
Abstract
Gastrointestinal stromal tumors (GISTs) are characterized by mutations in exons 9, 11, 13, and 17 of KIT or exons 12, 14, and 18 of PDGFRA gene. However, approximately 10 to 15 % of GISTs lack the mutations in KIT and PDGFRA, and these are referred to as wild-type GISTs which are less sensitive to tyrosine-kinase inhibitors. The aim of this study was to detect BRAF mutations in patients with wild-type GISTs. We applied a sensitive allele-specific PCR, which was optimized using the V600E mutation-harboring cell line RKO, followed by verification of the results by dideoxy sequencing. We selected 149 GIST patients without detectable mutations in KIT and PDGFRA genes from the Slovak national GIST register and analyzed biopsy specimens for the presence of BRAF mutations in exon 15. We identified nine patients with the V600E mutation. The BRAF-driven GISTs were primary gastric (n = 3), small intestinal (n = 3), colon (n = 1), and of uncertain origin (n = 1). We also included a liver metastasis of a patient with a simultaneous KIT exon 11-mutated intra-abdominal metastasis. We conclude that genome analysis of wild-type GISTs for mutations should include the BRAF gene, as its mutation status contributes to understanding of pathogenesis and might be important for decisions on therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract, essentially resistant to conventional chemotherapy and radiotherapy. GISTs are characterized by mutations in exons 9, 11, 13, and 17 of the KIT or exons 12, 14, and 18 of the PDGFRA gene. Activating mutations in these genes represent early molecular events and are considered to be mutually exclusive [1–3]. However, approximately 10 to 15 % of GISTs lack mutations in KIT and PDGFRA and are referred to as wild-type GISTs (WT GIST), which are less sensitive to tyrosine-kinase inhibitors [4]. The molecular characteristics of these GISTs are heterogeneous. They can be divided into two main groups according to immunohistochemical staining for succinate dehydrogenase subunit B (SDHB) [5]. GISTs characterized by SDHB loss carry germline or de novo mutations of any of the four SDH subunits [6]. SDHB-intact GISTs comprise neurofibromatosis 1 (NF1)-associated GISTs and a subset of sporadic adult GISTs with or without mutations in BRAF/RAS signaling associated genes [7].
BRAF is a downstream effector of the RAS protein in the RAS-RAF-MEK-ERK pathway, involved in cell cycle regulation and transcriptional activation. The BRAF gene is mutated in many cancers, such as malignant melanoma or colorectal carcinoma. The most frequent activating mutation is a nucleotide substitution at the position 1799 (A to T) in the kinase domain in exon 15, which results in the missense mutation V600E at protein level [8, 9]. Recently this BRAF mutation has been identified in between 3.5 and 13 % of WT GISTs [10–13]. For BRAF-mutated GISTs no specific pathological features have been reported. However, they may represent an alternative to the KIT and PDGFRA activation in GIST pathogenesis. GISTs with a BRAF mutation are mostly of non-gastric origin; however, the published studies seem to be small for definitive conclusions.
Accurate characterization of gene abnormalities in GISTs is important for proper administration of specific, molecular-targeted therapy with KIT/PDGFRA tyrosine-kinase inhibitors. Imatinib mesylate is very effective in GISTs harboring KIT exon 11 mutations as well as in GISTs with exon 9 mutation, be it at a higher dose. GISTs with KIT mutations in exon 13 or 17, the D842V mutation in PDGFRA exon 18 or WT GISTs are less sensitive to imatinib or imatinib resistant [4, 14]. Imatinib treatment would be ineffective in patients with BRAF mutant GISTs; but targeted drugs are in clinical trials for other BRAF-driven cancers, such as melanoma. A published case study has reported prolonged antitumor activity of the BRAF inhibitor dabrafenib in a BRAF-mutated GIST patient [15, 16].
Different methods can be employed for detection of BRAF mutations generally, notably in GISTs. DNA dideoxy sequencing is the “gold standard” and is the most frequently applied method for mutation detection [10, 11]. Often, a combination of screening methods, such as denaturing high-pressure liquid chromatography (DHPLC), is used [13]. Some studies applied allele-specific approaches [13, 17].
The aim of this study was to detect BRAF mutations in patients with WT GISTs using a sensitive allele-specific PCR (AS-PCR) and compare it with dideoxy sequencing. We also studied histopathological characteristics of newly identified BRAF-positive patients and compared them with published data.
Materials and methods
Patients and tumor characterization
We identified our patient cohort in the National Slovak GIST registry. GISTs had been analyzed for KIT/PDGFRA mutations and are part of international clinical studies focusing on the GIST recurrence [18, 19]. Collection of formalin-fixed paraffin-embedded (FFPE) tissue sections was included in the National GIST Registry, which is kept in the Department of Pathology of CU JFM and University Hospital in Martin. Molecular analysis of DNA isolated from FFPE sections included mutations in exons 9, 11, 13, and 17 of KIT and exons12, 14, and 18 of PDGRA [19]. Samples negative for mutations in these exons were tested for the presence of mutations in KIT exon 8 using a previously published protocol with the following primers: Ex 8 KIT-F 5′-TTTCCAGCACTCTGACATATGGC-3′ and Ex8KIT_R 5′-TCCCCTCTGCATTATAAGCAGTGC-3′ [19]. Patients without a mutation in these exons are referred to as WT GISTs, and in the cohort of 704 patients, registered in the National GIST Register between 2004 and 2014, 149 (21.1 %) WT GIST patients were identified. In 135 (19 %) cases, the patients were adults; in 15 (2.1 %) cases, age information was not available (Table 1).
The GIST diagnosis on the biopsy specimens were verified by two senior pathologists (LP and PS) using internationally accepted histological diagnostic, differential diagnostic, and risk stratification criteria as described in detail in WHO classification of GI tract and WHO classification of soft tissue tumors [20, 21] and included histomorphological GIST type, size, and primary localization of the tumor, mitotic activity index expressed as a number of mitotic figures/5 mm2. We also performed immunohistochemical staining with a standard panel of antibodies to detect expression of the GIST markers CD117, CD34, and DOG-1, as well as of S-100 protein and smooth muscle-specific actin, supplemented in individual cases by detection of other lineage-specific antigens for differential diagnostic purposes. As the DOG1 antibody was not yet available at the time of diagnosis of all cases, we performed immunostaining for DOG in cases missing this information when sufficient archival material was available. Details of our approach to classification of GIST have been described previously [19].
DNA extraction
FFPE sections were deparaffinized twice in xylene and rehydrated through a series of descending concentrations of alcohol. Genomic DNA was extracted with the DNeasy Blood and Tissue Kit (Qiagen, Germany) according to the manufacturer’s instructions, using silica membrane columns. Briefly, 180 μl of lysis buffer and 20 μl of proteinase K were added to the tube containing resuspended deparaffinized tumor tissue, and the samples were incubated at 56 °C for 48 h. The cell lysate was mixed with 200 μl of protein precipitation solution and 200 μl of absolute ethanol and purified through the column. After washing, DNA was eluted with 60 μl of elution buffer. The concentration of DNA was determined at 260 nm by spectrophotometry.
AS-PCR reaction for BRAF c.1799T˃A (V600E)
PCR amplification was performed in two separate tubes—one for amplification of wild type and one for the mutant variant. The first tube contained primers for wild-type BRAF BRAFEx15ASFwt 5′-GTGATTTTGGTCTAGCTACAGT-3′ and BRAFEx15R 5′-GGCCAAAATTTAATCAG TGGA-3′. The second tube contained the BRAF c.1799T˃A (V600E) mutation-specific primer BRAFEx15ASFmut 5′-GTGATTTTGCTCTAGCTACAGA-3′ and the reverse primer BRAFEx15R. The optimized reaction mix for both PCR reactions contained 4 mM MgCl2, 10 pmol/L of each primer (Sigma-Aldrich, USA), 0.1 mmol/L of each of four dNTPs (Applied Biosystems, USA), and 1 unit of FastStart Taq DNA Polymerase (Roche Diagnostics, Switzerland). The thermal cycling protocol consisted of initial denaturation at 95 °C for 10 min, followed by 45 cycles of amplification: 95 °C for 30 s, 64 °C (for both alleles) for 1 min, and 72 °C for 1 min. Final extension was performed at 72 °C for 5 min. DNA extracted from the RKO cell line harboring a heterozygous V600E mutation (kindly provided by Dr. N.A.P. Franken from Academic Medical Center, University of Amsterdam in Netherlands) was used as positive control and DNA from a healthy individual as negative control. Serial concentrations of mutated control were prepared by mixing DNA from the RKO cell line with wild-type DNA, to reach concentrations of 10, 5, 2.5, 1.25, and 0.625 %. These were used to assess sensitivity and specificity of the AS-PCR. The amplification products were separated by electrophoresis on 1.75 % agarose gel, stained with GelRed Nucleic Acid (Biotinum Inc., USA) and visualized on an UV transilluminator.
DNA sequencing was performed on PCR products obtained by amplification with the BRAFEx15F 5′-TCATAATGCTTGCTCTGATAGGA-3′and the BRAFEx15R 5′-GGCCAAAATTTAATCAGTGGA-3′ primers. The PCR conditions were identical to those described above for AS-PCR and was performed as described previously [19]. We initially dideoxy sequenced 42 FFPE samples of WT GISTs, which was subsequently employed to confirm results obtained by AS-PCR.
Results
Characterization of the WT GIST cohort
Histopathological parameters of the cases, 67 male and 67 female patients and 15 of which gender information was missing, including tumor size, histological grade, localization, morphological subtype, and mitotic count, are listed in Table 1. Tumor size was less than 2 cm in 29 (19 %) cases, between 2 and 5 cm in 48 cases (32 %) and larger than 5 cm in 48 cases (32 %). In 24 (16 %) cases, tumor size was not available. Most tumors (58 %) showed spindle cell morphology, while epithelioid and spindle-epithelioid morphology each represented 18 %. In 5 % of cases, tumor morphology was not known (Table 1). Of WT GISTs, 65 (44 %) occurred in the stomach, while 44 cases (30 %) were localized in the small intestine, 4 (3 %) were localized in the colon, 1 (1 %) in the rectum, 5 (3 %) were liver metastases, and the location was unknown for 8 cases (5 %). In 22 (15 %) cases, neither the surgical protocol nor the biopsy examination allowed identification of the GIST origin, and these were considered GISTs of uncertain origin. Most GISTs were primary tumors (129, 87 %); five were liver metastases and four omentum or mesentery metastases. CD117 positive were 122 (82 %) GISTs. Strong CD 117 staining was noted in 76 % and weak staining in 6 % of cases. In 5 % of cases, a CD 117 staining result was not available. CD34 staining was positive in 93 (62 %) cases.
BRAF mutation detection by dideoxy sequencing
The FFPE sections from the first 42 WT GIST patients were analyzed for the mutations in exon 15 of the BRAF gene. However, the no-BRAF exon 15 mutation was identified in these samples.
BRAF mutation detection by AS-PCR and confirmation by sequencing
The dilution series of mutated RKO and non-mutated DNA was amplified with primers specific for the wild-type and mutated sequences. The PCR primers generate a 125-bp amplicon (Fig. 1a). Allele-specific primer BRAFEx15Fmut PCR detected mutated DNA with a sensitivity of 0.63 %.

a Ethidiumbromide stained agarose gel showing the results of AS-PCR of the primer for the BRAF c.1799T˃A (V600E) mutation for the serial dilutions of the RKO cell line DNA. The size of the amplicon comprises 125 bp compared with the 100-bp ladder in the first lane. b Results of the AS-PCR on patient samples performed in two tubes, one with allele-specific wild type (wt) and a second with allele-specific mutation primer (mut), compared with the 100-bp ladder (first line on the left). Samples 1, 7, and 12 are referred to as positive and the remaining samples as negative for the BRAF c.1799T˃A (V600E) mutation
Figure 1b shows representative results of AS-PCR for detection of the nucleotide substitution c.1799T˃A in BRAF in 149 WT GISTs. Lanes 1, 7, and 12 show the presence of wild-type and mutated alleles in the same sample. Lanes 2 to 6 and 8 to 11 show the presence of wild-type alleles only. Using this approach, 9 of 149 samples (6 %) tested positive for the BRAF V600E mutation (Table 2). However, subsequent dideoxy sequencing confirmed the mutation in four cases only (3 %) (Fig. 2a, electropherogram on the bottom). The proportion of patients with a BRAF-mutated (V600E) GIST with known KIT, and PDGFRA mutation status was 1.3 % (9/704) and in the WT GIST subset 6.04 % (9/149).

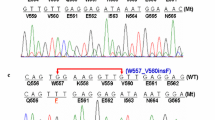
a DNA sequence of BRAF wild-type sequence (the electropherogram on the top), a patient with the c.1799T˃A substitution (on the bottom). The presence of the mutations was confirmed by both forward and reverse sequencing. b Aligned sequences of V600E patient and wild-type exon 15 BRAF. Changes are shown in red (nucleotides are indicated by capital letters above, amino acids by capital letters under the nucleotides). V600E mutations were detected as confirmation of the AS-PCR
Clinicopathological characterisitics of patients with a BRAF-mutated GIST
Clinicopathological data are summarized in Table 2. Average age of patients was 61.9 years. Five patients were females and four males with a mean age of 60.4 and 63.8 years, respectively. Average tumor size was 59.9 mm. Eight tumors were of spindle cell and one of epithelioid morphology. They were localized in the stomach (three cases), small intestine (three cases), and colon (one case); two tumors were multifocal. In four cases, the tumors showed low mitotic activity (˂5 mitoses/50 HPF). In seven cases, the primary tumors were analyzed; in one case, the (primary or metastatic) nature was not clear and one case was a liver metastasis. The liver metastasis was KIT and PDGFRA wild type but V600E positive and was positive for both CD117 and DOG1 antigens (Fig. 3a–d). Interestingly, another metastasis originating from the perisplenic region of the same patient showed a mutation in exon 11 of KIT gene (D579del), was BRAF wild type and CD117 positive (Fig. 3E, F).

Histopathological and immunohistochemical examination of liver metastasis of BRAF V600E-positive case 2 (a–e). a GIST liver metastasis with spindle cell morphology (HE, ×10). b Detail from (a; HE, ×40). c CD117 positivity of the liver metastasis (×20). d DOG1 positivity of the liver metastasis (×20). e Perisplenic infiltration by GIST (HE, ×10). f Detail from (e; HE, ×63). g CD117-positive perisplenic infiltrate (×20)
Discussion
BRAF mutations trigger RAS-BRAF-MEK-MAPK signaling and represent a very common mutational event in different types of cancer, such as colorectal cancer or melanoma. This makes them a favored object for the development of targeted therapies. In WT GISTs, the BRAF mutation V600E has been reported with a low frequency between 3.5 and 13.4 % [10–13, 15]. Our study represents the largest WT GIST cohort analyzed for the presence of BRAF mutation published as yet. We studied 149 patients with a KIT and PDGFRA WT GIST for the presence of a BRAF mutation in exon 15 in two phases. Initially, we used dideoxy sequecing on 42 patients, but these were all negative for the c.1799T˃A mutation resulting in V600E amino acid substitution. We concluded that dideoxy sequecing is not sensitive enough and decided to apply AS-PCR, which is more sensitive. Using this approach, we detected a V600E BRAF mutation in 9 of the 149 WT GISTs, in one of which the mutation status of the primary tumor was not known. This represents 6 % of the WT GIST cohort and is within the frequency range of earlier published data [10, 13, 17].
The first report identifying a V600E mutation in a GIST concerned three middle-aged female patients with a primary GIST in the small bowel, with spindle cell morphology and high risk of malignancy [10]. Agaimy et al. [11] found BRAF mutations in two male patients with an early-stage GISTs in the gastric corpus and jejunum, also with spindle cell morphology but mitotically inactive. A study of 26 WT GISTs reported one case with V600E mutation in a male patient with a small intestinal GIST [12]. Hostein et al. [13] reported as many as 13.4 % BRAF mutations in GISTs localized in small intestine, stomach, and peritoneum. BRAF-positive GISTs in our cohort had spindle cell morphology as previously reported, but occurred in various sites including stomach, small intestine, colon, rectum, and extra-intestinal. Low mitotic activity was found in only four cases, which is less than in previous reports. Our data confirm that BRAF-mutated GISTs tend to show spindle cell morphology, as reported by Daniels et al. [17] and others. We found the small intestine and stomach to be the most frequent site for BRAF-mutated GISTs.
Treatment with imatinib mesylate, which mainly targets KIT-driven GISTs, has improved outcome, but is less effective in patients with a KIT WT BRAF-mutated GIST. For BRAF mutation analysis in GISTs, dideoxy sequencing, heteroduplex analysis by VAWE, or allele-specific approaches have been used [13, 17]. The gold standard is dideoxy sequencing, but its sensitivity is limited (estimated at about 10 %) which may provide false-negative results, even though usually sufficient to detect a KIT or PDGFRA mutation. Allele-specific approaches have significantly higher sensitivity than dideoxy sequencing [22]. We therefore applied two methods, AS-PCR (which showed a sensitivity of 0.63 %) and dideoxy sequencing, for the detection of BRAF mutations. Of the nine cases positive for V600E by AS-PCR, only four were detected by dideoxy sequencing. This suggest that GISTs are molecularly heterogeneous, consisting of subclones driven by successive or different mutational events. This is supported by our finding of a BRAF mutation in a liver metastasis, but a KIT mutation in a spleen metastasis from the same patient. Unfortunately, it was impossible to test the primary tumor of this patient. Agaimy et al. [11] reported two patients, one of which had two tumors in the gastric corpus, one with a BRAF mutation and the other a WT GIST. GISTs are usually solitary tumors, multifocal disease occurring mostly in familial or pediatric GISTs even though some reports suggested the existence of sporadic multifocal primary GISTs [23–25]. Gasperatto et al. [25] reported 26 patients presenting metastatic disease at the time of diagnosis, 5 with up to three distinct GIST nodules. In one case, one primary tumor harbored a KIT mutation while the second was KIT/PDGFRA WT, similar to our patient with different mutations in two metastases. In a case of melanoma, a concomitant KIT K624E and BRAF mutation has been reported. Functional studies in this case suggested that K624E represents a weakly active KIT form, requiring an additional directly interacting alteration in order to provide a significant oncogenic signal [26]. This hypothesis is supported by the finding of concomitant KIT/PDGFRA and BRAF/KRAS mutations in patients with GIST. A patient with D579del in KIT with spleen metastasis showed concomitant KRAS G12A/G13D mutations while BRAF WT [15], which confirms that mutations in these two genes are mutually exclusive, as reported for colorectal cancer [27]. In KIT-only mutated cell lines responding to imatinib, introduction of an activating KRAS or BRAF mutation showed that imatinib was able to switch off the mutated KIT receptor but not downstream signaling triggered by RAS-BRAF [15]. Our finding of a KIT and a BRAF mutation respectively in two different metastases in one patient suggests that these represent independent events, even though originating from a single primary, which unfortunately was unavailable for molecular analysis. While clonal heterogeneity between primary colorectal carcinoma and its metastases has been reported, a high concordance in mutation status of driver genes such as KRAS or BRAF between primaries and their metastases is found. In colorectal cancer, approximately 10 % of cases were discordant, but this might be higher when lung or brain metastasis would be included [28]. The existence in a GIST case of different metastases with a mutation in different driver genes calls for further study, in order to better understand the mechanisms involved and its impact on treatment.
Some patients with a KIT exon 11-mutated GIST do not respond to imatinib, which calls for further analysis of BRAF/KRAS not only in WT GIST but also in patients with a GIST with an imatinib-sensitive mutation who have not responded to the drug. Next-generation sequencing might identify new clinically relevant genes in WT GISTs [29] and improve sensitivity of mutation testing [30]. Our results contribute additional details on the mechanisms involved in the development of WT GIST and their resistance to treatment.
References
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishigur S, Kawano K, Handa M, Kurata A, Takeda M, Tunio GM, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y (1998) Gain-of function mutations of c-kit in human gastrointestinal stromal tumors. Science 279:577–580. doi:10.1126/science.279.5350.577
Heinrich MC, Corless CL, Duensing A, McGreevey L, Jie Chen C, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CDM, Fletcher JA (2003) PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299:708–710. doi:10.1126/science.1079666
Barnett CM, Corless CL, Heinrich MC (2013) Gastrointestinal stromal tumors: molecular markers and genetic subtypes. Hematol Oncol Clin N Am 27:871–888. doi:10.1016/j.hoc.2013.07.003
Heinrich MC, Owzar K, Corless CL et al (2008) Correlation of kinase genyotype and clinical outcome in the North America intergroup phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol 28:5360–5367. doi:10.1200/JCO.2008.17.4284
Gill AJ, Chou A, Vilain R et al (2010) Immunohistochemeistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol 3:636–644. doi:10.1097/PAS.0b013e3181d6150d
Pantaleo MA, Astolfi A, Urbini M et al (2014) Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur J Hum Genet 22:32–39. doi:10.1038/ejhg.2013.80
Nannini M, Astolfi A, Urbini M, Indio V, Santini D, Heinrich MC, Corless CL, Ceccarelli C, Saponara M, Mandrioli A, Lolli C, Ercolani G, Brandi G, Biasco G, Pantaleo MA (2014) Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST). BMC Cancer 14:685. doi:10.1186/1471-2407-14-685
Davies H, Bignell GR, Cox C, Stephens P et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954. doi:10.1038/nature00766
Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R, Cancer Genome Project (2004) Mechanisms of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116:855–867. doi:10.1016/S0092-8674(04)00215-6
Agaram NP, Wong GC, Guo T, Maki RG, Singer S, Dematteo RP, Besmer P, Antonescu CR (2008) Novel V600E mutations in imatinib naïve and imatinib resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 47:853–859. doi:10.1002/gcc.20589
Agaimy A, Terracciano LM, Dirnhofer S, Tornillo L, Foester A, Hartmann A, Bihl MP (2009) V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-Tye gastrointestinal stromal tumours. J Clin Pathol 62:613–616. doi:10.1136/jcp.2009.064550
Martinho O, Gouveia A, Viana-Pereira M, Silva P, Pimenta A, Reis RM, Lopes JM (2009) Low frequency of MAP kinase pathway alterations in KIT and PDFGRA wild-type GISTs. Histopathology 55:53–62. doi:10.1111/j.1365-2559.2009.03323.x
Hostein I, Faur N, Primois C, Boury F, Denard J, Emile JF, Bringuier PP, Scoazec JY, Coindre JM (2010) BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol 133:141–148. doi:10.1309/AJCPPCKGA2QGBJ1R
Debiec-Rychter M, Sciot R, Cesne A, Schlemmer M, Hohenberger P, Oosterom AT, Blay JY, Leyvraz S, Stul M, Casali PG, Zalcberg J, Verweij J, Glabbeke M, Hagemeijer A, Judson I KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 42:1093–1103. doi:10.1016/j.ejca.2006.01.030
Miranda C, Nucifora M, Molinari F, Conca E, Anania MC, Bordoni A, Saletti P, Mazzucchelli L, Pilotti S, Pierotti MA, Tamborini E, Greco A, Frattini M (2012) KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res 18:1769–1776. doi:10.1158/1078-0432.CCR-11-2230
Falchook GS, Trent JC, Heinrich MC, Beadling C, Patterson J, Bastida CC, Blackman SC, Kurzrock R (2013) BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomix sequencing analysis for acquired resistance. Oncotarget 4:310–315. doi:10.18632/oncotarget.864
Daniels M, Lurkin I, Pauli R, Erbstosser E et al (2011) Spectrum of KIT/PDGFRA/BRAF mutations and phosphatidylinositol −3-kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett 312:43–54. doi:10.1016/j.canlet.2011.07.029
Joensuu H, Vehtari A, Riihimäki J, Nishida T, Steigen SE, Brabec P, Plank L, Nilsson B, Cirilli C, Braconi C, Bordoni A, Magnusson MK, Linke Z, Sufliarsky J, Federico M, Jonasson JG, Tos AP, Rutkowski P (2012) Risk of recurrence of gastrointestinal stromal tumors after surgery: an analysis of pooled population-based cohorts. Lancet Oncol 121:539–549. doi:10.1016/S1470-2045(11)70299-6
Minarik G, Plank L, Lasabova Z, Szemes T, Burjanivova T, Szepe P, Buzalkova V, Porubsky D, Sufliarsky J (2013) Spectrum of mutations in gastrointestinal stromal tumor patients – a population based study from Slovakia. APMIS 121:539–548. doi:10.1111/apm.12019
Liegl-Atzwanger B, Fletcher JA, Fletcher CDM (2010) Gastrointestinal stromal tumors. Virchows Arch 456:111–127. doi:10.1007/s00428-008-0875-y
Fletcher C, Bridge J, Hogendoorn P, Mertens F (eds) (2013) ISBN-10 9283224345 World Health Organization classification of tumours of soft tissue and bone: pathology and genetics of tumours of soft tissue and bone, 4th edn. IARC Press, Lyon, France
Van Krieken JH, Jung A, Kirchner T et al (2008) KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch 453:417–431. doi:10.1007/s00428-008-0665-y
Haller F, Schulten HJ, Armbrust T et al (2007) Multicentric sporadic gastrointestinal stromal tumors (GISTs) of the stomach with distinct clonal origin: differential diagnosis to familial and syndromal GIST variants and peritoneal metastasis. Am J Surg Pathol 31:933–937. doi:10.1097/01.pas.0000213440.78407.27
Kang DY, Park CH, Choi JS et al (2007) Multiple gastrointestinal stromal tumors: clinicopathological and genetic analysis of 12 patients. Am J Surg Pathol 31:224–232. doi:10.1097/01.pas.0000213318.66800.94
Gasparotto D, Rossi S, Bearz I et al (2008) Multiple primary sporadic gastrointestinal stromal tumors in the adult: an underestimate entity. Clin Cancer Res 14:5715–5721. doi:10.1158/1078-0432.CCR-12-3863
Curtin JA, Busam K, Pinkel D, Bastian BC (2006) Somatic activation of KIT in distinct sunbtypes of melanoma. J Clin Oncol 10:4340–4346. doi:10.1200/JCO.2006.06.2984
Lamy A, Blanchard F, LePessot F, Sesboué R, DiFiore F, Bossut J, Fiant E, Frébourg T, Sabourin JC Metastatic colorectal cancer KRAS genotyping in routine practice: results and pitfalls. Mod Pathol 24:1090–1100. doi:10.1038/modpathol.2011.60
Mao C, Wu XY, Yang ZY, Threapleton DE, Yuan JQ, Yu YY, Tang JL (2015) Concordant analysis of KRAS, BRAF, PIK3CA mutations, and PTEN expression between primary colorectal cancer and matched metastases. Sci Rep 5:8065. doi:10.1038/srep08065
Schoppmann SF, Vinatzer U, Popitsch N, Mittlbock M, Leibman-Reindl S, Jomrich G, Streubel B, Bimer P (2013) Novel clinically relevant genes in gastrointestinal stromal tumors indetified by exome sequencing. Clin Canc Res 19:5329–5339. doi:10.1158/1078-0432.CCR-12-3863
Gleeson FC, Kipp BR, Kerr SE, Voss JS, Graham RP, Campion MB, Minot DM, Tu ZJ, Klee EW, Lazaridis KN, Henry MR, Levy MJ (2015) Kinase genotype analysis of gastric gastrointestinal stromal tumor cytology samples using targeted next-generation sequencing. Clin Gastroenterol Hepatol 13:202–206. doi:10.1016/j.cgh.2014.06.024
Acknowledgements
This study was supported by the grant of Novartis Slovakia Ltd., s,r,o, by the project CEPV II (ITMS 26220120036) as well as by the project of Slovak Research and Development Agency no. APVV-14-0273. We thank Mrs. A. Vanochova for her excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest statement
We declare that we have no conflict of interest.
Compliance of ethical standards
The study is in compliance with ethical standards.
Additional information
Lukas Plank and Zora Lasabova senior co-authors
Rights and permissions
About this article

Cite this article
Jasek, K., Buzalkova, V., Minarik, G. et al. Detection of mutations in the BRAF gene in patients with KIT and PDGFRA wild-type gastrointestinal stromal tumors. Virchows Arch 470, 29–36 (2017). https://doi.org/10.1007/s00428-016-2044-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-016-2044-4