
Abstract
Since the 2016 WHO update, progress has been made in understanding the biology of Burkitt lymphoma (BL) and the concept of high-grade B-cell lymphomas (HGBCL) that allows some degree of refinement. The summary presented here reviews in detail the discussions of the Clinical Advisory Committee and expands upon the newly published 2022 International Consensus Classification for lymphoid malignancies (Campo et al. Blood, 2022). BL remains the prototypic HGBCL and diagnostic criteria are largely unchanged. HGBCL with MYC and BCL2 and HGBCL with MYC and BCL6 rearrangements are now separated to reflect biologic and pathologic differences. HGBCL, NOS remains a diagnosis of exclusion that should be used only in rare cases. FISH strategies for diffuse large B-cell lymphoma (DLBCL) and HGBCL are discussed in detail for these diseases. Advances in integrative analysis of mutations, structural abnormalities, copy number, and gene expression signatures allow a more nuanced view of the heterogeneity of DLBCL, NOS as well as definitions of HGBCL and point to where the future may be headed for classification of these diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the 2016 WHO (World Health Organization) update, progress has been made in understanding the biology of Burkitt lymphoma (BL) and the concept of high-grade B-cell lymphoma (HGBCL) that allows some degree of refinement. Advances in integrative analysis of mutations, structural abnormalities, copy number, and gene expression signatures allow a more nuanced view of the heterogeneity of DLBCL, NOS as well as definitions of high-grade B-cell lymphomas. Relationships to specific types of normal counterparts in the germinal center are also being refined. In BL and HGBCL, the expression of TdT has taken on less prominence in driving classification based on recent molecular findings. The segregation of MYC and BCL2 from MYC and BCL6 double-hit lymphomas reflects studies that suggest important differences and the need for further study. Strategies for identifying MYC, BCL2, and BCL6 rearrangements and the importance of partner genes for MYC rearrangements are areas of active investigation. Finally, the criteria for identifying HGBCL, NOS are still problematic due to their heavy reliance on subjective assessment of the cytologic appearance of the neoplastic cells. The following reflects the conclusions the Clinical Advisory Committee (CAC) members reached following the discussions held in the drafting of the 2022 International Consensus Classification (ICC) revision to the 2016 WHO classification (Fig. 1).

International Consensus Classification approach to large B-cell/high-grade B-cell lymphomas. Lymphomas with a Burkitt-like, blastoid, or large-cell morphology that have MYC with BCL2 and/or BCL6 rearrangements are categorized as HGBCL, with either MYC and BCL2 (with or without BCL6R) or MYC and BCL6 R. HGBCL, NOS lacks these double hits and may have either Burkitt-like or blastoid morphology. TdT expression, but not CD34, may be seen in HGBCL. Burkitt lymphomas typically have a MYC R without BCL2 or BCL6 and a mature GC B-cell phenotype. B-lymphoblastic lymphomas are blastoid-appearing, usually TdT + and frequently but not always CD34 + . Cases with a large B-cell morphology that lack a DH, generally, are categorized as one of the DLBCL, NOS or as one of the more specific types of large B-cell lymphoma. The possibility of a large B-cell lymphomas with 11q aberrations should be considered particularly with a Burkitt-like morphologic appearance but with coarse apoptotic material in the tingible body macrophages and lack of a MYC R. Some cases, however, will more closely resemble a DLBCL. DLBCL, diffuse large B-cell lymphoma; R, rearrangement; HGBCL: high-grade B-cell lymphoma; DH, double hit
Burkitt lymphoma
BL is a highly aggressive B-cell lymphoma related to dark zone germinal center B-cells with three recognized clinical variants, namely sporadic (sBL), endemic (eBL, occurring in equatorial areas in sub-Saharan-Africa and South America), and immunodeficiency-associated (iBL) typically seen in the setting of acquired immunodeficiency syndrome (AIDS). Given the near universal association of endemic BL with EBV infection and association with endemic malaria, host and environmental factors likely impact pathogenesis of BL and are areas of ongoing investigation [1, 2]. Clinical presentations differ somewhat based on clinical scenario. In eBL, the peak incidence occurs in children (age 6–8) with a male predominance. Tumors present as rapidly growing masses with a predilection for extranodal sites such as the jaw in younger children and abdomen in older children. A decline in jaw tumors has been observed [3,4,5]. Sporadic tumors demonstrate age-dependent incidence rates with peaks in young children (around age 10), younger adults (age 30–40), and older adults (age 70) in more recently diagnosed cases [6]. Again, there is a male predominance with abdominal and other extranodal sites being involved. In HIV-associated iBL, cases occur early in infection and appear to still occur in the era of highly active anti-retroviral therapy [7, 8]. Patients present in advanced stage with frequent bone marrow and CNS involvement [9].
Regardless of the clinical variant, pathologic features are similar with a relatively uniform population of intermediate-sized cells with round nuclei and multiple small nucleoli. There is scant to moderate amount of cytoplasm that, on Wright-Giemsa-stained smear preparations, is basophilic with small lipid vacuoles. A background “starry-sky” pattern is seen and numerous mitotic figures reflect the high proliferative index. Reports focusing on HIV-associated BL have noted an increased frequency of plasmacytoid differentiation, usually in the setting of EBV positivity and which may be associated with IRF4/MUM1 and cytoplasmic Ig expression (Fig. 2) [10, 11]. Rare cases of BL will have a florid background proliferation of epithelioid histiocytes and granulomas (Fig. 3) [12, 13]. In some cases, these may obscure the tumor, especially on small biopsies, and could lead to misdiagnosis as an infectious process or sarcoidosis. These cases appear to be nearly 100% EBV positive (Fig. 3) [13]. Interestingly, this feature has been found to portend a favorable prognosis, and even, in rare cases, spontaneous remission [12,13,14].

Burkitt lymphoma with plasmacytic differentiation in a HIV patient. (A) H&E shows more pleomorphism than is typical for Burkitt lymphoma, including cells with plasmacytoid features. (B) MUM1 staining highlights plasmacytic differentiation. Both images, 1000 × oil

Burkitt lymphoma, EBV + , associated with granulomas in parotid biopsy (CD20 + , CD10 + , BCL2-, FISH studies showed IGH::MYC but no BCL2 or BCL6 rearrangement). (A) There is a diffuse lymphoid infiltrate associated with scattered granulomas. (B) At higher magnification note the relatively uniform intermediate-sized cells associated with mitotic figures and tingible body macrophages. The granulomatous areas also included smaller T-cells. (C) The tumor is EBER positive (A H&E, 100 × , B H&E, 1000 × oil, C EBER in situ hybridization with hematoxylin counterstain, 400 ×)
The characteristic immunophenotype is CD19 + , CD20 + , CD5-, CD10 + , BCL6 + , BCL2 negative (or very weak), surface immunoglobulin (sIg) positive, and TdT negative. The molecular hallmark of BL is the translocation of MYC with an IG [2] gene, mostly commonly IGH (approximately 80%), followed by IGK (approximately 15%) and IGL (approximately 5%) [1]. As noted above, EBV is found in essentially 100% of eBL and in approximately 30% of sBL and iBL.
While these basic features remain unchanged, the 2022 ICC update notes that cases previously reported as TdT + BL should be distinguished from BL based on several genetic findings including (1) recent molecular analysis demonstrating IG::MYC rearrangements due to aberrant VDJ recombination rather than class switch recombination (CSA) or somatic hypermutation (SHM), (2) lack of mutations commonly seen in BL but rather recurrent NRAS/KRAS mutations, and (3) lack of functional B-cell receptor [15, 16]. Such TdT + cases should be designated as B-lymphoblastic lymphoma/leukemia with MYC rearrangement to recognize their biology and allow clinicians to consider appropriate treatment options.
Studies characterizing the molecular genetic landscape of BL at the genomic and transcriptomic levels have provided insight into the physiological counterpart and important cellular pathways targeted by recurrent genetic alterations. With regard to the IG::MYC rearrangement, sequence analysis in sBL showed class switch recombination, particular into the IGHA switch region (an uncommon event in other MYC-rearranged GC B-cell derived lymphomas) to be most common (CSR, 73%) followed by SHM, 27%, with the SHM being seen most frequently in translocations with IGL partner gene, compared to IGH [17]. Thus, in line with other studies, BL derives from a mature GC-derived B-cell. These MYC rearrangements also result in loss of repressive BCL6 binding sites in the MYC promoter and, along with mutations in MYC that alter protein phosphorylation sites involved in proteosomal degradation, account for aberrant BCL6 and MYC protein co-expression. Recurrent mutations in coding genes affect a limited set of genes in BL including MYC, ID3, CCND3, TP53, SMARCA4, FBXO11, ARID1A, and DDX3X being seen in more than 20% of tumors [17]. Important pathways that are genetically or epigenetically altered in addition to MYC include constitutive activation of the BCR/PI3K/AKT (TCF3, ID3, FOXO1), cell cycle regulation (CCND3, CDKN2B, TP53), RAS, and Gα13 signaling (GNA13, ARHGEF1, S1PR2, RHOA, and P2RY8) [17]. Genomic studies have also suggested the greater role of EBV compared to geographic origin in distinguishing different subsets of BL with potential therapeutic implications [18].
Treatment of pediatric BL is similar to pediatric DLBCL with favorable outcomes using risk-adapted intensive chemotherapy regimens, with addition of rituximab in some settings [19]. Treatment of adult BL consists also of intensive immunochemotherapy regimens. Outcomes are generally good (75–85% overall survival) with these strategies but challenges remain for patients with CNS involvement and older patients (> 60 years), and in the relapse setting [20].
High-grade B-cell lymphoma
The 2016 WHO classification introduced the category HGBCL with MYC and BCL2 and/or BCL6 rearrangements, frequently termed double-hit (DH) and triple-hit (TH) lymphomas (DHL/THL) depending on whether or not both BCL2 and BCL6 rearrangements are present [2, 21]. This category included many of the cases previously classified under the WHO 2008 as “B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma” as well as a subset of DLBCL. The 2016 DHL/THL category thus unified cases with varying morphology based on their genetic findings and generally poor prognosis [21,22,23,24,25,26,27,28]. These DHL/THL may occur de novo, or as transformation of a prior low-grade lymphoma, usually follicular lymphoma (FL) [29].
Morphologically, DH/TH cases may resemble blastoid cells of lymphoblastic lymphoma, centroblasts of DLBCL, intermediate-sized, uniform cells of BL, and those that overlap between the three (Fig. 4). The 2016 WHO indicates that whether a given case has high-grade cytology (Burkitt-like and lymphoblastoid) or large-cell (DLBCL-like) cytology should be noted in the pathology report, because data from several studies have suggested that, among DHL/THL, those with high-grade cytology have a worse prognosis than those with large-cell cytology [26, 27, 30]. However, there is widespread acknowledgement in the field that there is poor reproducibility in identifying what represents “high-grade” cytology [31].

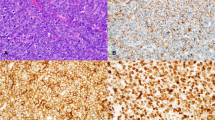
High-grade cytology compared with large-cell cytology in B-cell lymphomas. Panels (A) and (B) demonstrate two cases with high-grade cytology showing intermediate-sized cells with open chromatin and inconspicuous nucleoli. These cases may or may not have tingible body macrophages imparting a starry-sky appearance similar to what would be expected in Burkitt lymphoma. In contrast, panels (C) and (D) show two cases of large B-cell lymphoma cytology which should not be considered “high-grade” in the absence of “double-hit” (DH) cytogenetics. Panel (C) demonstrates frequent tingible body macrophages and tumor cells with open chromatin and amphophilic cytoplasm which may be mistaken for Burkitt lymphoma; however, the cells are unequivocally large. In the absence of DH, this would be best considered diffuse large B-cell lymphoma. Panel (D) also shows large-cell cytology but with coarse chromatin. Although there are frequent mitoses and apoptotic bodies, this case should again be diagnosed as large B-cell lymphoma in the absence of a DH. All H&E, 600 × oil
In the 2022 ICC, the DHL/THL group is split into two distinct categories: (1) HGBCL with MYC and BCL2 rearrangements (HGBCL-DH-BCL2) and (2) HGBCL with MYC and BCL6 rearrangements (HGBCL-DH-BCL6) based on biologic differences [15]. The prior recommendation to note whether they have high-grade or large-cell cytology remains given its potential prognostic impact. There is substantial evidence to maintain the HGBCL-DH-BCL2 cases as distinct from other DLBCLs even if they have some overlapping features with a subset of DLBCLs. As explained below, there is debate as to whether the HGBCL-DH-BCL6 cases are a uniform and distinct disease group. As such, HGBCL-DH-BCL6 is considered a provisional entity, maintained to encourage further study of these tumors. Cases with MYC and both BCL2 and BCL6 rearrangements (TH) should be categorized as HGBCL-DH-BCL2, as the role of BCL6 rearrangement in these cases remains uncertain [32].
High-grade B-cell lymphoma with MYC and BCL2 rearrangements
HGBCL with MYC and BCL2 rearrangements (HGBCL-DH-BCL2) accounts for 80–90% of DH/TH lymphoma cases but only 2% of non-Hodgkin lymphomas overall. The median patient age is approximately 60 years, but cases in teenagers and young adults have been reported. Cases in younger children are exceptionally rare. There is a slight male predominance (60%). Most patients present with advanced-stage (III/IV) disease. These are aggressive lymphomas, with poor prognosis using conventional R-CHOP chemotherapy [23,24,25,26,27,28, 33, 34]. There are small subsets of patients with DHL, defined based on selected clinical and laboratory findings, however, who do well [30, 35, 36].
As described above, the morphology is variable, and the genetic findings alone are sufficient to classify cases as HGBCL-DH-BCL2 with two exceptions. First, cases of otherwise typical grade 1–2 or grade 3A follicular lymphoma with MYC and BCL2 rearrangements have been reported [37, 38], but should still be categorized as FLs and not as a HGBCL. Although some studies have suggested a more aggressive course than typical for indolent FL, data is limited and variable and, as a group, they are clearly not as aggressive as classic HGBCL-DH-BCL2 [37,38,39]. Whether grade 3B FL with these rearrangements are more like a HGBCL-DH-BCL2 is uncertain. Second, there are very rare cases of what are believed to be true B-lymphoblastic leukemia/lymphoma (B-ALL/LBL) with DH cytogenetics. However, very strict clinical and pathologic criteria must be applied to identify these cases, as HGBCL-DH-BCL2 lymphomas can have phenotypic overlap with ALL and can even express TdT (discussed below) [40,41,42,43].
HGBCL-DH-BCL2 are mature lymphomas, which virtually always express pan B-cell markers PAX5 and CD19, and are usually CD20 positive. By flow cytometry, surface light chain expression is found in up to 80% of cases [44]. Bright CD38 is a distinctive feature of some cases [44]. CD34 is absent and if present should prompt strong consideration for B-ALL/LBL.
TdT expression has been reported in 11–14% of DHL/THL [40, 41]. Expression of TdT, however, does not provide sufficient support for reclassification to B-ALL/LBL, in contrast to the WHO 2016 recommendation. Although commonly considered a marker only of immature cells, the majority of TdT-positive HGBCL-DH represent either transformations from prior follicular lymphoma, relapses of aggressive B-cell lymphomas that did not previously express TdT, or co-occurrence with a similar mature aggressive lymphoma without TdT expression [30, 40, 41, 43, 45]. Importantly, these cases never express CD34, and many have other features of maturity such as surface light chain and CD20 expression [30, 40, 41, 43, 45]. Each of these scenarios supports these TdT-positive cases to have originated from a mature B-cell, with acquisition of TdT expression as a secondary event in the course of clonal evolution. Limited mutational studies have suggested a frequently branching clonal evolution [40, 42]. Additionally, targeted mutation analysis performed on 6 cases of TdT-positive LBCL shows mutation profiles similar to GCB-type DLBCL and distinct from B-ALL/LBL [40]. Furthermore, the presence of somatic hypermutation in the small number of cases tested also supports these to be mature lymphomas [42]. Rare reports of true, otherwise clinically and pathologically classic, B-ALL/LBL with DH cytogenetics are challenging to reevaluate retrospectively. However, TdT alone should not be used as a de facto evidence of immaturity, and the overall clinicopathologic features should be considered before rendering a diagnosis of B-ALL/LBL with DH cytogenetics.
HGBCL-DH-BCL2 cases are almost 100% of germinal center B-cell (GCB) origin as classified by either the Hans algorithm immunohistochemistry (IHC) or gene expression profiling (GEP) methods [46]. CD10, BCL6, and BCL2 are positive in over 90% of cases, while IRF4/MUM1 is usually negative. MYC is usually expressed using an IHC cutoff of ≥ 40%. However, importantly, neither expression of MYC nor BCL2 is sensitive or specific enough to identify these rearrangements. Several mechanisms accounting for MYC negativity in tumors with MYC rearrangement have been described including low MYC mRNA levels and a MYC variant (NS11S) which disrupts the epitope of the antibody used for IHC, thereby making MYC expression appear low albeit with maintained mRNA and protein levels [47] (see further discussion of the significance of protein expression of MYC and BCL2 in DLBCL in Dirnhofer et al. from this article series) [46, 48]. The relatively uniform phenotype of the HGBCL-DH-BCL2 cases hints at their underlying uniform biology which, as discussed below, supports their derivation from a follicular lymphoma-like clone.
With recognition of HGBCL-DH-BCL2 as a distinct, aggressive entity, an active area of study has been the unique biology of these tumors, and their relationship to both BL and DLBCL, NOS. HGBCL-DH-BCL2 typically have a complex karyotype consistent with an underlying biologic complexity beyond the constitutive expression of MYC and BCL2 [49]. Mutations in BCL2 (~ 80%), CREBBP (~ 50%), EZH2 (~ 50%), and TNFRSF14 (~ 45%) are frequently seen in HGBCL-DH-BCL2 and overlap with mutations typical of FL and GCB DLBCL [29, 32, 50]. In addition, HGBCL-DH-BCL2 also harbor mutations in ID3 (20–40%), CCND3 (10–20%), which are common also in BL, and FOXO1 (~ 25%) which is known to regulate the germinal center dark zone along with CCND3 [29, 32, 51, 52]. MYC mutations are frequent (~ 40%) [29]. In addition to these well-established recurrent mutations, whole exome sequencing studies (WES) performed by Künstner et al. identified mutational drivers which were not previously identified in panel-based studies. TP53 mutations (25–30%) are commonly seen in HGBCL-DH-BCL2, in striking contrast to the HGBCL-DH-BCL6 (discussed below). Künstner et al. also compared their results to the molecular DLBCL clusters from Chapuy et al. and Schmitz et al. and report significant overlap between the EZB/C3 groups and the HGBCL-DH-BCL2 [32, 53,54,55]. These results continue to support a FL-like clone as the origin of the HGBCL-DH-BCL2 tumors. Finally, triple-hit lymphoma cases in general show mutational similarity to HGBCL-DH-BCL2, but with overlap to HGBCL-DH-BCL6 cases as well, possibly suggesting alternative routes of clonal evolution [29, 32].
High-grade B-cell lymphoma with MYC and BCL6 rearrangements
HGBCL-DH-BCL6 accounts for only 10–20% of DH/TH and in the 2022 ICC is considered a provisional entity [15]. In general, the molecular features of HGBCL-DH-BCL6 are less well-established and clinicopathologic features are less uniform than those of the HGBCL-DH-BCL2 group. The decision to separate these cases from HGBCL-DH-BCL2 stems primarily from their distinct biology [26, 46, 56]. The demographic data of patients with HGBCL-DH-BCL6 are similar to the HGBCL-DH-BCL2 cases, although there may be a particular predilection for extranodal disease in this group [57, 58]. Because of their relative rarity, and because they have typically been combined with the more numerous HGBCL-DH-BCL2 cases for clinical trials, data on their prognosis is challenging to establish. While several recent studies have suggested a very poor prognosis, similar to HGBCL-DH-BCL2, others suggest such lymphomas have similar prognosis to DLBCL, NOS [22, 26, 27, 56, 57].
Morphologically, the majority of HGBCL-DH-BCL6 have DLBCL morphology with a smaller subset showing high-grade morphology [57]. As in HGBCL-DH-BCL2, the cells are phenotypically mature B-cells (express CD19, CD20, CD79a, PAX5), which do not express CD34, and only rarely express TdT, a scenario which again should not independently prompt reclassification as B-ALL/LBL. In significant distinction from HGBCL-DH-BCL2, by GEP, COO is GCB in only about half of HGBCL-DH-BCL6 cases, and ABC/non-GC in the remaining half although by Hans algorithm higher percentages of GCB cases are seen [46, 57, 58]. CD10 is less common than in HGBCL-DH-BCL2 cases, and IRF4/MUM1 expression is seen in 40–90%. MYC expression is seen in up to 90%, and BCL2 expression in 20–80% [46, 57, 58].
The MYC gene partner in HGBCL-DH-BCL6 includes both IG and non-IG partners as with HGBCL-DH-BCL2 (discussed below). However, unlike in HGBCL-DH-BCL2 cases, HGBCL-DH-BCL6 includes a subset of cases in which the BCL6 gene is actually the MYC partner gene resulting from a t(3;8)(q27;q24) which has been termed a “pseudo-double hit” [59, 60]. This MYC::BCL6 translocation has been found to account for up to 30% of cases of DH/TH with MYC and BCL6 rearrangements [61]. Thus, of the HGBCL-DH-BCL6 group, a substantial number harbor this “pseudo-DH” rearrangement, further supporting the separation of these cases from HGBCL-DH-BCL2 in the 2022 ICC [15]. Although there is some literature to support the aggressive behavior of these pseudo-DH tumors, data remains sparse and further study is needed before a recommendation can be made to separate these cases from HGBCL-DH-BCL6 [59]. Additionally, many of the cases (as many as ~ 70%) harboring the MYC::BCL6 also have BCL2 rearrangements (THL), making it challenging to draw conclusions about the significance of the MYC::BCL6 alone [59]. As such, FISH testing to determine whether a HGBCL-DH-BCL6 has MYC::BCL6 translocation is not required.
In contrast to HGBCL-DH-BCL2, the mutational landscape of HGBCL-DH-BCL6 is more heterogeneous and knowledge comes from limited case numbers. Although recurrent mutations in HGBCL-DH-BCL2 are seen in some cases of HGBCL-DH-BCL6, they are found at much lower frequencies: CREBBP (~ 14%), KMT2D (~ 18%), EZH2 (~ 5%) [29, 32, 52]. Overlap with BL mutations such as ID3 (~ 30%) and CCND3 (~ 30%) is similar to what is seen in HGBCL-DH-BCL2, and emphasizes both an overlap with the HGBCL-DH-BCL2 and the distinction of these cases from other DLBCL, NOS [29, 32, 52]. Compared to HGBCL-DH-BCL2, HGBCL-DH-BCL6 shows infrequent BCL2 mutations [32, 62]. While some HGBCL-DH-BCL6 tumors cluster with EZB lymphomas, others cluster with the BN2 subtype reflective of marginal zone lymphoma-like biology, suggesting that there may be two distinct pathways of transformation (one from low-grade FL and one from marginal zone lymphoma) leading to the same HGBCL-DH-BCL6 endpoint. Finally, nearly half fall into other groups or remain unclassified by these algorithms [32] highlighting the heterogeneity within this group and distinction from HGBCL-DH-BCL2 and from DLBCL, NOS.
FISH probe strategy in high-grade and large B-cell lymphoma
A sensitive, efficient, and cost-effective FISH probe strategy to identify DH/TH lymphomas in the clinical setting has been the subject of much debate since the WHO 2016 publication highlighted the need to identify these aggressive lymphomas. Prior to the establishment of the DH/TH category, FISH for MYC was typically performed at the pathologist’s discretion only in cases that resembled BL or those with high Ki-67 proliferation index or mitotic rate. However, it has now been widely established that these “high-grade” morphologic features are inadequate for screening, as approximately 10% of otherwise typical DLBCL harbor MYC rearrangements [26, 46]. Screening by COO has been proposed as an efficient and cost-saving measure, as GCB lymphomas account for virtually 100% of the HGBCL-DH-BCL2 cases using GEP; however, this strategy will miss approximately 50% of the HGBCL-DH-BCL6 cases that are of non-GCB origin and a small number (up to 4%) of HGBCL-DH-BCL2 which may be misclassified by the Hans algorithm [22, 26, 27, 46]. Finally, neither expression of MYC, nor BCL2, nor BCL6 is sufficient to screen for their respective rearrangements. As such, the 2022 ICC recommends screening all large B-cell and high-grade B-cell lymphomas for the possibility of HGBCL-DH of either type [15].
The approach to screening typically begins with a sensitive MYC FISH breakapart probe (BAP). However, available commercial MYC BAP probes vary in sensitivity depending on the extent of the breakpoint region covered in the probe design [61, 63, 64]. Even the most sensitive MYC BAP, however, will miss a subset of rearrangements due to cryptic insertions or other complex genomic rearrangements that are too small or out of the coverage area of the probes. Performing MYC::IGH dual-color, dual-fusion (DF) FISH in addition to MYC BAP can detect the 4–12% of MYC rearrangements that are missed by BAP alone [30, 65]. The addition of MYC::IGL and MYC::IGK DF probes likely also increases sensitivity, although data are sparse due to the lack of widespread availability of these probes.
The use of MYC::IGH, MYC::IGL, and MYC::IGK probes to detect MYC rearrangements, however, belies the diversity of MYC gene partners and complexity of possible rearrangements, especially in the non-BL setting [63]. In BL, the MYC partner is virtually always an IG gene, whereas in non-BL DLBCL/HGBCL the partner genes are non-IG in at least 40% of cases and include a variety of genes such as BCL6, PAX5, IRF4, BCL11A, IKZF1, IMMP2L, and RFTN1 among others [22, 26, 61, 63, 66]. The true “false negative” rate of current MYC FISH strategies is almost certainly underestimated with one estimate being 20%, and increasingly, in the research setting, molecular methods are allowing for detection of rearrangements cryptic to FISH [65, 67].
The clinical significance of the MYC partner gene has also been the subject of much debate. Whether the canonical concept of juxtaposition of IG enhancers to the MYC gene leading to overexpression of MYC protein occurs in a similar fashion with enhancers of non-IG genes is uncertain. Several studies have suggested that, in cases with DLBCL morphology, the prognosis of MYC::IG translocations is inferior to those with MYC-non-IG translocations, and in fact that DH/TH lymphomas with MYC-non-IG have no difference in prognosis to DLBCL, NOS [22, 27]. However, others have shown discrepant results, and especially in a cohort enriched with high-grade cytology tumors, there was no difference in survival among DH/TH lymphomas with IG vs non-IG MYC rearrangement [26]. In summary, although many in the CAC group felt compelled by the data supporting worse prognosis in MYC::IG cases, this remains an unresolved question. Importantly, the lack of widespread availability of IGK and IGL probes precludes routine testing for all IG rearrangements in clinical practice, and, as such, the 2022 ICC does not require identification of the MYC partner [15].
The WHO 2017 Blue Book specifically states that copy number alterations (CNA) of MYC, BCL2, or BCL6 are not equivalent to rearrangement for classification of DH/TH lymphomas and the 2022 ICC retains this concept [2, 15]. FISH testing invariably leads to identification of CNA in tested genes. The majority are copy number gains of 3–10 copies per cell, many due to extra copies of whole chromosomes common in lymphoma in general [68]. Difficulty in elucidating the significance of these CNA is in part due to the lack of consistency as to how such gains are reported by different laboratories and the likely variable significance of whole chromosome versus single gene locus gains in altering gene expression [48]. In addition, studies looking at the significance of “amplification” have used various definitions. Interestingly, at least one study has shown that while BCL2 CNA were reported to be strongly associated with increased BCL2 expression, MYC CNA were not associated with such an increase [47]. This finding further casts doubt on the significance of such CNA. While some studies have found CNA of MYC to yield poor prognosis similar to rearrangement, others do not support this assertion [68,69,70,71,72,73,74,75]. There are some data to suggest that high amplification of MYC (as defined by uncountable FISH probe signals) is significant; however, these cases are rare and there are insufficient data to draw definitive conclusions [47, 71].
Gene expression signatures to identify high-grade B-cell lymphoma
Recently, two separate groups have identified gene expression signatures of “high-grade” lymphomas which encompass most, but not all, HGBCL-DH-BCL2 along with BL and some GCB-DLBCL. While termed “double hit signature” by Ennishi et al. [76] and “molecular high grade” by Sha et al. [77]), the patterns of gene expression in these signatures are reminiscent of dark zone (versus light zone) germinal center B-cells and could be consider dark zone signatures. The double hit signature (DHITsig) was developed on the gene expression features distinguishing HGBCL-DH-BCL2 from GCB-DLBCL [76]. Only half of the DHITsig cases identified by the classifier were HGBCL-DH-BCL2 by FISH [76]. However, in 30% of non-HGBCL-DH-BCL2, DHITsig tumors were subsequently found to have MYC or BCL2 rearrangements that were cryptic to FISH [67]. Interestingly, in spite of many not harboring the rearrangements, the DHITsig patients have significantly shorter time to progression, overall survival, and disease-specific survival than the DHITsig-negative GCB-DLBCL cases suggesting aggressive biology may be more closely tied to the signature than DH cytogenetic status [76]. The mutation profile of the DHITsig cases suggests enrichment of mutations in MYC and BCL2, as well as chromatin modifier genes including CREBBP, EZH2, and KMT2D along with DDX3X and TP53 and with low expression of major histocompatability (MHC) genes. A subsequent study applied the DHITsig to a series of HGBCL, NOS and found the DHITsig in 55% of these cases [31] further highlighting the heterogeneity of HGBCL, NOS.
In a parallel study, “molecular high grade” (MHG) signature was developed from the molecular Burkitt lymphoma gene expression signatures [78, 79]. Their MHG classifier also identifies a group of predominantly GCB tumors with GEP similar to centroblast (dark zone) germinal center B-cells which show poor prognosis that extends beyond the effect of the DH/TH cytogenetics [77]. Again, 51% of MHG cases lacked a MYC rearrangement and only 36% were DH. MHG lymphomas were enriched in similar mutations to those in the DHITsig group, and showed similar downregulation of MHC genes. Both the DHITsig and MHG classifiers identify groups which show considerable overlap with the molecular signature-based EZB/C3 clusters of DLBCL published by Chapuy et al. [53]. and Schmitz et al. [54] and discussed in more detail in regard to DLBCL by Dirnhofer et al. within this article series.
These novel findings identified by two independent groups underscore the common germinal center dark zone biology shared not only by the HGBCL-DH-BCL2 lymphomas but also by a subset GCB-DLBCLs with equally poor prognosis to the DH cases and BL. They raise the obvious possibility of adapting such an assay to the clinical realm, whether that be a GEP-based approach or a sequencing approach, reflecting the mutations characteristic of this subset. Regardless of the approach, it is clear that our current ability to identify both HGBCL-DH and HGBCL, NOS is insufficient to identify all of the tumors with aggressive biology that may benefit from more intensive therapy. The potential role and usage of MHG and DHITsig profiles remain to be determined as the dark zone biology is shared by a diverse group of tumors with different biology and clinical behavior, requiring interpretation in the context of other parameters. For example, BL, a biologically and clinically very different type of lymphoma, also displays this gene expression profile [80, 81]. In fact, when applied to a cohort of R-CHOP-treated GCB DLBCL, DHITsig only predicted poor survival if combined with TP53 abnormality, whereas a subset of DHITsig-positive tumors which lacked EZH2 and BCL2 abnormalities actually had an excellent prognosis [82].
High-grade B-cell lymphoma, not otherwise specified
With the dissolution of the WHO 2008 category BCL, unclassifiable, with features intermediate between DLBCL and BL, there remained cases which did not fall into the HGBCL-DH category but also did not fit neatly into DLBCL, NOS, BL, or B-ALL/LBL. The WHO 2016 created the high-grade B-cell lymphoma, not otherwise specified (HGBCL, NOS) category for these cases [21], and the 2022 ICC retains this category to be used as a diagnosis of exclusion [15]. HGBCL, NOS cases typically have intermediate-sized cells, often with blastoid or Burkitt-like cytology (Fig. 4A, B) [2, 21]. DLBCL with starry-sky morphology and/or high proliferation index are not recategorized as HGBCL, NOS (Fig. 4C, D). Identification of HGBCL, NOS is subjective and requires well-fixed tissue biopsies as the distinction between intermediate and large cell size is imprecise, and cell size, as well as chromatin characteristics, can both be affected by differences in specimen type, tissue fixation, processing, section thickness, and stain quality among other pre-analytic factors. It is a diagnosis to be used very sparingly and there is limited data concerning these cases. In a multi-institutional study from the Lymphoma/Leukemia Molecular Profiling Project (LLMPP) with central pathology review (CPR) on 61 tumors submitted as HGBCL, NOS, the reviewers reclassified 48% as DLBCL and 5% as BL [31] highlighting the challenging nature of these distinctions.
Data on HGBCL, NOS remains sparse, but unsurprisingly points toward heterogeneity within these tumors. Patients appear to be older adults (median age 70), many of whom have advanced-stage disease [83, 84]. Rates of response to standard DLBCL-like therapy remain difficult to ascertain because of the recent adoption of this category and lack of complete alignment with historical categories; however, one early study suggests superior outcomes with more aggressive therapy regimens [83].
In a German study, also with CPR, HGBCL, NOS showed frequent expression of CD10 (65%) and BCL6 (90%), but interestingly only one HGBCL, NOS had the characteristic phenotype/MYC rearrangement typical of BL [85]. However, this may reflect an increased emphasis on phenotype/genotype vs morphology in this study. In the LLMPP HGBCL, NOS study, COO results showed 57% GCB, 25% ABC, and 18% unclassifiable tumors by GEP [31]. Only 54% of HGBCL, NOS cases showed a DHITsig profile [31]. In fact, the mutation profile, GEP, and phenotypic features were similar between cases classified as HGBCL, NOS and those reclassified as DLBCL emphasizing the heterogeneity of this category and potential flaw in using morphology to distinguish these tumors [31]. The German series and LLMPP series showed 13% and 46% of HGBCL, NOS harbored a single-hit MYC rearrangement, respectively, emphasizing likely differences in morphologic criteria used for inclusion [31, 85]. Of note, in spite of the higher percentage of cases with MYC rearrangement in the LLMPP series, this was the same percentage as was seen in their cases reclassified as DLBCL, emphasizing that MYC rearrangement alone does not explain the high-grade morphologic features and also highlighting the limits of classification based strictly on subjective morphologic features.
In summary, the 2022 ICC retains the HGBCL, NOS category with the acknowledgement that these cases likely do not represent a true, uniform biologic entity. However, until the field progresses to a molecular-based classification scheme, the consensus among CAC participants was that this category should remain but be used very sparingly.
Conclusion
Several updates to the 2016 WHO classification of BL and HGBCLs are recommended by the ICC. These include recognizing that TdT expression in DHL/THLs does not necessarily warrant a diagnosis of B-lymphoblastic leukemia/lymphoma. While HGBCL-DH-BCL2 remain relative well-defined, the best FISH strategies remain open to debate, realizing that cases will be missed if only MYC breakapart or MYC::IGH dual-fusion probes are used. Variation in performance of commercially available probe sets exists and laboratorians should be aware of their limitations. The so-called pseudo HGBCL-DH-BCL6 exist due to the t(3;8) translocation and the 2022 ICC segregates HGBCL-DH-BCL2 from HGBCL-DH-BCL6. HGBCL, NOS remains problematic due to an imprecise definition that relies heavily on morphologic features. While identifying molecular Burkitt/high-grade gene expression signatures may become a part of our future diagnostic armamentarium, their precise application in conjunction with our other morphologic, phenotypic, and genetic tools remains to be determined.
References
Pasqualucci L (2019) Molecular pathogenesis of germinal center-derived B cell lymphomas. Immunol Rev 288:240–261. https://doi.org/10.1111/imr.12745
Swerdlow SH CE, Harris NL et al (2017) WHO classification of tumors of haematopoetic and lymphoid tissues (Revised 4th edn). IARC: Lyon 2017
Gebauer N, Witte HM, Merz H, Oschlies I, Klapper W, Caliebe A, Tharun L, Spielmann M, von Bubnoff N, Feller AC, MurgaPenas EM (2021) Aggressive B-cell lymphoma cases with 11q aberration patterns indicate a spectrum beyond Burkitt-like lymphoma. Blood Adv 5:5220–5225. https://doi.org/10.1182/bloodadvances.2021004635
Offor UT, Akyea RK, Neequaye JE, Renner LA, Segbefia CI (2018) The changing clinical pattern of endemic Burkitt lymphoma in Western Africa: experience from a tertiary center in Ghana. Pediatr Blood Cancer 65:e27275. https://doi.org/10.1002/pbc.27275
Rochford R (2021) Reframing Burkitt lymphoma: virology not epidemiology defines clinical variants. Ann Lymphoma. https://doi.org/10.21037/aol-21-18
Mbulaiteye SM, Anderson WF, Ferlay J, Bhatia K, Chang C, Rosenberg PS, Devesa SS, Parkin DM (2012) Pediatric, elderly, and emerging adult-onset peaks in Burkitt’s lymphoma incidence diagnosed in four continents, excluding Africa. Am J Hematol 87:573–578. https://doi.org/10.1002/ajh.23187
Gibson TM, Morton LM, Shiels MS, Clarke CA, Engels EA (2014) Risk of non-Hodgkin lymphoma subtypes in HIV-infected people during the HAART era: a population-based study. AIDS 28:2313–2318. https://doi.org/10.1097/qad.0000000000000428
Lim ST, Karim R, Nathwani BN, Tulpule A, Espina B, Levine AM (2005) AIDS-related Burkitt’s lymphoma versus diffuse large-cell lymphoma in the pre-highly active antiretroviral therapy (HAART) and HAART eras: significant differences in survival with standard chemotherapy. J Clin Oncol 23:4430–4438. https://doi.org/10.1200/jco.2005.11.973
Alderuccio JP, Olszewski AJ, Evens AM, Collins GP, Danilov AV, Bower M, Jagadeesh D, Zhu C, Sperling A, Kim SH, Vaca R, Wei C, Sundaram S, Reddy N, Dalla Pria A, D’Angelo C, Farooq U, Bond DA, Berg S, Churnetski MC, Godara A, Khan N, Choi YK, Kassam S, Yazdy M, Rabinovich E, Post FA, Varma G, Karmali R, Burkart M, Martin P, Ren A, Chauhan A, Diefenbach C, Straker-Edwards A, Klein A, Blum KA, Boughan KM, Mian A, Haverkos BM, Orellana-Noia VM, Kenkre VP, Zayac A, Maliske SM, Epperla N, Caimi P, Smith SE, Kamdar M, Venugopal P, Feldman TA, Rector D, Smith SD, Stadnik A, Portell CA, Lin Y, Naik S, Montoto S, Lossos IS, Cwynarski K (2021) HIV-associated Burkitt lymphoma: outcomes from a US-UK collaborative analysis. Blood Adv 5:2852–2862. https://doi.org/10.1182/bloodadvances.2021004458
Gloghini A, Dolcetti R, Carbone A (2013) Lymphomas occurring specifically in HIV-infected patients: from pathogenesis to pathology. Semin Cancer Biol 23:457–467. https://doi.org/10.1016/j.semcancer.2013.08.004
Gualco G, Queiroga EM, Weiss LM, Klumb CE, Harrington WJ Jr, Bacchi CE (2009) Frequent expression of multiple myeloma 1/interferon regulatory factor 4 in Burkitt lymphoma. Hum Pathol 40:565–571. https://doi.org/10.1016/j.humpath.2008.07.021
Haralambieva E, Rosati S, van Noesel C, Boers E, van Marwijk KM, Schuuring E, Kluin P (2004) Florid granulomatous reaction in Epstein-Barr virus-positive nonendemic Burkitt lymphomas: report of four cases. Am J Surg Pathol 28:379–383. https://doi.org/10.1097/00000478-200403000-00011
Schrager JA, Pittaluga S, Raffeld M, Jaffe ES (2005) Granulomatous reaction in Burkitt lymphoma: correlation with EBV positivity and clinical outcome. Am J Surg Pathol 29:1115–1116
Li JN, Gao LM, Wang WY, Chen M, Li GD, Liu WP, Zhang WY (2014) HIV-related Burkitt lymphoma with florid granulomatous reaction: an unusual case with good outcome Int J. Clin Exp Pathol 7:7049–7053
Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH, Anderson KC, Brousset P, Cerroni L, de Leval L, Dirnhofer S, Dogan A, Feldman A, Fend F, Friedberg JW, Gaulard P, Ghia P, Horwitz SM, King RL, Salles GA, San-Miguel JF, Seymour JF, Treon SP, Vose J, Zucca E, Advani R, Ansell SM, Au WY, Barrionuevo C, Bergsagel PL, Chan WC, Cohen JI, d'Amore F, Davies AJ, Falini B, Ghobrial IM, Goodlad JR, Gribben JG, Hsi ED, Kahl BS, Kim WS, Kumar SK, LaCasce AS, Laurent C, Lenz G, Leonard JP, Link MP, López-Guillermo A, Mateos MV, Macintyre EA, Melnick AM, Morschhauser F, Nakamura S, Narbaitz M, Pavlovsky A, Pileri SA, Piris MA, Pro B, Rajkumar SVV, Rosen ST, Sander B, Sehn LH, Shipp MA, Smith SM, Staudt LM, Thieblemont C, Tousseyn T, Wilson WH, Yoshino T, Zinzani PL, Dreyling M, Scott DW, Winter JN, Zelenetz AD (2022) The international consensus classification of mature lymphoid neoplasms: a report from the Clinical Advisory Committee. Blood. https://doi.org/10.1182/blood.2022015851
Wagener R, López C, Kleinheinz K, Bausinger J, Aukema SM, Nagel I, Toprak UH, Seufert J, Altmüller J, Thiele H, Schneider C, Kolarova J, Park J, Hübschmann D, MurgaPenas EM, Drexler HG, Attarbaschi A, Hovland R, Kjeldsen E, Kneba M, Kontny U, de Leval L, Nürnberg P, Oschlies I, Oscier D, Schlegelberger B, Stilgenbauer S, Wössmann W, Schlesner M, Burkhardt B, Klapper W, Jaffe ES, Küppers R, Siebert R (2018) IG-MYC (+) neoplasms with precursor B-cell phenotype are molecularly distinct from Burkitt lymphomas. Blood 132:2280–2285. https://doi.org/10.1182/blood-2018-03-842088
López C, Kleinheinz K, Aukema SM, Rohde M, Bernhart SH, Hübschmann D, Wagener R, Toprak UH, Raimondi F, Kreuz M, Waszak SM, Huang Z, Sieverling L, Paramasivam N, Seufert J, Sungalee S, Russell RB, Bausinger J, Kretzmer H, Ammerpohl O, Bergmann AK, Binder H, Borkhardt A, Brors B, Claviez A, Doose G, Feuerbach L, Haake A, Hansmann ML, Hoell J, Hummel M, Korbel JO, Lawerenz C, Lenze D, Radlwimmer B, Richter J, Rosenstiel P, Rosenwald A, Schilhabel MB, Stein H, Stilgenbauer S, Stadler PF, Szczepanowski M, Weniger MA, Zapatka M, Eils R, Lichter P, Loeffler M, Möller P, Trümper L, Klapper W, Hoffmann S, Küppers R, Burkhardt B, Schlesner M, Siebert R (2019) Genomic and transcriptomic changes complement each other in the pathogenesis of sporadic Burkitt lymphoma. Nat Commun 10:1459. https://doi.org/10.1038/s41467-019-08578-3
Grande BM, Gerhard DS, Jiang A, Griner NB, Abramson JS, Alexander TB, Allen H, Ayers LW, Bethony JM, Bhatia K, Bowen J, Casper C, Choi JK, Culibrk L, Davidsen TM, Dyer MA, Gastier-Foster JM, Gesuwan P, Greiner TC, Gross TG, Hanf B, Harris NL, He Y, Irvin JD, Jaffe ES, Jones SJM, Kerchan P, Knoetze N, Leal FE, Lichtenberg TM, Ma Y, Martin JP, Martin MR, Mbulaiteye SM, Mullighan CG, Mungall AJ, Namirembe C, Novik K, Noy A, Ogwang MD, Omoding A, Orem J, Reynolds SJ, Rushton CK, Sandlund JT, Schmitz R, Taylor C, Wilson WH, Wright GW, Zhao EY, Marra MA, Morin RD, Staudt LM (2019) Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 133:1313–1324. https://doi.org/10.1182/blood-2018-09-871418
National Cancer Institute (US) (2002) Childhood non-Hodgkin lymphoma treatment (PDQ®): health professional version. PDQ Cancer Information Summaries. National Cancer Institute (US), Bethesda (MD)
Crombie J, LaCasce A (2021) The treatment of Burkitt lymphoma in adults. Blood 137:743–750. https://doi.org/10.1182/blood.2019004099
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES (2016) The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127:2375–2390. https://doi.org/10.1182/blood-2016-01-643569
Copie-Bergman C, Cuillière-Dartigues P, Baia M, Briere J, Delarue R, Canioni D, Salles G, Parrens M, Belhadj K, Fabiani B, Recher C, Petrella T, Ketterer N, Peyrade F, Haioun C, Nagel I, Siebert R, Jardin F, Leroy K, Jais JP, Tilly H, Molina TJ, Gaulard P (2015) MYC-IG rearrangements are negative predictors of survival in DLBCL patients treated with immunochemotherapy: a GELA/LYSA study. Blood 126:2466–2474. https://doi.org/10.1182/blood-2015-05-647602
Horn H, Ziepert M, Becher C, Barth TF, Bernd HW, Feller AC, Klapper W, Hummel M, Stein H, Hansmann ML, Schmelter C, Moller P, Cogliatti S, Pfreundschuh M, Schmitz N, Trumper L, Siebert R, Loeffler M, Rosenwald A, Ott G, German High-Grade Non-Hodgkin Lymphoma Study G (2013) MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 121:2253–2263. https://doi.org/10.1182/blood-2012-06-435842
Johnson NA, Savage KJ, Ludkovski O, Ben-Neriah S, Woods R, Steidl C, Dyer MJ, Siebert R, Kuruvilla J, Klasa R, Connors JM, Gascoyne RD, Horsman DE (2009) Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood 114:2273–2279. https://doi.org/10.1182/blood-2009-03-212191
Klapper W, Stoecklein H, Zeynalova S, Ott G, Kosari F, Rosenwald A, Loeffler M, Trümper L, Pfreundschuh M, Siebert R (2008) Structural aberrations affecting the MYC locus indicate a poor prognosis independent of clinical risk factors in diffuse large B-cell lymphomas treated within randomized trials of the German High-Grade Non-Hodgkin’s Lymphoma Study Group (DSHNHL). Leukemia 22:2226–2229. https://doi.org/10.1038/leu.2008.230
McPhail ED, Maurer MJ, Macon WR, Feldman AL, Kurtin PJ, Ketterling RP, Vaidya R, Cerhan JR, Ansell SM, Porrata LF, Nowakowski GS, Witzig TE, Habermann TM (2018) Inferior survival in high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements is not associated with MYC/IG gene rearrangements. Haematologica 103:1899–1907. https://doi.org/10.3324/haematol.2018.190157
Rosenwald A, Bens S, Advani R, Barrans S, Copie-Bergman C, Elsensohn MH, Natkunam Y, Calaminici M, Sander B, Baia M, Smith A, Painter D, Pham L, Zhao S, Ziepert M, Jordanova ES, Molina TJ, Kersten MJ, Kimby E, Klapper W, Raemaekers J, Schmitz N, Jardin F, Stevens WBC, Hoster E, Hagenbeek A, Gribben JG, Siebert R, Gascoyne RD, Scott DW, Gaulard P, Salles G, Burton C, de Jong D, Sehn LH, Maucort-Boulch D (2019) Prognostic significance of MYC rearrangement and translocation partner in diffuse large B-cell lymphoma: a study by the Lunenburg Lymphoma Biomarker Consortium. J Clin Oncol 37:3359–3368. https://doi.org/10.1200/jco.19.00743
Savage KJ, Johnson NA, Ben-Neriah S, Connors JM, Sehn LH, Farinha P, Horsman DE, Gascoyne RD (2009) MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood 114:3533–3537. https://doi.org/10.1182/blood-2009-05-220095
Cucco F, Barrans S, Sha C, Clipson A, Crouch S, Dobson R, Chen Z, Thompson JS, Care MA, Cummin T, Caddy J, Liu H, Robinson A, Schuh A, Fitzgibbon J, Painter D, Smith A, Roman E, Tooze R, Burton C, Davies AJ, Westhead DR, Johnson PWM, Du MQ (2020) Distinct genetic changes reveal evolutionary history and heterogeneous molecular grade of DLBCL with MYC/BCL2 double-hit. Leukemia 34:1329–1341. https://doi.org/10.1038/s41375-019-0691-6
Moore EM, Aggarwal N, Surti U, Swerdlow SH (2017) Further exploration of the complexities of large B-cell lymphomas with MYC abnormalities and the importance of a blastoid morphology. Am J Surg Pathol 41:1155–1166. https://doi.org/10.1097/pas.0000000000000818
Collinge BJ, Hilton LK, Wong J, Ben-Neriah S, Rushton CK, Slack GW, Farinha P, Cook JR, Ott G, Rosenwald A, Campo E, Amador C, Greiner TC, Raess PW, Song JY, Inghirami G, Jaffe ES, Weisenburger DD, Chan WC, Holte H, Beiske K, Fu K, Delabie J, Pittaluga S, Feldman AL, Savage KJ, Mungall AJ, Staudt LM, Steidl C, Rimsza LM, Morin RD, Scott DW (2021) Characterization of the genetic landscape of high-grade B-cell lymphoma, NOS– an LLMPP project. Hematol Oncol 39. https://doi.org/10.1002/hon.13_2880
Künstner A, Witte HM, Riedl J, Bernard V, Stölting S, Merz H, Olschewski V, Peter W, Ketzer J, Busch Y, Trojok P, Bubnoff NV, Busch H, Feller AC, Gebauer N (2021) Mutational landscape of high-grade B-cell lymphoma with MYC-, BCL2 and/or BCL6 rearrangements characterized by whole-exome sequencing. Haematologica. 107(8):1850–1863. https://doi.org/10.3324/haematol.2021.279631
Merron B, Davies A (2018) Double hit lymphoma: how do we define it and how do we treat it? Best Pract Res Clin Haematol 31:233–240. https://doi.org/10.1016/j.beha.2018.07.012
Oki Y, Noorani M, Lin P, Davis RE, Neelapu SS, Ma L, Ahmed M, Rodriguez MA, Hagemeister FB, Fowler N, Wang M, Fanale MA, Nastoupil L, Samaniego F, Lee HJ, Dabaja BS, Pinnix CC, Medeiros LJ, Nieto Y, Khouri I, Kwak LW, Turturro F, Romaguera JE, Fayad LE, Westin JR (2014) Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 166:891–901. https://doi.org/10.1111/bjh.12982
Petrich AM, Gandhi M, Jovanovic B, Castillo JJ, Rajguru S, Yang DT, Shah KA, Whyman JD, Lansigan F, Hernandez-Ilizaliturri FJ, Lee LX, Barta SK, Melinamani S, Karmali R, Adeimy C, Smith S, Dalal N, Nabhan C, Peace D, Vose J, Evens AM, Shah N, Fenske TS, Zelenetz AD, Landsburg DJ, Howlett C, Mato A, Jaglal M, Chavez JC, Tsai JP, Reddy N, Li S, Handler C, Flowers CR, Cohen JB, Blum KA, Song K, Sun HL, Press O, Cassaday R, Jaso J, Medeiros LJ, Sohani AR, Abramson JS (2014) Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: a multicenter retrospective analysis. Blood 124:2354–2361. https://doi.org/10.1182/blood-2014-05-578963
Snuderl M, Kolman OK, Chen YB, Hsu JJ, Ackerman AM, Dal Cin P, Ferry JA, Harris NL, Hasserjian RP, Zukerberg LR, Abramson JS, Hochberg EP, Lee H, Lee AI, Toomey CE, Sohani AR (2010) B-cell lymphomas with concurrent IGH-BCL2 and MYC rearrangements are aggressive neoplasms with clinical and pathologic features distinct from Burkitt lymphoma and diffuse large B-cell lymphoma. Am J Surg Pathol 34:327–340. https://doi.org/10.1097/PAS.0b013e3181cd3aeb
Miao Y, Hu S, Lu X, Li S, Wang W, Medeiros LJ, Lin P (2016) Double-hit follicular lymphoma with MYC and BCL2 translocations: a study of 7 cases with a review of literature. Hum Pathol 58:72–77. https://doi.org/10.1016/j.humpath.2016.07.025
Miyaoka M, Kikuti YY, Carreras J, Ikoma H, Hiraiwa S, Ichiki A, Kojima M, Ando K, Yokose T, Sakai R, Hoshikawa M, Tomita N, Miura I, Takata K, Yoshino T, Takizawa J, Bea S, Campo E, Nakamura N (2018) Clinicopathological and genomic analysis of double-hit follicular lymphoma: comparison with high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements. Mod Pathol 31:313–326. https://doi.org/10.1038/modpathol.2017.134
Chaudhary S, Brown N, Song JY, Yang L, Skrabek P, Nasr MR, Wong JT, Bedell V, Murata-Collins J, Kochan L, Li J, Zhang W, Chan WC, Weisenburger DD, Perry AM (2021) Relative frequency and clinicopathologic characteristics of MYC-rearranged follicular lymphoma. Hum Pathol 114:19–27. https://doi.org/10.1016/j.humpath.2021.04.014
Bhavsar S, Liu YC, Gibson SE, Moore EM, Swerdlow SH (2022) Mutational landscape of TdT+ large B-cell lymphomas supports their distinction from B-lymphoblastic neoplasms: a multiparameter study of a rare and aggressive entity. Am J Surg Pathol 46:71–82. https://doi.org/10.1097/pas.0000000000001750
Khanlari M, Medeiros LJ, Lin P, Xu J, You MJ, Tang G, Yin CC, Wang W, Qiu L, Miranda RN, Bueso-Ramos CE, Li S (2021) Blastoid high-grade B-cell lymphoma initially presenting in bone marrow: a diagnostic challenge. Mod Pathol (2022 Mar) 35(3):419–426. https://doi.org/10.1038/s41379-021-00909-4
Nie K, Redmond D, Eng KW, Zhang T, Cheng S, Mathew S, Elemento O, Tam W (2021) Mutation landscape, clonal evolution pattern, and potential pathogenic pathways in B-lymphoblastic transformation of follicular lymphoma. Leukemia 35:1203–1208. https://doi.org/10.1038/s41375-020-01014-2
Ok CY, Medeiros LJ, Thakral B, Tang G, Jain N, Jabbour E, Pierce SA, Konoplev S (2019) High-grade B-cell lymphomas with TdT expression: a diagnostic and classification dilemma. Mod Pathol 32:48–58. https://doi.org/10.1038/s41379-018-0112-9
Alsuwaidan A, Pirruccello E, Jaso J, Koduru P, Garcia R, Krueger J, Doucet M, Chaudhry R, Fuda F, Chen W (2019) Bright CD38 expression by flow cytometric analysis is a biomarker for double/triple hit lymphomas with a moderate sensitivity and high specificity. Cytometry B Clin Cytom 96:368–374. https://doi.org/10.1002/cyto.b.21770
Moench L, Sachs Z, Aasen G, Dolan M, Dayton V, Courville EL (2016) Double- and triple-hit lymphomas can present with features suggestive of immaturity, including TdT expression, and create diagnostic challenges. Leuk Lymphoma 57:2626–2635. https://doi.org/10.3109/10428194.2016.1143939
Scott DW, King RL, Staiger AM, Ben-Neriah S, Jiang A, Horn H, Mottok A, Farinha P, Slack GW, Ennishi D, Schmitz N, Pfreundschuh M, Nowakowski GS, Kahl BS, Connors JM, Gascoyne RD, Ott G, Macon WR, Rosenwald A (2018) High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood 131:2060–2064. https://doi.org/10.1182/blood-2017-12-820605
Collinge B, Ben-Neriah S, Chong L, Boyle M, Jiang A, Miyata-Takata T, Farinha P, Craig JW, Slack GW, Ennishi D, Mottok A, Meissner B, Chavez EA, Gerrie AS, Villa D, Freeman C, Savage KJ, Sehn LH, Morin RD, Mungall AJ, Gascoyne RD, Marra MA, Connors JM, Steidl C, Scott DW (2021) The impact of MYC and BCL2 structural variants in tumors of DLBCL morphology and mechanisms of false-negative MYC IHC. Blood 137:2196–2208. https://doi.org/10.1182/blood.2020007193
Chisholm KM, Bangs CD, Bacchi CE, Molina-Kirsch H, Cherry A, Natkunam Y (2015) Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am J Surg Pathol 39:294–303. https://doi.org/10.1097/pas.0000000000000365
Aukema SM, Siebert R, Schuuring E, van Imhoff GW, Kluin-Nelemans HC, Boerma EJ, Kluin PM (2011) Double-hit B-cell lymphomas. Blood 117:2319–2331. https://doi.org/10.1182/blood-2010-09-297879
Evrard SM, Péricart S, Grand D, Amara N, Escudié F, Gilhodes J, Bories P, Traverse-Glehen A, Dubois R, Brousset P, Parrens M, Laurent C (2019) Targeted next generation sequencing reveals high mutation frequency of CREBBP, BCL2 and KMT2D in high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements. Haematologica 104:e154–e157. https://doi.org/10.3324/haematol.2018.198572
Gebauer N, Bernard V, Feller AC, Merz H (2013) ID3 Mutations are recurrent events in double-hit B-cell lymphomas. Anticancer Res 33:4771–4778
Momose S, Weißbach S, Pischimarov J, Nedeva T, Bach E, Rudelius M, Geissinger E, Staiger AM, Ott G, Rosenwald A (2015) The diagnostic gray zone between Burkitt lymphoma and diffuse large B-cell lymphoma is also a gray zone of the mutational spectrum. Leukemia 29:1789–1791. https://doi.org/10.1038/leu.2015.34
Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, Staiger AM, Wala JA, Ducar MD, Leshchiner I, Rheinbay E, Taylor-Weiner A, Coughlin CA, Hess JM, Pedamallu CS, Livitz D, Rosebrock D, Rosenberg M, Tracy AA, Horn H, van Hummelen P, Feldman AL, Link BK, Novak AJ, Cerhan JR, Habermann TM, Siebert R, Rosenwald A, Thorner AR, Meyerson ML, Golub TR, Beroukhim R, Wulf GG, Ott G, Rodig SJ, Monti S, Neuberg DS, Loeffler M, Pfreundschuh M, Trümper L, Getz G, Shipp MA (2018) Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 24:679–690. https://doi.org/10.1038/s41591-018-0016-8
Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, Hodson DJ, Xiao W, Yu X, Yang Y, Zhao H, Xu W, Liu X, Zhou B, Du W, Chan WC, Jaffe ES, Gascoyne RD, Connors JM, Campo E, Lopez-Guillermo A, Rosenwald A, Ott G, Delabie J, Rimsza LM, TayKuang Wei K, Zelenetz AD, Leonard JP, Bartlett NL, Tran B, Shetty J, Zhao Y, Soppet DR, Pittaluga S, Wilson WH, Staudt LM (2018) Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med 378:1396–1407. https://doi.org/10.1056/NEJMoa1801445
Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, Wang JQ, Schmitz R, Morin RD, Tang J, Jiang A, Bagaev A, Plotnikova O, Kotlov N, Johnson CA, Wilson WH, Scott DW, Staudt LM (2020) A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell 37:551-568.e514. https://doi.org/10.1016/j.ccell.2020.03.015
Ye Q, Xu-Monette ZY, Tzankov A, Deng L, Wang X, Manyam GC, Visco C, Montes-Moreno S, Zhang L, Dybkær K, Chiu A, Orazi A, Zu Y, Bhagat G, Richards KL, Hsi ED, Choi WW, van Krieken JH, Huh J, Ponzoni M, Ferreri AJ, Parsons BM, Møller MB, Piris MA, Winter JN, Medeiros LJ, Hu S, Young KH (2016) Prognostic impact of concurrent MYC and BCL6 rearrangements and expression in de novo diffuse large B-cell lymphoma. Oncotarget 7:2401–2416. https://doi.org/10.18632/oncotarget.6262
Li S, Desai P, Lin P, Yin CC, Tang G, Wang XJ, Konoplev SN, Khoury JD, Bueso-Ramos CE, Medeiros LJ (2016) MYC/BCL6 double-hit lymphoma (DHL): a tumour associated with an aggressive clinical course and poor prognosis. Histopathology 68:1090–1098. https://doi.org/10.1111/his.12884
Pillai RK, Sathanoori M, Van Oss SB, Swerdlow SH (2013) Double-hit B-cell lymphomas with BCL6 and MYC translocations are aggressive, frequently extranodal lymphomas distinct from BCL2 double-hit B-cell lymphomas. Am J Surg Pathol 37:323–332. https://doi.org/10.1097/PAS.0b013e31826cebad
Johnson SM, Umakanthan JM, Yuan J, Fedoriw Y, Bociek RG, Kaiser-Rogers K, Sanmann JN, Montgomery ND (2018) Lymphomas with pseudo-double-hit BCL6-MYC translocations due to t(3;8)(q27;q24) are associated with a germinal center immunophenotype, extranodal involvement, and frequent BCL2 translocations. Hum Pathol 80:192–200. https://doi.org/10.1016/j.humpath.2018.06.006
Ryan RJH, Drier Y, Whitton H, Cotton MJ, Kaur J, Issner R, Gillespie S, Epstein CB, Nardi V, Sohani AR, Hochberg EP, Bernstein BE (2015) Detection of enhancer-associated rearrangements reveals mechanisms of oncogene dysregulation in B-cell lymphoma Cancer. Discovery 5:1058–1071. https://doi.org/10.1158/2159-8290.Cd-15-0370
Chong LC, Ben-Neriah S, Slack GW, Freeman C, Ennishi D, Mottok A, Collinge B, Abrisqueta P, Farinha P, Boyle M, Meissner B, Kridel R, Gerrie AS, Villa D, Savage KJ, Sehn LH, Siebert R, Morin RD, Gascoyne RD, Marra MA, Connors JM, Mungall AJ, Steidl C, Scott DW (2018) High-resolution architecture and partner genes of MYC rearrangements in lymphoma with DLBCL morphology. Blood Adv 2:2755–2765. https://doi.org/10.1182/bloodadvances.2018023572
Stengel A, Kern W, Meggendorfer M, Haferlach T, Haferlach C (2019) Detailed molecular analysis and evaluation of prognosis in cases with high grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements. Br J Haematol 185:951–954. https://doi.org/10.1111/bjh.15653
Bertrand P, Bastard C, Maingonnat C, Jardin F, Maisonneuve C, Courel MN, Ruminy P, Picquenot JM, Tilly H (2007) Mapping of MYC breakpoints in 8q24 rearrangements involving non-immunoglobulin partners in B-cell lymphomas. Leukemia 21:515–523. https://doi.org/10.1038/sj.leu.2404529
Muñoz-Mármol AM, Sanz C, Tapia G, Marginet R, Ariza A, Mate JL (2013) MYC status determination in aggressive B-cell lymphoma: the impact of FISH probe selection. Histopathology 63:418–424. https://doi.org/10.1111/his.12178
King RL, McPhail ED, Meyer RG, Vasmatzis G, Pearce K, Smadbeck JB, Ketterling RP, Smoley SA, Greipp PT, Hoppman NL, Peterson JF, Baughn LB (2019) False-negative rates for MYC fluorescence in situ hybridization probes in B-cell neoplasms. Haematologica 104:e248–e251. https://doi.org/10.3324/haematol.2018.207290
Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM (1982) Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A 79:7824–7827. https://doi.org/10.1073/pnas.79.24.7824
Hilton LK, Tang J, Ben-Neriah S, Alcaide M, Jiang A, Grande BM, Rushton CK, Boyle M, Meissner B, Scott DW, Morin RD (2019) The double-hit signature identifies double-hit diffuse large B-cell lymphoma with genetic events cryptic to FISH. Blood 134:1528–1532. https://doi.org/10.1182/blood.2019002600
Landsburg DJ, Falkiewicz MK, Petrich AM, Chu BA, Behdad A, Li S, Medeiros LJ, Cassaday RD, Reddy NM, Bast MA, Vose JM, Kruczek KR, Smith SE, Patel P, Hernandez-Ilizaliturri F, Karmali R, Rajguru S, Yang DT, Maly JJ, Blum KA, Zhao W, Vanslambrouck C, Nabhan C (2016) Sole rearrangement but not amplification of MYC is associated with a poor prognosis in patients with diffuse large B cell lymphoma and B cell lymphoma unclassifiable. Br J Haematol 175:631–640. https://doi.org/10.1111/bjh.14282
Krull JE, Wenzl K, Hartert KT, Manske MK, Sarangi V, Maurer MJ, Larson MC, Nowakowski GS, Ansell SM, McPhail E, Habermann TM, Link BK, King RL, Cerhan JR, Novak AJ (2020) Somatic copy number gains in MYC, BCL2, and BCL6 identifies a subset of aggressive alternative-DH/TH DLBCL patients. Blood Cancer J 10:117. https://doi.org/10.1038/s41408-020-00382-3
Li S, Seegmiller AC, Lin P, Wang XJ, Miranda RN, Bhagavathi S, Medeiros LJ (2015) B-cell lymphomas with concurrent MYC and BCL2 abnormalities other than translocations behave similarly to MYC/BCL2 double-hit lymphomas. Mod Pathol 28:208–217. https://doi.org/10.1038/modpathol.2014.95
Schieppati F, Balzarini P, Fisogni S, Re A, Pagani C, Bianchetti N, Micheli L, Passi A, Ferrari S, Maifredi A, Bottelli C, Leopaldo R, Pellegrini V, Facchetti F, Rossi G, Tucci A (2020) An increase in MYC copy number has a progressive negative prognostic impact in patients with diffuse large B-cell and high-grade lymphoma, who may benefit from intensified treatment regimens. Haematologica 105:1369–1378. https://doi.org/10.3324/haematol.2019.223891
Sermer D, Bobillo S, Dogan A, Zhang Y, Seshan V, Lavery JA, Batlevi C, Caron P, Hamilton A, Hamlin P, Horwitz S, Joffe E, Kumar A, Matasar M, Noy A, Owens C, Moskowitz A, Palomba ML, Straus D, von Keudell G, Rodriguez-Rivera I, Falchi L, Zelenetz A, Yahalom J, Younes A (2020) Extra copies of MYC, BCL2, and BCL6 and outcome in patients with diffuse large B-cell lymphoma. Blood Adv 4:3382–3390. https://doi.org/10.1182/bloodadvances.2020001551
Stasik CJ, Nitta H, Zhang W, Mosher CH, Cook JR, Tubbs RR, Unger JM, Brooks TA, Persky DO, Wilkinson ST, Grogan TM, Rimsza LM (2010) Increased MYC gene copy number correlates with increased mRNA levels in diffuse large B-cell lymphoma. Haematologica 95:597–603. https://doi.org/10.3324/haematol.2009.012864
Testoni M, Kwee I, Greiner TC, Montes-Moreno S, Vose J, Chan WC, Chiappella A, Baldini L, Ferreri AJ, Gaidano G, Mian M, Zucca E, Bertoni F (2011) Gains of MYC locus and outcome in patients with diffuse large B-cell lymphoma treated with R-CHOP. Br J Haematol 155:274–277. https://doi.org/10.1111/j.1365-2141.2011.08675.x
Yoon SO, Jeon YK, Paik JH, Kim WY, Kim YA, Kim JE, Kim CW (2008) MYC translocation and an increased copy number predict poor prognosis in adult diffuse large B-cell lymphoma (DLBCL), especially in germinal centre-like B cell (GCB) type. Histopathology 53:205–217. https://doi.org/10.1111/j.1365-2559.2008.03076.x
Ennishi D, Jiang A, Boyle M, Collinge B, Grande BM, Ben-Neriah S, Rushton C, Tang J, Thomas N, Slack GW, Farinha P, Takata K, Miyata-Takata T, Craig J, Mottok A, Meissner B, Saberi S, Bashashati A, Villa D, Savage KJ, Sehn LH, Kridel R, Mungall AJ, Marra MA, Shah SP, Steidl C, Connors JM, Gascoyne RD, Morin RD, Scott DW (2019) Double-Hit gene expression signature defines a distinct subgroup of germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol 37:190–201. https://doi.org/10.1200/jco.18.01583
Sha C, Barrans S, Cucco F, Bentley MA, Care MA, Cummin T, Kennedy H, Thompson JS, Uddin R, Worrillow L, Chalkley R, van Hoppe M, Ahmed S, Maishman T, Caddy J, Schuh A, Mamot C, Burton C, Tooze R, Davies A, Du MQ, Johnson PWM, Westhead DR (2019) Molecular high-grade B-cell lymphoma: defining a poor-risk group that requires different approaches to therapy. J Clin Oncol 37:202–212. https://doi.org/10.1200/jco.18.01314
Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ, Greiner TC, Weisenburger DD, Rosenwald A, Ott G, Müller-Hermelink HK, Gascoyne RD, Delabie J, Rimsza LM, Braziel RM, Grogan TM, Campo E, Jaffe ES, Dave BJ, Sanger W, Bast M, Vose JM, Armitage JO, Connors JM, Smeland EB, Kvaloy S, Holte H, Fisher RI, Miller TP, Montserrat E, Wilson WH, Bahl M, Zhao H, Yang L, Powell J, Simon R, Chan WC, Staudt LM (2006) Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med 354:2431–2442. https://doi.org/10.1056/NEJMoa055759
Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TF, Bernd HW, Cogliatti SB, Dierlamm J, Feller AC, Hansmann ML, Haralambieva E, Harder L, Hasenclever D, Kühn M, Lenze D, Lichter P, Martin-Subero JI, Möller P, Müller-Hermelink HK, Ott G, Parwaresch RM, Pott C, Rosenwald A, Rosolowski M, Schwaenen C, Stürzenhofecker B, Szczepanowski M, Trautmann H, Wacker HH, Spang R, Loeffler M, Trümper L, Stein H, Siebert R (2006) A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med 354:2419–2430. https://doi.org/10.1056/NEJMoa055351
Mlynarczyk C, Fontán L, Melnick A (2019) Germinal center-derived lymphomas: the darkest side of humoral immunity. Immunol Rev 288:214–239. https://doi.org/10.1111/imr.12755
Tripodo C, Zanardi F, Iannelli F, Mazzara S, Vegliante M, Morello G, Di Napoli A, Mangogna A, Facchetti F, Sangaletti S, Chiodoni C, VanShoiack A, Jeyasekharan AD, Casola S, Colombo MP, Ponzoni M, Pileri SA (2020) A Spatially resolved dark- versus light-zone microenvironment signature subdivides germinal center-related aggressive B cell lymphomas. iScience 23:101562. https://doi.org/10.1016/j.isci.2020.101562
Song JY, Perry AM, Herrera AF, Chen L, Skrabek P, Nasr MR, Ottesen RA, Nikowitz J, Bedell V, Murata-Collins J, Li Y, McCarthy C, Pillai R, Wang J, Wu X, Zain J, Popplewell L, Kwak LW, Nademanee AP, Niland JC, Scott DW, Gong Q, Chan WC, Weisenburger DD (2021) Double-hit signature with TP53 abnormalities predicts poor survival in patients with germinal center type diffuse large B-cell lymphoma treated with R-CHOP. Clin Cancer Res 27:1671–1680. https://doi.org/10.1158/1078-0432.Ccr-20-2378
Li J, Liu X, Yao Z, Zhang M (2020) High-grade B-cell lymphomas, not otherwise specified: a study of 41 cases. Cancer Manag Res 12:1903–1912. https://doi.org/10.2147/cmar.S243753
Olszewski A, Kurt H, Evens AM (2021) Defining and treating high-grade B-cell lymphoma. NOS Blood. https://doi.org/10.1182/blood.2020008374
Hüttl KS, Staiger AM, Richter J, Ott MM, Kalmbach S, Klapper W, Biesdorf AS, Trümper L, Rosenwald A, Ziepert M, Horn H, Ott G (2021) The “Burkitt-like” immunophenotype and genotype is rarely encountered in diffuse large B cell lymphoma and high-grade B cell lymphoma. NOS Virchows Arch 479:575–583. https://doi.org/10.1007/s00428-021-03050-4
Author information
Authors and Affiliations
Contributions
All authors contributed to the gathering of supporting data during the Clinical Advisory Committee meeting (9/2021) and afterwards. RLK and EDH drafted the manuscript. SHS, EDH, and RLK created the figures. All authors edited the manuscript.
Compliance with all ethical standards was undertaken for this work. No research involving human participants and/or animals was performed for this work. No informed consent was required.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare the following conflicts of interest.
DWS: Consulting: Abbvie, AstraZeneca, Incyte, Janssen.
Research funding: Janssen, NanoString Technologies, Roche.
Patents: named inventor on patents describing using gene expression to subtype B-cell lymphomas, including one licensed to NanoString Technologies.
MAP: has served on the advisory board for Millenium/Takeda, Celgene, Gilead, Jansen, Nanotring, and Kyowa Kirin; received lecture fees from Millenium/Takeda, Kyowa Kirin, EUSA and Jansen; and received research funding from Millenium/Takeda, Gilead, and Kura.
EDH: Research funding: Eli Lilly, Virtuoso. Advisory Boards: Abcon, Cytomx, Astellas, Novartis.
RLK, SHS, JRC, and WCC have no conflicts or competing interests to disclose.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
King, R.L., Hsi, E.D., Chan, W.C. et al. Diagnostic approaches and future directions in Burkitt lymphoma and high-grade B-cell lymphoma. Virchows Arch 482, 193–205 (2023). https://doi.org/10.1007/s00428-022-03404-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-022-03404-6