazine (10 μM), light stimulation still resulted in large direct inward ChR2-mediated currents, but no sIPSCs were present before or after the light stimulation, confirming that all events are GABAAR-mediated (n = 5, data not shown). Bath application of atropine (5 μM) completely abolished the optically induced increase in sIPSC frequency, confirming that cholinergic-induced increases in GABA release in the MS/DBB are predominately mediated by muscarinic receptors (Fig. 7e).

Activation of cholinergic neurons in the MS/DBB increases GABA release onto cholinergic neurons. a Cholinergic cells in the MS/DBB of ChAT-Cre mice-expressing ChR2 responded to 470 nm light simulation by firing action potentials that followed light pulses at 10 Hz. b A sample trace from a MS/DBB cholinergic cell showing that 4 bursts of 10 Hz/1 s light stimulation resulted in an increase in sIPSC frequency. c Mean data showing sIPSC frequency and amplitude before and after light stimulation, n = 17. d Individual cell data showing light-induced changes in sIPSC frequency. e In the presence of atropine (5 µM), light stimulation had no significant effect on sIPSC frequency or amplitude, n = 9. Data presented ± S.E.M. Asterisk indicates p < 0.05
To determine if galanin could disrupt the endogenous cholinergic-induced increases in GABA release in the same way that it blocked carbachol-induced GABA release, we performed our optogenetic stimulation protocol in the presence of galanin (Fig. 8a). In slices that were exposed to galanin prior to and during our light stimulation protocol, light stimulation did not significantly alter the frequency or amplitude of sIPSCs recorded from cholinergic neurons (Fig. 8b). Compared to what was observed under control conditions, there were very few cells that responded to the stimulation with an increase in frequency; 3/15 cells (20 %) had at least a 20 % increase in frequency, and only 2/15 cells (13 %) had at least a 50 % increase in frequency (Fig. 8c). These results indicate that galanin is dampening the ability of endogenous cholinergic activation to increase GABA release onto cholinergic neurons.

Galanin blunts acetylcholine-induced increases in GABA release onto cholinergic neurons in the MS/DBB. a A sample trace recorded from a MS/DBB cholinergic neuron showing sIPSCs before and after 470 nm light stimulation in the presence of galanin (500 nM). b Mean data showing sIPSC frequency and amplitude before and after light stimulation in the presence of galanin, n = 16. c Individual cell data showing light-induced changes in sIPSC frequency in the presence of galanin. Data presented ± SEM. Asterisk indicates p < 0.05
Discussion
Understanding circuitry within the BF is critical to determining how the BF regulates activity in the hippocampus and cortex, and the overall impact of the cholinergic dysfunction that occurs during AD. The overexpression of galanin in the BF of patients with late-stage AD (Chan-Palay 1988; Mufson et al. 1993; Bowser et al. 1997) points to a potentially critical role for this peptide in regulating BF function during the progression of the disease. However, little is known about the function of galanin in the BF, and whether it acts in a neuroprotective or detrimental manner in AD. Here, we show that galanin significantly decreases TTX-dependent GABA release via activation of the GAL1 receptor and disrupts a local cholinergic-GABAergic circuit in the MS/DBB. Conversely, Aβ1–42 significantly decreases glutamate release in the MS/DBB, and does not interfere with the galanin-mediated decrease in GABA release. Together, these data suggest that galanin works in the BF to disinhibit cholinergic neurons by reducing inhibitory input and preventing cholinergic-induced increases in GABA release.
Anatomical studies have shown that projecting cholinergic neurons in the BF possesses local axon collaterals (Zaborszky and Duque 2000; Duque et al. 2007), but the functional connectivity of these cholinergic fibers has only recently been explored. Recent studies using optogenetic techniques have shown that selective activation of cholinergic neurons can elicit responses in BF GABAergic and glutamatergic neurons (Dannenberg et al. 2015; Xu et al. 2015). Here, we show that not only do MS/DBB cholinergic neurons synapse onto local GABAergic neurons, they also increase GABA release back onto cholinergic neurons, forming a functional circuit within the BF. This circuit will allow cholinergic neurons to effectively inhibit themselves, thereby preventing prolonged and excessive periods of elevated excitation. This balance may be critical for maintaining normal firing levels and for regulating activity during periods of elevated or rhythmic firing, such as gamma and theta rhythms, which are critical for memory encoding and retrieval (Hasselmo et al. 2002; Lisman and Buzsaki 2008; Colgin et al. 2009) and are altered in AD (Jelic et al. 2000; van Deursen et al. 2008). In the MS/DBB, projecting PV+ GABAergic neurons can rhythmically fire at theta frequency, while cholinergic neurons fire at slow, non-rhythmic rates (Griffith and Matthews 1986; Sotty et al. 2003; Varga et al. 2008). A recent study reported that MS/DBB neurons regulate theta rhythmicity via an intra-septal relay that is dependent on mAChR activation and leads to the recruitment of PV+ neurons (Dannenberg et al. 2015). The cholinergic-GABAergic interactions described here may be part of this intra-septal relay and aid in the generation of theta by maintaining the low firing rate of cholinergic neurons while allowing for increased activation of PV+ GABAergic neurons.
An interplay between cholinergic and galaninergic signaling was first proposed when it was suggested that galanin may be co-expressed in cholinergic neurons of the BF (Melander et al. 1985). However, the extent to which this co-expression occurs has subsequently come into question (Miller et al. 1998; Perez et al. 2001; Keimpema et al. 2014). Here, we show that galanin fibers are found throughout the MS/DBB of the adult mouse. These fibers are intermingled with both cholinergic and GABAergic neurons, putting them in ideal position to modulate BF function. We did not note co-localization of galanin in cholinergic or GABAergic cell bodies in the BF, suggesting that if galanin is expressed in GABAergic terminals in the BF, as previously shown (Keimpema et al. 2014), these processes may originate from neurons outside the BF. It is also possible that blocking axonal transport of galanin with colchicine treatment may have revealed some co-localization of galanin in cell bodies of the MS/DBB (Melander et al. 1985). However, colchicine has been shown to elevate galanin levels in the brain while reducing ChAT levels (Cortes et al. 1990), which could potentially confound data acquired using this method. Further studies will need to be conducted to definitively say from where galanin fibers in the MS/DBB originate. The diffuse expression of galanin fibers throughout the MS/DBB shown here is similar to what has been observed in higher apes and humans (Benzing et al. 1993; Mufson et al. 1993). These results indicate that the role that galanin plays in regulating BF function in mice may closely reflect the role that galanin plays in the human BF.
Several studies have shown that galanin can regulate acetylcholine release in the hippocampus in a manner that is highly dependent on the region of the brain in which galanin is expressed. For instance, galanin given intracerebroventricularly or infused directly into the hippocampus can lead to a decrease in acetylcholine release in the ventral hippocampus (Fisone et al. 1987; Dutar et al. 1989; Yoshitake et al. 2011). Conversely, galanin infused directly into the MS increases acetylcholine release in the hippocampus (Elvander et al. 2004; Elvander and Ogren 2005). The mechanisms for this increase in acetylcholine release are not known, but may be due to a direct activation of cholinergic neurons (Jhamandas et al. 2002) or a disinhibition of cholinergic neurons. In support of the latter possibility, we show that galanin significantly decreases action potential-dependent GABA release in the MS/DBB. This could be due, in part, to decreased firing of BF GABAergic neurons, which have relatively high intrinsic firing rates (Ovsepian et al. 2012; McKenna et al. 2013), and provide inhibitory synaptic input to cholinergic and non-cholinergic neurons in the BF (Colom 2006; Xu et al. 2015). The dichotomy of effects of galanin on the cholinergic system would make it difficult to interpret results from galanin overexpressing or knockout mice. Consequently, both galanin overexpressing and knockout mice display deficits in cognitive tasks (Crawley 2008). To tease apart the localized effects of galanin, there is a need for conditional knockout or overexpressing mice that have galanin altered only in a subset of cells, or within a certain brain region.
The ability of galanin to block mAChR-induced GABA release in the MS/DBB described here suggests an additional level of control over the cholinergic-GABAergic circuit within the BF. These results are in line with the previous studies showing that blocking mAChRs in conjunction with infusing galanin into the MS causes a dramatic increase in acetylcholine release in the hippocampus (Elvander et al. 2004). In the hippocampus, galanin has also been shown to inhibit the actions of mAChR activation (Fisone et al. 1991; Palazzi et al. 1991). Similar to these studies, we found that this inhibition of mAChR function does not appear to be a result of a direct antagonism of the mAChRs, as indicated by the lack of effect of atropine on galanin-induced decreases in GABA release. It is possible that this inhibition of mAChR function is due to an effect of galanin on voltage-gated Ca2+ entry or an opening of G protein-coupled inwardly rectifying potassium channels (Palazzi et al. 1991; Smith et al. 1998; Endoh et al. 2008; Anselmi et al. 2009). Interestingly, long-term exposure to galanin can upregulate mAChR expression in primary cortical neuron cultures (Cheng and Yu 2015) and can preserve mAChR expression in the hippocampus following increases in ventricular pressure (Barreda-Gomez et al. 2015). Thus, there could be a dual level of control of mAChRs by galanin; an initial and acute inhibition of mAChR-mediated responses followed by a chronic upregulation of mAChRs. It remains to be determined if these mAChRs are functional in nature, or what the long-term consequences of this upregulation may be.
The overexpression of galanin during AD (Chan-Palay 1988; Mufson et al. 1993; Bowser et al. 1997) points to a potentially critical role for this peptide during the progression of the disease. While there have been several studies suggesting that galanin may exacerbate AD symptoms by impairing learning and memory (Steiner et al. 2001; Crawley 2008), the majority of recent evidence suggests a neuroprotective role for galanin (Ding et al. 2006; Counts et al. 2008, 2009; Elliott-Hunt et al. 2011; Li et al. 2013). The results presented here show that galanin can decrease GABAergic transmission in the BF and prevent cholinergic-induced increases in GABA release onto cholinergic neurons. Although further studies would need to be conducted to see what, if any, impact this has on BF function during AD, it is possible that galanin may work to allow surviving cholinergic neurons to fire and release ACh in regions of the brain, such as the hippocampus and cortex, without the negative feedback in the BF.
Because galanin and Aβ are both found in high levels during AD, it is important to take into consideration any potential interactions with Aβ that may alter galanin’s actions. Recent studies have shown that Aβ may significantly alter the function of glutamatergic neurons in the BF, leading to changes in synaptic transmission (Chin et al. 2007; Santos-Torres et al. 2007), and firing rate (Leao et al. 2012). Confirming a role for Aβ in regulating excitatory transmission in the BF, we found that Aβ1-42 by itself can significantly decrease glutamatergic transmission without altering GABAergic transmission. These data show that the effects of Aβ1-42 on synaptic transmission in the BF are distinct from the actions of galanin. Furthermore, because Aβ1-42 decreases excitation and galanin decreases inhibition in the BF, it is possible that the galanin-induced decreases in GABA release described here could, to some extent, compensate for the Aβ-induced decrease in glutamate release and partially restore the balance of inputs onto cholinergic neurons.
Interestingly, we found that Aβ1-42 did not interfere with carbachol-induced GABA release. This is in contrast to the previous reports demonstrating an antagonism of mAChR function by Aβ (Santos-Torres et al. 2007; Preda et al. 2008). Conversely, other studies have shown that Aβ interactions with mAChRs are more pronounced following prolonged, rather than acute, exposure (Kelly et al. 1996; Janickova et al. 2013). It is possible that longer periods of Aβ exposure could disrupt mAChR function, and impair acetylcholine-stimulated GABA release in the BF. If this is the case, it would be interesting to determine how this would affect the acute actions of galanin on mAChR-mediated GABA release described here, or the enhancement of mAChR expression induced by chronic galanin described previously (Barreda-Gomez et al. 2015; Cheng and Yu 2015).
Because of the proposed impact of galanin on cholinergic transmission in the brain, the primary focus of this study was on the effects of galanin on synaptic transmission onto cholinergic neurons in the BF. Accordingly, our optogenetic data clearly show an impact of galanin on GABAergic input onto cholinergic neurons. However, our initial recordings were done on neurons that likely represented a mixture of neuronal cell types. In these experiments, galanin reduced sIPSC frequency by at least 15 % onto 10 of 11 recorded neurons, suggesting that in addition to decreasing GABA release onto cholinergic neurons, galanin is likely also reducing GABAergic input onto other neuronal cell types found in the MD/DBB (e.g., GABAergic and glutamatergic neurons). This could have a profound effect on synaptic transmission within the BF, as well as on the hippocampus and cortex, which receive GABAergic, glutamatergic and cholinergic input from the BF (Gritti et al. 1997; Manns et al. 2001; Sotty et al. 2003). Further examination of galanin-induced changes in synaptic transmission onto non-cholinergic neurons within the BF is warranted to understand the full extent of galanin’s effects on BF function.
In conclusion, we show that inhibitory input onto cholinergic neurons in the MS/DBB is, in part, mediated by local cholinergic input onto GABAergic neurons. Exploring this circuit may be key to understanding the intricate circuitry within the BF, and how intra-BF synaptic transmission may affect output to regions of the brain, such as the cortex and hippocampus. We further show that this BF circuit is disrupted by galanin, resulting in a decrease in GABA release onto cholinergic neurons and a blunting of mAChR-mediated increases in GABA release. The resulting disinhibition of cholinergic neurons is likely to have an effect on intra-BF activity as well as on BF output. Continuing to examine galanin’s role in BF function will be critical for understanding how galanin affects learning and memory, and determining the functional significance of galanin overexpression in the BF during AD.
References
Alreja M, Wu M, Liu W, Atkins JB, Leranth C, Shanabrough M (2000) Muscarinic tone sustains impulse flow in the septohippocampal GABA but not cholinergic pathway: implications for learning and memory. J Neurosci Off J Soc Neurosci 20(21):8103–8110
Anselmi L, Stella SL Jr, Brecha NC, Sternini C (2009) Galanin inhibition of voltage-dependent Ca(2 +) influx in rat cultured myenteric neurons is mediated by galanin receptor 1. J Neurosci Res 87(5):1107–1114. doi:10.1002/jnr.21923
Barreda-Gomez G, Lombardero L, Giralt MT, Manuel I, Rodriguez-Puertas R (2015) Effects of galanin subchronic treatment on memory and muscarinic receptors. Neuroscience 293:23–34. doi:10.1016/j.neuroscience.2015.02.039
Bartus RT, Dean RL, Beer B, Lippa AS (1982) The cholinergic hypothesis of geriatric memory dysfunction. Science 217(4558):408–417. doi:10.1126/science.7046051
Beach TG, Kuo YM, Spiegel K, Emmerling MR, Sue LI, Kokjohn K, Roher AE (2000) The cholinergic deficit coincides with Abeta deposition at the earliest histopathologic stages of Alzheimer disease. J Neuropathol Exp Neurol 59(4):308–313
Benzing WC, Kordower JH, Mufson EJ (1993) Galanin immunoreactivity within the primate basal forebrain: evolutionary change between monkeys and apes. J Comp Neurol 336(1):31–39. doi:10.1002/cne.903360103
Bowser R, Kordower JH, Mufson EJ (1997) A confocal microscopic analysis of galaninergic hyperinnervation of cholinergic basal forebrain neurons in Alzheimer’s disease. Brain Pathol 7(2):723–730
Chan-Palay V (1988) Galanin hyperinnervates surviving neurons of the human basal nucleus of Meynert in dementias of Alzheimer’s and Parkinson’s disease: a hypothesis for the role of galanin in accentuating cholinergic dysfunction in dementia. J Comp Neurol 273(4):543–557. doi:10.1002/cne.902730409
Cheng Y, Yu LC (2015) Galanin up-regulates the expression of M1 muscarinic acetylcholine receptor via the ERK signaling pathway in primary cultured prefrontal cortical neurons. Neurosci Lett 590:161–165. doi:10.1016/j.neulet.2015.02.011
Chin JH, Ma L, MacTavish D, Jhamandas JH (2007) Amyloid beta protein modulates glutamate-mediated neurotransmission in the rat basal forebrain: involvement of presynaptic neuronal nicotinic acetylcholine and metabotropic glutamate receptors. J Neurosci Off J Soc Neurosci 27(35):9262–9269. doi:10.1523/JNEUROSCI.1843-07.2007
Colgin LL, Denninger T, Fyhn M, Hafting T, Bonnevie T, Jensen O, Moser MB, Moser EI (2009) Frequency of gamma oscillations routes flow of information in the hippocampus. Nature 462(7271):353–357. doi:10.1038/nature08573
Colom LV (2006) Septal networks: relevance to theta rhythm, epilepsy and Alzheimer’s disease. J Neurochem 96(3):609–623. doi:10.1111/j.1471-4159.2005.03630.x
Cortes R, Ceccatelli S, Schalling M, Hokfelt T (1990) Differential effects of intracerebroventricular colchicine administration on the expression of mRNAs for neuropeptides and neurotransmitter enzymes, with special emphasis on galanin: an in situ hybridization study. Synapse 6(4):369–391. doi:10.1002/syn.890060410
Counts SE, He B, Che S, Ginsberg SD, Mufson EJ (2008) Galanin hyperinnervation upregulates choline acetyltransferase expression in cholinergic basal forebrain neurons in Alzheimer’s disease. Neuro-degenerative diseases 5(3–4):228–231. doi:10.1159/000113710
Counts SE, He B, Che S, Ginsberg SD, Mufson EJ (2009) Galanin fiber hyperinnervation preserves neuroprotective gene expression in cholinergic basal forebrain neurons in Alzheimer’s disease. J Alzheimer’s Dis JAD 18(4):885–896. doi:10.3233/JAD-2009-1196
Crawley JN (2008) Galanin impairs cognitive abilities in rodents: relevance to Alzheimer’s disease. Cell Molecul Life Sci CMLS 65(12):1836–1841. doi:10.1007/s00018-008-8158-3
Dannenberg H, Pabst M, Braganza O, Schoch S, Niediek J, Bayraktar M, Mormann F, Beck H (2015) Synergy of direct and indirect cholinergic septo-hippocampal pathways coordinates firing in hippocampal networks. J Neurosci Off J Soc Neurosci 35(22):8394–8410. doi:10.1523/JNEUROSCI.4460-14.2015
Davies P, Maloney AJ (1976) Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2(8000):1403
Ding X, MacTavish D, Kar S, Jhamandas JH (2006) Galanin attenuates beta-amyloid (Abeta) toxicity in rat cholinergic basal forebrain neurons. Neurobiol Dis 21(2):413–420. doi:10.1016/j.nbd.2005.08.016
Duque A, Tepper JM, Detari L, Ascoli GA, Zaborszky L (2007) Morphological characterization of electrophysiologically and immunohistochemically identified basal forebrain cholinergic and neuropeptide Y-containing neurons. Brain Struct Funct 212(1):55–73. doi:10.1007/s00429-007-0143-3
Dutar P, Lamour Y, Nicoll RA (1989) Galanin blocks the slow cholinergic EPSP in CA1 pyramidal neurons from ventral hippocampus. Eur J Pharmacol 164(2):355–360
Dutar P, Bassant MH, Senut MC, Lamour Y (1995) The septohippocampal pathway: structure and function of a central cholinergic system. Physiol Rev 75(2):393–427
Elliott-Hunt CR, Holmes FE, Hartley DM, Perez S, Mufson EJ, Wynick D (2011) Endogenous galanin protects mouse hippocampal neurons against amyloid toxicity in vitro via activation of galanin receptor-2. J Alzheimer’s Dis JAD 25(3):455–462. doi:10.3233/JAD-2011-110011
Elvander E, Ogren SO (2005) Medial septal galanin and acetylcholine: influence on hippocampal acetylcholine and spatial learning. Neuropeptides 39(3):245–248. doi:10.1016/j.npep.2004.12.018
Elvander E, Schott PA, Sandin J, Bjelke B, Kehr J, Yoshitake T, Ogren SO (2004) Intraseptal muscarinic ligands and galanin: influence on hippocampal acetylcholine and cognition. Neuroscience 126(3):541–557. doi:10.1016/j.neuroscience.2004.03.058
Endoh T, Sato D, Wada Y, Shibukawa Y, Ishihara K, Hashimoto S, Yoshinari M, Matsuzaka K, Tazaki M, Inoue T (2008) Galanin inhibits calcium channels via Galpha(i)-protein mediated by GalR1 in rat nucleus tractus solitarius. Brain Res 1229:37–46. doi:10.1016/j.brainres.2008.06.036
Fisone G, Wu CF, Consolo S, Nordstrom O, Brynne N, Bartfai T, Melander T, Hokfelt T (1987) Galanin inhibits acetylcholine release in the ventral hippocampus of the rat: histochemical, autoradiographic, in vivo, and in vitro studies. Proc Natl Acad Sci USA 84(20):7339–7343
Fisone G, Bartfai T, Nilsson S, Hokfelt T (1991) Galanin inhibits the potassium-evoked release of acetylcholine and the muscarinic receptor-mediated stimulation of phosphoinositide turnover in slices of monkey hippocampus. Brain Res 568(1–2):279–284
Griffith WH, Matthews RT (1986) Electrophysiology of AChE-positive neurons in basal forebrain slices. Neurosci Lett 71(2):169–174
Griffith WH, Dubois DW, Fincher A, Peebles KA, Bizon JL, Murchison D (2014) Characterization of age-related changes in synaptic transmission onto F344 rat basal forebrain cholinergic neurons using a reduced synaptic preparation. J Neurophysiol 111(2):273–286. doi:10.1152/jn.00129.2013
Gritti I, Mainville L, Mancia M, Jones BE (1997) GABAergic and other noncholinergic basal forebrain neurons, together with cholinergic neurons, project to the mesocortex and isocortex in the rat. J Comp Neurol 383(2):163–177
Gu Z, Yakel JL (2011) Timing-dependent septal cholinergic induction of dynamic hippocampal synaptic plasticity. Neuron 71(1):155–165. doi:10.1016/j.neuron.2011.04.026
Habert-Ortoli E, Amiranoff B, Loquet I, Laburthe M, Mayaux JF (1994) Molecular cloning of a functional human galanin receptor. Proc Natl Acad Sci USA 91(21):9780–9783
Hasselmo ME (2006) The role of acetylcholine in learning and memory. Curr Opin Neurobiol 16(6):710–715. doi:10.1016/j.conb.2006.09.002
Hasselmo ME, Sarter M (2011) Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 36(1):52–73. doi:10.1038/npp.2010.104
Hasselmo ME, Bodelon C, Wyble BP (2002) A proposed function for hippocampal theta rhythm: separate phases of encoding and retrieval enhance reversal of prior learning. Neural Comput 14(4):793–817. doi:10.1162/089976602317318965
He B, Counts SE, Perez SE, Hohmann JG, Koprich JB, Lipton JW, Steiner RA, Crawley JN, Mufson EJ (2005) Ectopic galanin expression and normal galanin receptor 2 and galanin receptor 3 mRNA levels in the forebrain of galanin transgenic mice. Neuroscience 133(2):371–380. doi:10.1016/j.neuroscience.2005.01.068
Howard AD, Tan C, Shiao LL, Palyha OC, McKee KK, Weinberg DH, Feighner SD, Cascieri MA, Smith RG, Van Der Ploeg LH, Sullivan KA (1997) Molecular cloning and characterization of a new receptor for galanin. FEBS Lett 405(3):285–290
Huerta PT, Lisman JE (1993) Heightened synaptic plasticity of hippocampal CA1 neurons during a cholinergically induced rhythmic state. Nature 364(6439):723–725. doi:10.1038/364723a0
Janickova H, Rudajev V, Zimcik P, Jakubik J, Tanila H, El-Fakahany EE, Dolezal V (2013) Uncoupling of M1 muscarinic receptor/G-protein interaction by amyloid beta(1–42). Neuropharmacology 67:272–283. doi:10.1016/j.neuropharm.2012.11.014
Jasek MC, Griffith WH (1998) Pharmacological characterization of ionotropic excitatory amino acid receptors in young and aged rat basal forebrain. Neuroscience 82(4):1179–1194
Jelic V, Johansson SE, Almkvist O, Shigeta M, Julin P, Nordberg A, Winblad B, Wahlund LO (2000) Quantitative electroencephalography in mild cognitive impairment: longitudinal changes and possible prediction of Alzheimer’s disease. Neurobiol Aging 21(4):533–540
Jhamandas JH, Harris KH, MacTavish D, Jassar BS (2002) Novel excitatory actions of galanin on rat cholinergic basal forebrain neurons: implications for its role in Alzheimer’s disease. J Neurophysiol 87(2):696–704
Keimpema E, Zheng K, Barde SS, Berghuis P, Dobszay MB, Schnell R, Mulder J, Luiten PG, Xu ZD, Runesson J, Langel U, Lu B, Hokfelt T, Harkany T (2014) GABAergic terminals are a source of galanin to modulate cholinergic neuron development in the neonatal forebrain. Cereb Cortex 24(12):3277–3288. doi:10.1093/cercor/bht192
Kelly JF, Furukawa K, Barger SW, Rengen MR, Mark RJ, Blanc EM, Roth GS, Mattson MP (1996) Amyloid beta-peptide disrupts carbachol-induced muscarinic cholinergic signal transduction in cortical neurons. Proc Natl Acad Sci USA 93(13):6753–6758
Kinney GA, Emmerson PJ, Miller RJ (1998) Galanin receptor-mediated inhibition of glutamate release in the arcuate nucleus of the hypothalamus. The Journal of neuroscience: the official journal of the Society for Neuroscience 18(10):3489–3500
Lang R, Gundlach AL, Holmes FE, Hobson SA, Wynick D, Hokfelt T, Kofler B (2015) Physiology, signaling, and pharmacology of galanin peptides and receptors: three decades of emerging diversity. Pharmacol Rev 67(1):118–175. doi:10.1124/pr.112.006536
Leao RN, Colom LV, Borgius L, Kiehn O, Fisahn A (2012) Medial septal dysfunction by Abeta-induced KCNQ channel-block in glutamatergic neurons. Neurobiol Aging 33(9):2046–2061. doi:10.1016/j.neurobiolaging.2011.07.013
Li L, Yu L, Kong Q (2013) Exogenous galanin attenuates spatial memory impairment and decreases hippocampal beta-amyloid levels in rat model of Alzheimer’s disease. Int J Neurosci 123(11):759–765. doi:10.3109/00207454.2013.800976
Lim YY, Maruff P, Schindler R, Ott BR, Salloway S, Yoo DC, Noto RB, Santos CY, Snyder PJ (2015) Disruption of cholinergic neurotransmission exacerbates Abeta-related cognitive impairment in preclinical Alzheimer’s disease. Neurobiol Aging 36(10):2709–2715. doi:10.1016/j.neurobiolaging.2015.07.009
Lisman J, Buzsaki G (2008) A neural coding scheme formed by the combined function of gamma and theta oscillations. Schizophr Bull 34(5):974–980. doi:10.1093/schbul/sbn060
Manns ID, Mainville L, Jones BE (2001) Evidence for glutamate, in addition to acetylcholine and GABA, neurotransmitter synthesis in basal forebrain neurons projecting to the entorhinal cortex. Neuroscience 107(2):249–263
McKenna JT, Yang C, Franciosi S, Winston S, Abarr KK, Rigby MS, Yanagawa Y, McCarley RW, Brown RE (2013) Distribution and intrinsic membrane properties of basal forebrain GABAergic and parvalbumin neurons in the mouse. J Comp Neurol 521(6):1225–1250. doi:10.1002/cne.23290
McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 46(6):860–866
Melander T, Staines WA, Hokfelt T, Rokaeus A, Eckenstein F, Salvaterra PM, Wainer BH (1985) Galanin-like immunoreactivity in cholinergic neurons of the septum-basal forebrain complex projecting to the hippocampus of the rat. Brain Res 360(1–2):130–138
Mennicken F, Hoffert C, Pelletier M, Ahmad S, O’Donnell D (2002) Restricted distribution of galanin receptor 3 (GalR3) mRNA in the adult rat central nervous system. J Chem Neuroanat 24(4):257–268
Mesulam MM, Mufson EJ, Levey AI, Wainer BH (1983) Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol 214(2):170–197. doi:10.1002/cne.902140206
Miller MA, Kolb PE, Raskind MA (1997) GALR1 galanin receptor mRNA is co-expressed by galanin neurons but not cholinergic neurons in the rat basal forebrain. Brain Res Mol Brain Res 52(1):121–129
Miller MA, Kolb PE, Planas B, Raskind MA (1998) Few cholinergic neurons in the rat basal forebrain coexpress galanin messenger RNA. J Comp Neurol 391(2):248–258
Mufson EJ, Cochran E, Benzing W, Kordower JH (1993) Galaninergic innervation of the cholinergic vertical limb of the diagonal band (Ch2) and bed nucleus of the stria terminalis in aging. Alzheimer’s disease and Down’s syndrome. Dementia 4(5):237–250
Mufson EJ, Deecher DC, Basile M, Izenwasse S, Mash DC (2000) Galanin receptor plasticity within the nucleus basalis in early and late Alzheimer’s disease: an in vitro autoradiographic analysis. Neuropharmacology 39(8):1404–1412
Mufson EJ, Counts SE, Perez SE, Ginsberg SD (2008) Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev Neurother 8(11):1703–1718. doi:10.1586/14737175.8.11.1703
Nava-Mesa MO, Jimenez-Diaz L, Yajeya J, Navarro-Lopez JD (2014) GABAergic neurotransmission and new strategies of neuromodulation to compensate synaptic dysfunction in early stages of Alzheimer’s disease. Front Cell Neurosci 8:167. doi:10.3389/fncel.2014.00167
O’Donnell D, Ahmad S, Wahlestedt C, Walker P (1999) Expression of the novel galanin receptor subtype GALR2 in the adult rat CNS: distinct distribution from GALR1. J Comp Neurol 409(3):469–481
Ovsepian SV, Anwyl R, Rowan MJ (2004) Endogenous acetylcholine lowers the threshold for long-term potentiation induction in the CA1 area through muscarinic receptor activation: in vivo study. Euro J Neurosci 20(5):1267–1275. doi:10.1111/j.1460-9568.2004.03582.x
Ovsepian SV, Dolly JO, Zaborszky L (2012) Intrinsic voltage dynamics govern the diversity of spontaneous firing profiles in basal forebrain noncholinergic neurons. J Neurophysiol 108(2):406–418. doi:10.1152/jn.00642.2011
Palazzi E, Felinska S, Zambelli M, Fisone G, Bartfai T, Consolo S (1991) Galanin reduces carbachol stimulation of phosphoinositide turnover in rat ventral hippocampus by lowering Ca2+ influx through voltage-sensitive Ca2+ channels. J Neurochem 56(3):739–747
Papas S, Bourque CW (1997) Galanin inhibits continuous and phasic firing in rat hypothalamic magnocellular neurosecretory cells. J Neurosci Off J Soc Neurosci 17(16):6048–6056
Perez SE, Wynick D, Steiner RA, Mufson EJ (2001) Distribution of galaninergic immunoreactivity in the brain of the mouse. J Comp Neurol 434(2):158–185
Preda S, Govoni S, Lanni C, Racchi M, Mura E, Grilli M, Marchi M (2008) Acute beta-amyloid administration disrupts the cholinergic control of dopamine release in the nucleus accumbens. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 33(5):1062–1070. doi:10.1038/sj.npp.1301485
Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, Choi MJ, Lauzon D, Lowell BB, Elmquist JK (2011) Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab 13(2):195–204. doi:10.1016/j.cmet.2011.01.010
Santos-Torres J, Fuente A, Criado JM, Riolobos AS, Heredia M, Yajeya J (2007) Glutamatergic synaptic depression by synthetic amyloid beta-peptide in the medial septum. J Neurosci Res 85(3):634–648. doi:10.1002/jnr.21150
Schliebs R, Arendt T (2011) The cholinergic system in aging and neuronal degeneration. Behav Brain Res 221(2):555–563. doi:10.1016/j.bbr.2010.11.058
Smith KE, Walker MW, Artymyshyn R, Bard J, Borowsky B, Tamm JA, Yao WJ, Vaysse PJ, Branchek TA, Gerald C, Jones KA (1998) Cloned human and rat galanin GALR3 receptors. Pharmacology and activation of G-protein inwardly rectifying K+ channels. J Biol Chem 273(36):23321–23326
Sotty F, Danik M, Manseau F, Laplante F, Quirion R, Williams S (2003) Distinct electrophysiological properties of glutamatergic, cholinergic and GABAergic rat septohippocampal neurons: novel implications for hippocampal rhythmicity. J Physiol 551(Pt 3):927–943. doi:10.1113/jphysiol.2003.046847
Steiner RA, Hohmann JG, Holmes A, Wrenn CC, Cadd G, Jureus A, Clifton DK, Luo M, Gutshall M, Ma SY, Mufson EJ, Crawley JN (2001) Galanin transgenic mice display cognitive and neurochemical deficits characteristic of Alzheimer’s disease. Proc Natl Acad Sci USA 98(7):4184–4189. doi:10.1073/pnas.061445598
Tatemoto K, Rokaeus A, Jornvall H, McDonald TJ, Mutt V (1983) Galanin: a novel biologically active peptide from porcine intestine. FEBS Lett 164(1):124–128
Toneff T, Funkelstein L, Mosier C, Abagyan A, Ziegler M, Hook V (2013) Beta-amyloid peptides undergo regulated co-secretion with neuropeptide and catecholamine neurotransmitters. Peptides 46:126–135. doi:10.1016/j.peptides.2013.04.020
Tsai HC, Zhang F, Adamantidis A, Stuber GD, Bonci A, de Lecea L, Deisseroth K (2009) Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science 324(5930):1080–1084. doi:10.1126/science.1168878
Unal CT, Golowasch JP, Zaborszky L (2012) Adult mouse basal forebrain harbors two distinct cholinergic populations defined by their electrophysiology. Front Behav Neurosci 6:21. doi:10.3389/fnbeh.2012.00021
van Deursen JA, Vuurman EF, Verhey FR, van Kranen-Mastenbroek VH, Riedel WJ (2008) Increased EEG gamma band activity in Alzheimer’s disease and mild cognitive impairment. J Neural Trans 115(9):1301–1311. doi:10.1007/s00702-008-0083-y
Varga V, Hangya B, Kranitz K, Ludanyi A, Zemankovics R, Katona I, Shigemoto R, Freund TF, Borhegyi Z (2008) The presence of pacemaker HCN channels identifies theta rhythmic GABAergic neurons in the medial septum. J Phys 586(Pt 16):3893–3915. doi:10.1113/jphysiol.2008.155242
Wang S, He C, Hashemi T, Bayne M (1997) Cloning and expressional characterization of a novel galanin receptor. Identification of different pharmacophores within galanin for the three galanin receptor subtypes. J Biol Chem 272(51):31949–31952
Wang J, Dickson DW, Trojanowski JQ, Lee VM (1999) The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol 158(2):328–337. doi:10.1006/exnr.1999.7085
Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR (1982) Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215(4537):1237–1239
Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, Gradinaru V, Ramakrishnan C, Deisseroth K (2010) Cholinergic interneurons control local circuit activity and cocaine conditioning. Science 330(6011):1677–1681. doi:10.1126/science.1193771
Wu M, Shanabrough M, Leranth C, Alreja M (2000) Cholinergic excitation of septohippocampal GABA but not cholinergic neurons: implications for learning and memory. J Neurosci Off J Soc Neurosci 20(10):3900–3908
Xu M, Chung S, Zhang S, Zhong P, Ma C, Chang WC, Weissbourd B, Sakai N, Luo L, Nishino S, Dan Y (2015) Basal forebrain circuit for sleep-wake control. Nat Neurosci 18(11):1641–1647. doi:10.1038/nn.4143
Yang C, McKenna JT, Zant JC, Winston S, Basheer R, Brown RE (2014) Cholinergic neurons excite cortically projecting basal forebrain GABAergic neurons. J Neurosci Off J Soc Neurosci 34(8):2832–2844. doi:10.1523/JNEUROSCI.3235-13.2014
Yoshitake T, Yoshitake S, Savage S, Elvander-Tottie E, Ogren SO, Kehr J (2011) Galanin differentially regulates acetylcholine release in ventral and dorsal hippocampus: a microdialysis study in awake rat. Neuroscience 197:172–180. doi:10.1016/j.neuroscience.2011.09.035
Zaborszky L, Duque A (2000) Local synaptic connections of basal forebrain neurons. Behav Brain Res 115(2):143–158
Zaborszky L, Pang K, Somogyi J, Nadasdy Z, Kallo I (1999) The basal forebrain corticopetal system revisited. Ann N Y Acad Sci 877:339–367
Acknowledgments
We thank Patricia Lamb for plasmid preparation, Dr. Bernd Gloss for virus packaging, and Charles J. Tucker for assistance with confocal microscopy. We also thank Dr. Guohong Cui for critical reading of this manuscript. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Damborsky, J.C., Smith, K.G., Jensen, P. et al. Local cholinergic-GABAergic circuitry within the basal forebrain is modulated by galanin. Brain Struct Funct 222, 1385–1400 (2017). https://doi.org/10.1007/s00429-016-1283-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-016-1283-0