
Abstract
Chalcone synthase (CHS) related type III plant polyketide synthases (PKSs) are likely to be involved in the biosynthesis of diarylheptanoids (e.g. curcumin and polycyclic phenylphenalenones), but no such activity has been reported. Root cultures from Wachendorfia thyrsiflora (Haemodoraceae) are a suitable source to search for such enzymes because they synthesize large amounts of phenylphenalenones, but no other products that are known to require CHSs or related enzymes (e.g. flavonoids or stilbenes). A homology-based RT-PCR strategy led to the identification of cDNAs for a type III PKS sharing only approximately 60% identity with typical CHSs. It was named WtPKS1 (W. thyrsiflora polyketide synthase 1). The purified recombinant protein accepted a large variety of aromatic and aliphatic starter CoA esters, including phenylpropionyl- and side-chain unsaturated phenylpropanoid-CoAs. The simplest model for the initial reaction in diarylheptanoid biosynthesis predicts a phenylpropanoid-CoA as starter and a single condensation reaction to a diketide. Benzalacetones, the expected release products, were observed only with unsaturated phenylpropanoid-CoAs, and the best results were obtained with 4-coumaroyl-CoA (80% of the products). With all other substrates, WtPKS1 performed two condensation reactions and released pyrones. We propose that WtPKS1 catalyses the first step in diarylheptanoid biosynthesis and that the observed pyrones are derailment products in the absence of downstream processing proteins.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Type III polyketide synthases (PKS III) play a major role in the biosynthesis of many plant and bacterial secondary metabolites (Schröder 2000; Thomas 2001; Austin and Noel 2003). Chalcone synthases (CHSs), the prototype of the plant PKS III superfamily, produce chalcones and thus are involved in the production of flavonoids, which are multifunctional plant defence and UV protective agents ubiquitously distributed in the plant kingdom. CHSs condense 4-coumaroyl-CoA with three malonyl-CoA units to form tetraketides followed by a Claisen-type cyclization. The very closely related stilbene synthases (STSs) also mainly prefer 4-coumaroyl-CoA as starter and malonyl-CoA as extender substrates but, in contrast to CHSs, catalyse cyclization in a different manner through an aldol condensation including loss of CO2. A variety of related enzymes such as 2-pyrone synthase (2PS) (Eckermann et al. 1998), stilbenecarboxylate synthase (STCS) (Eckermann et al. 2003) and others (Schröder 2000; Austin and Noel 2003), condensing various aromatic and aliphatic starter substrates with one or more malonyl-CoA-derived acetate units and different ring foldings, have been identified. Type III PKS enzymes have in common to be dimeric proteins of approximately 42 kDa with a catalytic triad Cys-His-Asn in the active centre. This facilitates the application of molecular techniques, such as using degenerate oligonucleotide primers or screening cDNA libraries, to identify further candidate genes from plants forming secondary metabolites, proposed to be formed by a CHS-related protein.
Some additional candidates for plant secondary metabolites derived from PKS III-catalysed biosynthesis have been discussed previously (Schröder 1997). Among them, diarylheptanoids are one of the most interesting groups. Linear diarylheptanoids, especially curcumin, increasingly attract attention as food additives due to their anti-oxidative, anti-inflammatory and cancer-protective activities (Chattopadhyay et al. 2004). Polycyclic aromatic diarylheptanoids of the phenylphenalenone type are phytoalexins of Musaceae and are involved in pathogen defence of the Haemodoraceae as well. The available evidence indicates that curcumin and other linear diarylheptanoids are formed from two phenylpropanoids and a one-carbon unit derived from malonate. Labelling studies with phenylphenalenone-producing root cultures of Anigozanthos preissii (Haemodoraceae) (Hölscher 1996, 1995) and Musa plants (Kamo et al. 2000) clearly showed incorporation of two phenylpropanoid units and C-2 of the acetate or malonate. Intramolecular cycloaddition of a diarylheptanoid resulting in phenylphenalenones (Bazan et al. 1978; Hölscher and Schneider 1995) indicated that diarylheptanoids are intermediates of phenylphenalenone biosynthesis and that phenylphenalenones are a subgroup of diarylheptanoids.
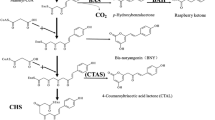
Although some details are still hypothetical (Hölscher 1996), diarylheptanoids and phenylphenalenones are clearly derived from the phenylpropanoid pathway (Fig. 1). In plants, due to the occurrence of diarylheptanoids and phenylphenalenones bearing differently substituted aryl moieties at the terminal positions of the C-7 chain, a branching relatively early in the pathway has to be hypothesized. In one branch, cinnamic acid is modified by hydroxylation followed by the formation of the CoA ester. The next step postulates a chain extension by a malonyl-CoA-derived C-2 unit, leading to a diketidyl-CoA intermediate. The diketide is anticipated to undergo a second condensation with a phenylpropanoid-CoA unit derived from the other biosynthetic branch. The details are not understood, but the reaction(s) to the diarylheptanoid must involve the removal of the terminal carboxyl group of the diketidyl intermediate.

Proposed biosynthetic pathway to diarylheptanoids and phenylphenalenones. Variations in the aryl substitution, phenylpropanoid side chain and by post-processing enhance the structural diversity of the products. Reduction of the double bond at C-2 of the diarylheptanoid is required prior to cyclization to phenylphenalenones
The condensation step between a phenylpropanoid-CoA and malonyl-CoA (see box in Fig. 1) is typical for a type III PKS reaction. Thus, the strategy was to search for CHS-related cDNAs using degenerate primers, followed by heterologous expression, characterization of the recombinant protein and in vitro investigations about whether the protein carried out a reaction that would be compatible with a role in the initial step of the diarylheptanoid synthesis.
Plant-specific CHS-related proteins show highly conserved amino acid sequences with approximately 60–80% identity, and X-ray analysis of CHS (Ferrer et al. 1999) and related proteins (Jez et al. 2000a; Austin et al. 2004a, b) has provided more insight into the steric geometry of the active site and the catalytic mechanism of these enzymes. However, the prediction of the protein function from sequence data is still a challenge (Schröder 2000; Austin and Noel 2003; Austin et al. 2004a). Moreover, even assays with recombinant proteins often provide no clear-cut answers with respect to the physiological substrates because most type III PKSs are rather promiscuous in their substrate acceptance in vitro. It appears that substrate availability and protein environment rather than strict substrate specificities are the major factors in determining the function of these enzymes in vivo. In view of these difficulties it would therefore be a considerable advantage to use a plant or a plant culture that does not synthesize other products that might be synthesized via type III PKSs (e.g. flavonoids, stilbenes, etc.). Plants producing diarylheptanoids usually also contain other compounds synthesized via such enzymes. Both diarylheptanoids and flavonoids were found, for example, in dicotyledonous families such as Betulaceae and in some monocots (Zhu et al. 2000). Musaceae and some Anigozanthos species (Haemodoraceae) produce a cyclic form of diarylheptanoids (phenylphenalenones) and dimeric stilbenes (Hölscher and Schneider 1996). However, Wachendorfia thyrsiflora (Haemodoraceae) and, in particular, its root culture appeared a good choice because they are rich in phenylphenalenones while other CHS-derived compounds have not been identified. In this study we demonstrate the abundance of phenylphenalenones and the absence of other detectable compounds possibly derived from CHS-type enzymes in the root cultures, and we report the cloning and functional characterization of a type III PKS that appears to be a prime candidate for a function in diarylheptanoid/phenylphenalenone biosynthesis.
Materials and methods
Plant material
Root cultures of W. thyrsiflora Burm. were established from seeds (Chiltern Seeds, Ulverston, Cumbria, UK) as described for in vitro cultures of another Haemodoraceae (Hölscher and Schneider 1997). The sterile cultures were grown in liquid M3 (Murashige and Skoog 1962) medium (100 ml) in conical flasks (volume 300 ml) on a gyratory shaker (90 rpm) at 23°C under permanent diffuse light (4.4 μmol m−2 s−1). Root tips (2.2 g) of cultured roots were used for RNA preparation because of their enhanced levels of phenylphenalenones. Root cultures with and without elicitation with jasmonic acid (50 μM final concentration) were used for identification of compounds 1–7 and 11–14.
Isolation of RNA
The orange root tips (2.2 g) of cultivated root cultures were dissected, frozen in liquid nitrogen and ground. The powder was transferred into TRIzol®-Reagent (Invitrogen, Karlsruhe, Germany) (7.5 ml) and mixed. After 10 min at 4°C, CHCl3 (3 ml) was added, shaken and after another 10 min centrifuged (2,500×g, 10 min). The supernatant was extracted with CHCl3 for a second time. The RNA was precipitated with i-PrOH [1.875 ml, 0.937 ml sodium citrate (0.8 M), 0.937 ml NaCl (1.2 M)] at −20°C for 6 h. Centrifugation at 8,500×g for 30 min and washing of the RNA with 75% ethanol followed. The RNA was dissolved in 800 μl DEPC-water with slight warming (55–60°C, 10 min). A LiCl precipitation (200 μl, 10 M) was done overnight at 4°C. The RNA was pelleted by centrifugation (42,000×g, 45 min), washed with ethanol (75%) and dissolved in 150 μl DEPC-water.
cDNA cloning
cDNA was synthesized from RNA (4.7 μg) using the 5′/3′-RACE kit from Roche (Mannheim, Germany). An approximately 600 bp DNA fragment was amplified using the following degenerate primers: K180_oli: 5′-GCI AA (AG) GA (CT) ITI GCI GA(AG) AA(CT) AA(CT) AAI GG-3′ and FGFG_oli: 5′-CC(AT) GG(AT) CCG AA(GT) CCG AA(GT) AG(GT) AC(AT) CCC-3′. This fragment was cloned into the vector pCR® II-TOPO® using the TOPO TA Cloning® kit (Invitrogen) and sequenced. To amplify the full-length cDNA sequence 3′- and 5′-RACEs were performed according to the recommendations of the manufacturer (version 1, July 1999) of the 5′/3′-RACE kit. Primers used for 3′-RACE: dhs06_wdf3race: 5′-GCC TTT GAG CCG CTG AAC ATC-3′ and oligodT-anchor: 5′-GAC CAC GCG TAT CGA TGT CGA C(T)16(AGC)-3′. Primers used for 5′-RACE: dhs07_wdf5_2: 5′-CCG CCT GGG TGG ACG ATC C-3′ for cDNA synthesis and dhs08_wdf5_3: 5′-CAC TCA TCA TGT TGC CGT TCT CC-3′ and the provided oligodT-anchor primer for PCR. The full coding region (without the start-methionine) was cloned into the vector pCR® II-TOPO® with primers extended by a BamHI and a HindIII restriction site, respectively, as follows: dhs20_wdf_chs1_Bam: 5′-AAG GAT CCG CGA GCA CAG AAG GCA TCC AGG-3′ and dhs21_wdf_chs1_Hind: 5′-TAA AGC TTC TAA ATC GAG AGC GGC ACA CTG-3′.
Protein purification
The restricted fragment was subcloned into the expression vector pHis8 (Jez et al. 2000b) (kindly provided by J.P. Noel, La Jolla, USA). For expression of the WtPKS1 protein, Escherichia coli BL21-CodonPlus® (DE3)-RIL (Stratagene, La Jolla, USA) were used (750 ml culture volume). After growing to OD600 0.6–0.8 at 37°C the cells were induced by 0.5 mM IPTG for 3 h at 22°C. Cells (OD600 2.9) were harvested (4,000×g, 10 min), washed (25 mM Pi buffer, 150 mM NaCl, pH 7.4) and frozen at −80°C.
After resuspension (20 mM Pi buffer, 0.5 M NaCl, 20 mM imidazole, 0.02 mg ml−1 RNase A, 0.04 mg ml−1 DNase, 1 mM PMSF, 3 mM β-mercaptoethanol) the cells were disrupted in a French press (SLM Instruments, Urbana, USA) at 1,018 psi. After centrifugation (10,000×g, 20 min), the crude extract was loaded on a Ni-NTA column (HiTrap Chelating HP, Amersham, Buckinghamshire, UK) with Ni2+ as affinity ligand. The column was washed with 80 and 150 mM imidazole buffer (20 mM Pi buffer, 0.5 M NaCl, 3 mM β-mercaptoethanol and the indicated concentration of imidazole, pH 7.4) and the protein was eluted with imidazole buffer (500 mM). Imidazole was removed by gel filtration (PD10, Amersham). The final buffer (0.1 M Hepes, pH 7) contained 0.1 M NaCl, 10% glycerol and 2 mM DTT. Protein content was measured according to the method of Bradford (1976) (Bio-Rad Protein Assay) with bovine serum albumin as a standard. The purity of the protein was verified by SDS-PAGE electrophoresis. Typically, from this procedure 4.5 mg of purified protein (3 mg ml−1) was obtained. Immunoblots were carried out as described (Lanz et al. 1991).
nano-LC-ESI-MS/MS and MALDI-TOF-MS analysis of WtPKS1
The gel piece from SDS-PAGE electrophoresis containing the recombinant WtPKS1 was washed thrice with H2O, twice with 50 mM NH4HCO3 and finally with 50 mM NH4HCO3 in 50% MeCN. Then it was dried under a gentle stream of nitrogen and reswollen in 20 μl 50 mM NH4HCO3 (pH 8.0). The protein was digested overnight at 37°C by in-gel proteolysis using trypsin (Promega, Madison, WI, USA) or GluC bicarbonate (Princeton Separations, Adelphia, NJ, USA) in parallel experiments. After digestion, the peptides were extracted from the gel pieces and injected into a CapLC (Micromass, Manchester, UK) equipped with an autosampler, gradient and auxiliary pump. The sample (6 μl) was injected via “microlitre pickup” mode and desalted on-line through a 300 μm×5 mm C18 trapping cartridge (Dionex GmbH, Idstein, Germany) at a flow rate of 20 μl min−1 for 5 min. The peptides were separated on a 75 μm×15 cm, 3 μm, C18 100Å PepMapTM column (Dionex GmbH) prior to introduction into the mass spectrometer. The following gradient was used: solvent A for 5 min, followed by 5–60% B over 20 min and 60–95% B over 15 min at a flow rate of 300 nl min−1 (A: 5% MeCN/95% 0.1% HCOOH, B: 95% MeCN/5% 0.1% HCOOH). MS/MS experiments were performed on a Q-TOF 2 mass spectrometer (Micromass), equipped with a modified nano-ESI source to hold a pico-tip (New Objective, Cambridge, MA, USA). The collision energy was determined on the fly based on the mass and charge state of the peptide. Charge state recognition was used to switch into MS/MS mode on only doubly and triply charged ions. Per survey scan up to three components were monitored. Spectra were acquired in MS mode at 1 s scan−1 and in MS/MS mode at 3 s scan−1. A searchable peak list was generated and fed into the search engine MASCOT (Matrix Science, London, UK) (http://www.matrixscience.com) and searched against the MSDB protein sequence database. For MALDI-TOF MS, a saturated solution of α-cyano-4-hydroxycinnamic acid in acetone (0.5 μl) was deposited onto the sample target. The supernatant (1 μl) of the in-gel digested protein was injected into a small drop of trifluoroacetic acid (2%) previously deposited onto the matrix surface in order to prevent dissolution of the matrix layer by the basic pH of the digest solution. The sample was allowed to dry and the dried spot was rinsed with trifluoroacetic acid (3×) (10 μl, 0.1%). MALDI mass spectra were obtained on a Bruker REFLEX II mass spectrometer (Bruker-Daltonik, Bremen, Germany), upgraded with a gridless delayed extraction ion source (pulsed ion extraction). Mass spectra were calibrated using a peptide mix as external standard. The monoisotopic masses ([M + H]+) and sequences of tryptic peptides obtained from MALDI-TOF-MS and LC-MS/MS are as follows: m/z 2,277.07, TDITHMVVCTGAGVDVPGVDYK; m/z 1,873.84, ENPTLTTYVDASYDER; m/z 1,455.78, MMNLLGLPPTVNR; m/z 1,431.68, VTNSEHLSPEYR; m/z 1,193.70, HLVLTEQLLK; m/z 1,069.62, QSIVLDAVPK; m/z 1,043.55, AILDQVQEK. GluC digestion resulted in peptides of the following monoisotopic masses ([M + H]+): m/z 2,824.32, MKHHHHHHHHGGLVPRGSHGSASTE; m/z 2,200.06, QLLKENPTLTTYVDASYDE; m/z 1,836.87, KPIFQIFSASQMTLPE; m/z 1,478.77, WGVLFGFGPGLSIE; m/z 1,406.76, NKLDVSRYVLAE; m/z 1,266.59, GIQAYRNNMAE; m/z 1,239.62, VSVMFFRGPAE; m/z 1,217.64, GEHLVAGHLRE; m/z 1,031.57, HLVAGHLRE; m/z 801.48, AAAKAIKE; m/z 688.39, KIGLEE.
Enzyme assay
Starter CoA esters were prepared according to the method described by Wang et al. (2000), using RP-18 cartridges (LiChrolut®, VWR Darmstadt, Germany) in a modified purification (Beuerle and Pichersky 2002). N-Acetylcysteamine diketide thioesters were synthesized by the Meldrum’s acid method (Gilbert et al. 1995). The starter substrates (50 μM) and the recombinant WtPKS1 protein (20 μg) were added to the buffer (0.1 M Hepes, pH 6.85; total volume of the assay 100 μl) and mixed. The assay mixture was adjusted to 37°C in a thermostat. The reaction was started by adding [2-14C]malonyl-CoA (80 μM, 0.17 kBq nmol−1; Hartmann Analytics, Braunschweig, Germany). The reaction was stopped after 30 min by extraction with 2×200 μl EtOAc. The combined EtOAc extracts were evaporated in a vacuum concentrator, redissolved in MeOH (20 μl), and a part of the solution (15 μl) analysed by reversed-phase HPLC on a HP 1100 system (Agilent, Palo Alto, USA; column Supelcosil LC18, 250×2.1 mm i.d., Sigma-Aldrich, Taufkirchen, Germany), solvent A: trifluoroacetic acid (0.1%), solvent B: MeCN, gradient: 10–50% solvent B over 40 min at a flow rate of 0.25 ml min−1. Enzyme products were detected in UV light at 280 nm (Agilent VWD or PDA detector) and radioactivity monitoring. A flow scintillation analyser (Canberra-Packard, Dreieich, Germany) was coupled to the HPLC, and scintillation liquid Ultima-Flo AP (Perkin Elmer, Wellesley, USA) (1 ml min−1) was added post-column. Enzyme activities were calculated based on the radioactivity of [2-14C]malonyl-CoA-derived products. The radioactivity was measured using a HPLC-coupled flow scintillation analyser, which was calibrated to a standard amount of [2-14C]malonyl-CoA. Radio-HPLC peak areas were recalculated to the amount of malonyl-CoA used in the assays. From these values, based on the stoichiometry of the enzyme reactions, the amount of starter-CoA ester incorporated into the products was calculated. The conditions of the assay varied with the parameter to be determined as indicated in the text. For MS and nuclear magnetic resonance (NMR) analysis, the assays were scaled up 15-fold, with the appropriate concentration of unlabelled malonyl-CoA. UV detection at 280 nm of HPLC runs was performed as described above. Fractions were pooled, evaporated and subjected to analysis. For measuring the kinetic parameters the substrate to be determined was varied between 2 and 40 μM; the other substrate concentration was kept constant at 100 μM. The reaction temperature was 37°C, the reaction buffer 0.1 M Hepes, pH 6.85. The incubation time varied between 30 and 40 min depending on the enzyme activity with the analysed substrate. In preliminary experiments it was shown that the enzyme activity was linear with time for more than 40 min.
Nuclear magnetic resonance spectroscopy
Nuclear magnetic resonance spectra for analysing enzyme products were measured on a Bruker AV 500 NMR spectrometer (Bruker, Rheinstetten, Germany) equipped with a CryoPlatformTM. 1H NMR, 1H–1H COSY and 1H–1H lrCOSY experiments were recorded using a 5 mm TXI CryoProbeTM. A Bruker DRX 500 NMR spectrometer was used for recording 1H and 13C NMR, 1H–1H COSY, HMBC and HMQC spectra of phenylphenalenones for structure elucidation, synthetic NAC diketide thioesters and analysis of biosynthetic incorporation of [1-13C]phenylalanine. Operating frequency was 500.13 MHz for acquisition of 1H and 125.75 MHz for 13C spectra. Acetone-d 6 was used as a solvent, if not otherwise mentioned in the text. Chemical shifts are given in δ values and referenced to trimethylsilane, which was used as an internal standard.
Electrospray mass spectrometry of enzyme products
Electrospray (ESI) mass spectra were recorded using a Micromass Quattro II (Waters, Micromass) tandem quadrupole mass spectrometer (geometry quadrupole–hexapole–quadrupole) equipped with an ESI source. The capillary and cone voltages in positive ESI mode were −3.0 kV and 25 V, in negative mode 4.0 kV and −15 V, respectively. Nitrogen for nebulization was applied at 15 l−1 h−1 and drying gas at 250 l−1 h−1. Source and electrospray capillary were heated at 60 and 150°C, respectively. The mass spectrometer was operated in conventional scanning mode using the first quadrupole. Full-scan mass spectra were recorded from m/z 90 to 450 (scanning time 1.5 s). Samples were injected (1.5 μl) as solutions in aqueous MeOH using an autosampler. Separations were achieved using a micro-bore Nucleodur 100-3 C18 column (100×1 mm, 3 μm, Macherey-Nagel, Düren, Germany). The column outlet was directly connected with a fused silica capillary (0.75 m×0.075 mm i.d.) to an ESI source with the Z-geometry (Micromass). Depending on the compound polarity several isocratic elution systems mixed from H2O and MeOH were used at 0.05 ml min−1 flow for 20 min. Temperatures of the column and the fused silica capillary were not regulated.
Analytical data of enzyme products
(E)-4-Phenyl-but-3-en-2-one (benzalacetone, 15): R t=33.1 min; ESI-MS: m/z 147 [M + H]+; R t and MS data were identical with those of the authentic reference. (E)-4-(4-Hydroxyphenyl)-but-3-en-2-one (4-hydroxy-benzalacetone, 16): R t=24.0 min; ESI-MS: m/z 163 [M + H]+ (rel. int. 3), 145 (4), 127 (51), 117 (9), 115 (24), 91 (13), 77 (3), 43 (100); 1H NMR (acetone-d 6): δ 7.57 (2H, d, J=8.7 Hz, H-2′/6′), 7.75 (1H, d, J=16.2 Hz, H-4), 6.91 (2H, d, J=8.7 Hz, H-3′/5′), 6.62 (1H, d, J=16.2 Hz, H-3), 2.29 (3H, s H3C-1); R t, MS and NMR data were identical with those of the authentic reference. (E)-6-Styryl-4-hydroxy-2H-pyran-2-one (17): R t=35.1 min; ESI-MS: m/z 215 [M + H]+ (rel. int. 14), 197 (10), 187 (27), 169 (49), 155 (20), 141 (96), 131 (100), 129 (16), 103 (10), 69 (25). (E)-6-(4-Hydroxystyryl)-4-hydroxy-2H-pyran-2-one (bisnoryangonin, 18): R t=26.0 min; ESI-MS: m/z 231 [M + 1]+ (rel. int. 3), 185 (6), 171 (23), 157 (24), 147 (100), 119 (25), 69 (23); 1H NMR (acetone-d 6): δ 7.54 (2H, d, J=8.6 Hz, H-2″/6″), 7.33 (1H, d, J=16.1 Hz, H-2), 6.89 (2H, d, J=8.6 Hz, H-3″/5″), 6.45 (1H, d, J=16.1 Hz, H-1), 6.14 (1H, brs, H-5′), 5.37 (1H, d, J=1.9 Hz, H-3′); R t, MS and NMR data were identical with those of the authentic reference. (E)-6-(1′-Hydroxy)-styryl-4-hydroxy-2H-pyran-2-one (19): R t = 24.4 min; ESI-MS: m/z 231 [M+H]+ (rel. int. 100), 213 (5.5), 190, 171, 156 (7), 139 (4), 129 (3), 111 (3), 101, 43 (3); 1H NMR (acetone-d 6): δ 7.29–7.27 (5H, m, phenyl-H), 6.57 (1H, s, H-2), 5.97 (1H, brs, H-5′), 5.35 (1H, brs, H-3′). 6-Phenethyl-4-hydroxy-2H-pyran-2-one (20): R t=28.0 min; ESI-MS: m/z 217 [M + H]+ (rel. int. 4), 175 (1), 126 (9), 105 (36), 91 (100); 1H NMR (acetone-d 6): δ 7.29 (4H, m, H-2″/6″ and H-3″/5″), 7.02 (1H, m, H-4″), 5.94 (1H, d, J=2.1 Hz, H-5′), 5.31 (1H, d, J=2.1 Hz, H-3′), 2.95 (H-2, from lrCOSY), 2.77 (H-1, from lrCOSY). 6-(4-Hydroxyphenethyl)-4-hydroxy-2H-pyran-2-one (dihydrobisnoryangonin, 21): R t=22.0 min; ESI-MS: m/z 233 [M + H]+ (rel. int. 100), 179 (8), 176 (12), 149 (7), 135 (14), 126 (16), 112 (11), 99 (6), 87 (14), 85 (14); 1H NMR (acetone-d 6): δ 7.06 (2H, d, J=8.6 Hz, H-2″/6″), 6.75 (2H, d, J=8.6 Hz, H-3″/5″), 5.91 (1H, brs, H-5′), 5.30 (1H, brs, H-3′), 2.84 (H-2, from lrCOSY), 2.70 (H-1, from lrCOSY). 6-(4-Hydroxy-3-methoxyphenethyl)-4-hydroxy-2H-pyran-2-one (22): R t=19.5 min; ESI-MS: m/z 263 [M + H]+ (rel. int. 4), 245 (0.5), 213 (1.4), 203 (1.1), 137 (100), 111 (1); 1H NMR (acetone-d 6): δ 6.88 (1H, d, J=1.9 Hz, H-2″), 6.74 (1H, d, J=8.0 Hz, H-5″), 6.69 (1H, dd, J=8.0, 1.9 Hz, H-6″), 5.97 (1H, brs, H-5′), 5.34 (1H, d, J=2.0 Hz, H-3′), 2.88 (H-2, from lrCOSY), 2.72 (H-1, from lrCOSY). 6-Benzyl-4-hydroxy-2H-pyran-2-one (23): R t=24.5 min; ESI-MS: m/z 203 [M + H]+ (rel. int. 100), 157 (16), 129 (31), 111 (17), 105 (14), 97 (14), 91 (38), 69 (8); 1H NMR (acetone-d 6): δ 7.34 (4H, m, H-2″/6″ and H-3″/5″), 7.23 (1H, m, H-4″), 5.98 (1H, brs, H-5′), 5.36 (1H, d, J=2.0 Hz, H-3′), 3.77 (2H, s, H-1). 4-Hydroxy-6-n-pentyl-2H-pyran-2-one (24): R t=29.0 min; ESI-MS: m/z 181 [M − H]- (rel. int. 100); 1H NMR (MeOH-d 4): δ 5.93 (1H, brs, H-5′), 5.27 (1H, brs, H-3′), 2.47 (2H, t, J=7.6 Hz, H-1), 1.65 (H-2, from COSY), 1.34 (H-2 and H-4, from COSY), 0.92 (2H, t, J=6.6 Hz, H-5).
Administration of [1-13C]phenylalanine
For biosynthetic feeding experiments, the in vitro cultured roots of W. thyrsiflora (approximately 10 g fresh weight) were transferred to fresh medium 2 days prior to administration of the labelled compound. [1-13C]Phenylalanine (5 mg, 99% 13C, Deutero GmbH, Castellaun, Germany) was dissolved in H2O (1 ml) and added to the root culture through a membrane filter to a final concentration of 290 μM and incubated for 5 days. Jasmonic acid (50 μM final concentration) was added to the medium simultaneously.
Isolation and identification of phenylphenalenones and related compounds
Cultured roots of W. thyrsiflora were separated from the medium, frozen in liquid N2, ground and extracted with MeOH at room temperature. After evaporation (<40°C) the extracts were partitioned between CH2Cl2–H2O and EtOAc–H2O. Compounds 2 to 8 and 14 were isolated according to recently described procedures and identified by NMR and MS. The data matched those of samples isolated before from the same or other plants (Opitz 2002; Opitz et al. 2002, Opitz and Schneider 2003). Haemodorin (1) (Bick and Blackman 1973) was isolated from root cultures as previously described and purified by HPLC. Isolation and structure elucidation of further phenylphenalenones from W. thyrsiflora including new natural products 12 and 13 will be reported elsewhere.
Results
Phytochemistry and precursor feeding experiments in Wachendorfia thyrsiflora
The first phytochemical study on W. thyrsiflora, a South African member of the Haemodoraceae, identified two phenylphenalenones, lachnanthoside aglycone (8) and haemocorin aglycone (9), from the roots of this plant (Edwards 1974) (Fig. 2). A compound with the putative structure 10, named thyrsiflorin, was found later in flowers of the same species (Dora 1991). In the course of studies on phenylphenalenones of the Haemodoraceae and other phenylphenalenone-producing species, we reinvestigated this plant, focusing on the intensely orange pigmented roots and in vitro root cultures. Several phenylphenalenones (for example, compounds 1 and 2 in Fig. 2), related oxabenzochrysenones (3–5) (Opitz 2002; Opitz et al. 2002) and their oxidative metabolites (phenylbenzoisochromenones, 11–14) (Opitz and Schneider 2003) were isolated from roots of intact plants and cultured roots and identified by NMR spectroscopic and mass spectrometric methods. Various fractions of very different polarities were thoroughly investigated during the isolation procedure, but apart from large amounts of phenylphenalenones only some phenylpropanoid carbohydrate conjugates could be identified (not shown). It is important to note that no flavonoids, stilbenes or other putative known products of CHS or related enzymes were detected. Treatment of root cultures with potent elicitors of plant secondary metabolite biosynthesis, jasmonic acid or coronalone, resulted in enhanced levels of phenylphenalenones, especially anigorufone (6) and methoxyanigorufone (7) (Schüler et al. 2004). Again, flavonoids or any other products of CHS-related enzymes were not observed. The high production of phenylphenalenones and the failure to find other products synthesized via CHS or related enzymes are strong arguments for the suitability of the Wachendorfia root culture system to search for the proposed new type of PKS.

Phenylphenalenones (1: haemodorin, 2: 6″-O-allophanyl-O-β-d-glucopyranosyl-2,5-dihydroxy-7-phenylphenalenone, 6: anigorufone, 7: methoxyanigorufone, 8: lachnanthoside aglycone, 9: haemocorin aglycone, 10: thyrsiflorin), oxabenzochrysenones (3–5) and phenylbenzoisochromenones (11–14) from Wachendorfia thyrsiflora roots. The diarylheptane skeleton (marked in bold) emphasizes that phenylphenalenones are a subgroup of diarylheptanoids. Oxabenzochrysenones (e.g. 3–5) are putative photochemical conversion products (Dora 1991). Oxalactone structures such as 11–14 are the result of oxidative post-processing of phenylphenalenones (Opitz and Schneider 2003)
The diarylheptane skeleton (aryl–C7–aryl) of phenylphenalenones and related structures depicted in Fig. 2 is marked in bold, emphasizing that these compounds are a subgroup of diarylheptanoids. The biosynthetic origin from the phenylpropanoid pathway has been demonstrated by a precursor feeding experiment. Compound 2 was isolated from a root culture supplied with [2-13C]phenylalanine and subjected to 13C NMR analysis. The 13C NMR spectrum (Fig. 3) exhibits two enhanced signals of C-2 (δ 151.2) and C-8 (δ 132.4), indicating incorporation of two intact phenylpropanoid units. This experiment confirmed results of previous precursor feeding experiments, which detected incorporation of phenylalanine and phenylpropanoids into phenylphenalenones in other Haemodoraceae (Hölscher 1996) and Musaceae species (Kamo et al. 2000) and into oxidatively modified phenylphenalenones in W. thyrsiflora (Opitz and Schneider 2003).

Partial 13C NMR spectra (125 MHz, MeOH-d 4) of the phenylphenalenone glycoside 2 from Wachendorfia thyrsiflora root cultures. a Spectrum of 2 isolated after biosynthetic incorporation of [2-13C]phenylalanine. b Spectrum of the unlabelled reference. R = 6″-O-allophanyl-O-β-d-glucopyranosyl; bold: phenylpropanoid units; filled circle, filled square = 13C-labelled carbon atoms incorporated from two phenylalanine-derived [2-13C]phenylpropanoid units. An illustration of the biosynthetic pathway is shown in Fig. 1
WtPKS1—a CHS-related protein from Wachendorfia thyrsiflora
According to the hypothesis that the condensation of a phenylpropanoid-CoA ester with malonyl-CoA should be the initial step of the diarylheptanoid biosynthesis, a homology-based approach with degenerate primers was employed to identify a CHS-related protein. RNA was prepared from tips of in vitro cultured roots of W. thyrsiflora. The mRNA was transcribed into cDNA and a partial sequence was amplified by PCR using degenerate primers (see Materials and methods), which were based on conserved regions of type III PKS proteins. A fragment of approximately 600 bp was obtained, cloned into the pCR® II-TOPO® vector and sequenced. Only few individual nucleotide differences were observed among 20 independent clones. Using 5′- and 3′-RACEs, a full-length cDNA sequence of 1,436 bp, containing a 132 bp 5′-non-coding region, a 1,185 bp open reading frame with start- and stop-codons encoding a protein with 394 amino acids (M r 43,081) and a 119 bp 3′-non-coding region with the polyA tail, was obtained.
The nucleotide sequence from W. thyrsiflora has been deposited in the EMBL/DDBJ/GenBankTM database under the accession number AY727928. Figure 4 shows that the deduced protein exhibited all common features of the type III PKS protein family (numbering of Medicago sativa CHS): Cys164, His303, Asn336 of the catalytic triad; most of the active site residues including Gly211, Gly216, Gly256, Pro375 and the “gatekeeper” phenylalanines Phe215 and Phe265) (Ferrer et al. 1999). These data clearly indicated that the protein belonged to the type III plant-specific PKSs, and therefore it was named WtPKS1.

Alignment of different polyketide synthase type III sequences. Gh2PS from Gerbera hybrida, GenBank accession no. CAA86219 (Helariutta et al. 1995); MsCHS from Medicago sativa, GenBank accession no. P30073 (Junghans et al. 1993); WtPKS1 from Wachendorfia thyrsiflora, GenBank accession no. AY727928. Sequenced fragments of the digested protein are marked by full lines (tryptic digestion, MALDI-TOF-MS and nano-LC-ESI-MS/MS analysis) and dotted lines (GluC digestion, MALDI-TOF-MS analysis) above the sequence of WtPKS1. The amino acids with a putative role in the substrate specificity (see Discussion and Fig. 9) are marked with an asterisk. Conserved amino acids of the catalytic triad (Cys167, His306 and Asn339, numbering in WtPKS1) are highlighted in grey
The deduced protein sequence shared about 60% identity with typical CHSs (e.g. the crystallized enzyme from M. sativa). The proteins with clearly established CHS functions are typically >85% identical, and thus the low values observed with WtPKS1 suggested already that it was not a CHS. The overall similarity with other proteins of the superfamily was investigated with a relationship tree established with 56 other sequences selected by the following criteria: (a) non-CHS, (b) a representative selection of CHS and (c) preference for plants that contain both CHS and non-CHS functions. The outgroup (root) for the tree was a PKS from the liverwort Marchantia polymorpha, the simplest plant from which type III PKS sequences had been established. Figure 5 shows that CHSs and non-CHSs formed two clearly separated main groups, except for the Leguminosae and Gymnosperms; in these cases the STSs were closer to the CHSs of the same plant or family than to other non-CHSs. WtPKS1 was clearly positioned in the group of non-CHSs, on a subbranch of its own, and without very closely related proteins. The main branch containing WtPKS1 contained a second subbranch with the CHSA and CHSB from the common morning glory (Ipomoea purpurea). Their physiological role is unknown, but it seems noteworthy that the recombinant proteins also carried out only two condensation reactions with 4-coumaroyl-CoA (H. Noguchi, personal communication), just like the maximum number of condensations performed by WtPKS1. It remains to be investigated whether WtPKS1 and those proteins share some specific structural properties.

Relationship tree of 57 selected type III plant PKSs. The selection focused on non-CHS functions and examples in which plants contain both CHS and non-CHS enzymes. Bold print: functions other than CHS. The tree was developed with the program TREECON for Windows (Van de Peer and De Wachter 1994), using the inbuilt matrix for amino acid sequences, and the neighbour-joining method for distance calculations (Saitou and Nei 1987). The outgroup (root) was the stilbenecarboxylate synthase 1 from Marchantia polymorpha (GenBank AAW30009). The length of the branches is a measure of the number of substitutions per site. The numbers at the forks are bootstrap values indicating the percent values for obtaining this particular branching in 1,000 repetitions of the analysis; only the values above 50% are shown
Escherichia coli BL21(DE3)RIL was selected as an expression system for WtPKS1 because it ensured that the correct amino acids, specifically Arg, Leu and Ile, were incorporated into the protein. The primers dhs20_wdf_chs1_Bam and dhs21_wdf_chs1_Hind (see Materials and methods) were used for cloning the protein encoding sequence into the pHis8 expression vector without the start codon to facilitate the expression of the protein. A western blot using antibodies against CHS/STS from Pinus sylvestris confirmed the structural similarity of WtPKS1 with plant-specific PKSs. The recombinant WtPKS1 was also checked by MALDI-TOF-MS and nano-LC-ESI-MS/MS analyses of trypsin and GluC-digested protein fragments. GluC digestion followed by MALDI-TOF-MS analysis was used to confirm the C-terminal region that is important for the activity of the protein. Eighteen peptides derived from both digestions were identical with the predictions from the cDNA sequences. They covered 184 out of 415 amino acids (44%) of the total His-tagged protein. The identified fragments are marked in the protein sequence (Fig. 4).
Products of recombinant WtPKS1
The catalytic activity of recombinant WtPKS1 was determined by incubation with [2-14C]malonyl-CoA and a series of aliphatic and aromatic CoA esters as starter substrates. The assay samples were analysed by HPLC using radioactivity and UV detectors. Assay conditions (0.1 M Hepes buffer, pH 6.85, 37°C) were optimized in experiments with phenylpropionyl-CoA as a starter substrate and then used with all other substrates. Typical radio-HPLC traces resulting from incubation with WtPKS1 are shown in Fig. 6.

HPLC analysis (flow scintillation detection) of the EtOAc extract from incubating WtPKS1 with a 4-coumaroyl-CoA and b 4-hydroxyphenylpropionyl-CoA (starter substrates) and [2-14C]malonyl-CoA (extender). The numbers refer to the substances; see Fig. 8
Depending on the starter substrate, one or two radioactive products were detected. Comparison with retention times of several synthetic diarylheptanoids (Baranovsky et al. 2003), commercially available standards and, if feasible, inspection of UV spectra suggested 2-ketones and pyrone-type products but no diarylheptanoids among the products. Unambiguous identification was achieved by LC-ESI-MS and NMR analyses of the products from 15-fold upscaled assays using non-labelled malonyl-CoA and the respective starter substrates. Examples of identification by 1H NMR spectroscopy are 4-hydroxybenzalacetone (16) and bisnoryangonin (18) as products of 4-coumaroyl-CoA (Fig. 7). A selection of structures identified as products of WtPKS1 is shown in Fig. 8.

1H NMR spectra (500 MHz, acetone-d6) of enzyme products obtained from incubation of WtPKS1 with 4-coumaroyl-CoA (starter) and malonyl-CoA (extender substrate). a 4-Hydroxybenzalacetone (16) (Rt=24.0 min), c bisnoryangonin (18) (Rt=26.0 min), b, d authentic references

Examples of products formed from various starter CoA esters and [2-14C]malonyl-CoA upon incubation with WtPKS1. Products of one condensation: (E)-4-phenylbut-3-en-2-one (benzalacetone, 15), (E)-4-(4-hydroxyphenyl)-but-3-en-2-one (4-hydroxy-benzalacetone, 16). Products of two condensations: (E)-6-styryl-4-hydroxy-2H-pyran-2-one (17), 6-(4-hydroxystyryl)-4-hydroxy-2H-pyran-2-one (bisnoryangonin, 18), (E)-6-(1-hydroxystyryl)-4-hydroxy-2H-pyran-2-one (19), 6-phenethyl-4-hydroxy-2H-pyran-2-one (20), 6-(4-hydroxyphenethyl)-4-hydroxy-2H-pyran-2-one (dihydrobisnoryangonin, 21), 6-(4-hydroxy-3-methoxyphenethyl)-4-hydroxy-2H-pyran-2-one (22), 6-benzyl-4-hydroxy-2H-pyran-2-one (23), 4-hydroxy-6-n-pentyl-2H-pyran-2-one (24). Carbon skeletons of starters are marked in bold. Filled circle, filled square = C-1 and C-2 derived from malonyl-CoA (extender)
The radioactive substances with shorter retention times, e.g. R t 24.0 in Fig. 6, were identified as 2-ketones and those eluting later from the RP18 column (e.g. R t 26.0 in Fig. 6) were pyrones. Hence, the protein WtPKS1 is catalysing condensation of the starter substrate with one or two malonyl-CoA units. While two condensations were observed with all accepted substrates (Fig. 8), products of one condensation were found only with cinnamoyl- and 4-coumaroyl-CoA. In typical experiments, approximately 20% benzalacetone (15) and 80% of the pyrone 17 were formed from cinnamoyl-CoA. The opposite ratio of products of one and two condensations was found using 4-coumaroyl-CoA as a starter. 4-Hydroxybenzalacetone (16) was the major product (80%) in these assays and bisnoryangonin (18), the product of two condensations, was the minor one (20%, Fig. 6). In addition to the structures shown in Fig. 8, which were fully characterized by NMR and mass spectral data, pyrone-type products were also found after incubation of WtPKS1 with dihydrocaffeoyl-CoA and the various aliphatic substrates n-octanoyl-CoA, n-heptanoyl-CoA, valeryl-CoA, isovaleryl-CoA, n-butyryl-CoA and isobutyryl-CoA by means of their characteristic retention times (not shown). No product formation was observed from benzoyl-CoA, 2- and 3-hydroxy-benzoyl-CoA, feruloyl-CoA and 2-coumaroyl-CoA. WtPKS1 revealed very high activities with some of the aliphatic CoA esters, but that was not unexpected in view of the broad substrate acceptance of most type III PKSs (Schröder 2000; Austin and Noel 2003). Such in vitro data do not permit conclusions on the physiological substrates if not supported by the presence of the products (or their derivatives) in the plants. Most efficient product formation with aromatic starters was found with phenylpropionyl-CoA (see Discussion below). The remarkable high product formation for phenylpyruvoyl-CoA may be the clue for the so far unresolved problem of the origin of the 4-hydroxyl group of tetraoxygenated phenylphenalenones 1–4, 8 and 9 (Fig. 2). Incorporation of an intact phenylpyruvoyl unit or one of the corresponding stereoisomeric phenyllactic acids would be a plausible alternative for the hydroxylation of diarylheptanoids or phenylphenalenones late in biosynthesis.
In order to investigate whether WtPKS1 accepts diketide substrates, which may be considered as intermediates of the formation of the pyrones shown in Fig. 8, N-acetylcysteamine (NAC) thioesters of diketide analogues of cinnamic acid, phenylpropionic acid, 4-coumaric acid and 4-hydroxyphenylpropionic acid were synthesized and assayed under the conditions used for aromatic and aliphatic CoA esters. The products detected by radio-HPLC exactly matched the retention times found for the pyrones 17 (35.3 min), 18 (26.0 min), 20 (33.2 min) and 21 (22.0 min) (Fig. 8), formed from the corresponding phenylpropanoid- and phenylpropionyl-CoA esters, indicating condensation of the NAC diketide thioesters with one malonyl-CoA. Products of condensation of NAC diketide derivatives with two or more malonyl-CoA units were not detected. Diketidyl derivatives are also considered as likely intermediates in diarylheptanoid biosynthesis (Fig. 1), and therefore incubations of NAC-thioesters without malonyl-CoA, but with phenylpropanoid- and phenylpropionyl-CoAs were investigated. No product(s) were detected, but small amounts might have escaped detection because the lack of radiolabelled substances precluded a high sensitivity assay.
Enzyme properties of WtPKS1
The pH optima of WtPKS1 with phenylpropionyl-CoA were determined as pH 6.9 in Hepes buffer (100 mM), pH 7.0 in bis-Tris/HCl (100 mM) and pH 6.5 in phosphate buffer (100 mM). The activity of WtPKS1 for the formation of 6-phenethyl-4-hydroxy-2H-pyran-2-one (20) in phosphate buffer was 15 pkat mg−1 protein, which was approximately 50% of the activity in bis-Tris/HCl (33 pkat mg−1) and Hepes (27 pkat min−1). Hepes buffer was used in the assays and for determining kinetic parameters. The optimum temperature with phenylpropionyl-CoA in Hepes was 37°C. As shown in Table 1, the K m and k cat values and the specificity constant k cat/K m values of some substrates indicated that in vitro phenylpropionyl-CoAs were better substrates than the corresponding unsaturated phenylpropanoid-CoAs.
As reported for other plant PKS III, aliphatic starters showed considerable or even higher activity than phenylpropionyl-CoAs, although they are not the physiological substrates (Jez et al. 2002). In the case of WtPKS1, this was found for n-hexanoyl-CoA (Table 1) and several other n-alkyl-CoA esters such as n-butyryl-CoA (rel. activity 12%), n-valeryl-CoA (70%), n-heptanoyl-CoA (90%), n-octanoyl-CoA (130%) and, in addition, also with branched alkyl-CoAs, for example isobutyryl-CoA (19%) and isovaleryl-CoA (15%).
Discussion
The abundant occurrence of phenylphenalenones and the absence of detectable chalcone derivatives, e.g. flavonoids, stilbenes, and other products that might be synthesized via CHS-related enzymes suggest that the protein encoded in WtPKS1 is a good candidate for the postulated type III PKS in the biosynthesis of phenylphenalenones. The proposal is also consistent with the fact that the enzyme is not closely related (i.e. <65% identity) with any other member of the protein family. The question is therefore whether the biochemical properties of the protein are compatible with the proposed function or whether they are clearly in conflict with that.
The substrate preference is compatible with the proposal because the expected substrates (phenylpropanoid units) are accepted, but otherwise the enzyme is as promiscuous as all other type III PKSs investigated, and the percent figures for the substrate preferences do not provide convincing clues. It is not surprising that linear CoA esters (e.g. n-octanoyl-CoA, n-hexanoyl-CoA) are the best substrates, and that pyrones obtained after two condensation reactions were the products. Similar findings have been reported for several CHSs and other CHS family proteins for which the physiological substrates are known not to be linear CoA esters (Schröder 2000; Austin and Noel 2003).
An important point concerns the question whether the side chain of the predicted starter phenylpropanoid unit is unsaturated (e.g. 4-coumarate) or saturated (phenylpropionate). This has not been investigated thoroughly with Wachendorfia, but precursor feeding studies with another member of the Haemodoraceae, A. preissii, provided evidence for the interconvertibility of unsaturated and saturated phenylpropanoids in vivo. Although in these competitive feeding experiments the incorporation rate is higher for the unsaturated than the saturated phenylpropanoid, a clear decision is not possible at present (Schmitt and Schneider 1999). However, reduction of a side chain double bond of one of the diarylheptanoid-forming phenylpropanoids is indispensable for the later cyclization to phenylphenalenones (Bazan et al. 1978; Schmitt and Schneider 1999), but it is not clear at which level this occurs. An attractive possibility is that it takes place at the diketide level after the initial condensation reaction because such enzyme activity has been described in the biosynthesis of 4-(4-hydroxyphenyl)-butan-2-one from 4-hydroxybenzalacetone in raspberries (Borejsza-Wysocki and Hrazdina 1994), but other possibilities (e.g. at the level of linear diarylheptanoids) are not excluded. The available in vivo data do not permit a definite conclusion, but they do not contradict the in vitro properties of WtPKS1, and actually these properties could possibly provide support for the hypothesis that unsaturated phenylpropanoids are the precursors in vivo.
Another crucial point for the interpretation is the number of condensation reactions carried out with the various substrates. The biosynthesis model predicts one condensation reaction (Fig. 1). There is in fact only one substrate that is satisfactorily in accordance with this prediction; it is 4-coumaroyl-CoA, with 80% of the product corresponding to the expected release product after one condensation (4-hydroxybenzalacetone). With cinnamoyl-CoA, the second unsaturated substrate, the percentage was much lower (20%).
All other substrates led exclusively to two condensations and the release of pyrones. The failure to find such compounds or likely derivatives in the thorough investigation of the secondary products in the plant and its organ culture makes it very unlikely that WtPKS1 has a corresponding activity in vivo. As noted above, pyrones are typical for in vitro incubations of type III PKSs with non-physiological substrates. If that is acceptable for the linear CoA esters, it is also not excluded that the same reasoning applies to the saturated phenylpropanoid starters which also lead to pyrone products. In common, both substrate groups are more flexible than 4-coumaroyl-CoA, suggesting that they may be more efficient in the use of the space available in the active site cavity, thus being more amenable to a second condensation than the rigid diketide derived from 4-coumaroyl-CoA. This starter was not one of the best substrates in vitro, but it could be argued that this was a consequence of the non-physiological accumulation of enzyme-bound diketide thioesters, i.e. that the intermediate “got stuck” in the absence of the missing downstream coupling enzyme, which presumably performs the condensation with the second phenylpropanoid-CoA. Taken together, the results with 4-coumaroyl-CoA are compatible with the proposed role of WtPKS1 in diarylheptanoid and phenylphenalenone biosyntheses.
WtPKS1 was modelled based on the known structures of CHS (Ferrer et al. 1999) and 2PS (Jez et al. 2000a), in order to get more insights into the reasons for the properties of the enzyme. The models showed that the overall 3D structures were in all cases very similar (Fig. 9). The active site cavities, however, revealed some differences that were probably significant. For example, Ser338 and Thr197 in the M. sativa CHS were replaced in WtPKS1 by the more bulky Met341 and Phe200 (Fig. 9). A similar change in 2PS (Ser338 to Ile343 and Thr197 to Leu202) had been shown to reduce the size of the active site cavity (Jez et al. 2000a), most likely a significant contribution to the fact that 2PS could not use phenylpropanoids, but only smaller substrates (Eckermann et al. 1998). This was not the effect seen with WtPKS1, but it might well limit the size or shape of the cavity to the extent that products larger than triketides could not be synthesized. There were also other differences to CHS in residues believed to be important for the substrate or the size and shape of the active site (Austin and Noel 2003). Examples were the exchanges of Thr132 in MsCHS to Gly135 of WtPKS1, of Ser133 to Ala136, of Met137 to Val140 and of Ile254 to Val257. Interestingly, these changes led mostly to less bulky residues and in two cases involved the loss of hydroxyl groups. One of them, the change of Thr, which is present both in the cavity of CHS (position 132) and 2PS (position 137), to Gly135 in WtPKS1 might not only provide additional rotational freedom but, due to the missing hydroxyl group, also reduce the possible hydrogen bond formation with the substrate. Although that remains to be investigated by structural studies, it is an interesting possibility that this compensated to some extent the size-reducing effect of the other changes, thus permitting the use of the large phenylpropanoid substrates without negating the limits imposed on the size of the products.

Upper row: ribbon models (Swiss-Pdb Viewer) of CHS from Medicago sativa (Ferrer et al. 1999), 2PS from Gerbera hybrida (Jez et al. 2000a) and WtPKS1 from Wachendorfia thyrsiflora. The 3D structures of 2PS and WtPKS1 were modelled based on the X-ray structure of CHS. Lower row: models of active sites of CHS from Medicago sativa, 2PS from Gerbera hybrida and WtPKS1 from Wachendorfia thyrsiflora. Amino acids involved in substrate binding are shown as colored balls. The models were developed with SWISS-MODEL, the automated protein homology-modelling server (Schwede et al. 2003)
The modelling could not explain satisfactorily why the expected one condensation was essentially obtained only with 4-coumaroyl-CoA, but not with any of the other substrates. Type III PKS were often found to be imperfect in performing in vitro the number of condensations expected from the products in vivo (Schröder 2000; Austin and Noel 2003), but usually the numbers were reduced rather than increased (e.g. only two instead of three). There was, however, at least one example of a plant type III PKS that carried out more condensations than expected. This was the recombinant BAS from Rheum palmatum that carried out two condensations instead of one when the pH of the assay was changed (Abe et al. 2003), demonstrating that minor changes may have drastic effects on the number of condensation reactions carried out by a type III PKS. An effect of the pH on the number of condensations was not seen with WtPKS1, but it is not excluded that the trigger condition for a switch to only one condensation with all substrates was not detected. Another interesting example was a type III PKS from Streptomyces griseus. It carried out four condensations with its physiological substrate malonyl-CoA to synthesize 1,3,6,8-tetrahydroxynaphthalene. With the unphysiological substrate n-octanoyl-CoA, however, it was capable of performing five condensations in vitro, releasing a pyrone [4-hydroxy-6-(2,4,6-trioxotridecyl)-2-pyrone] as product (Funa et al. 2002). It appears from such data that in vitro products are sometimes more revealing about the maximum size and capacity of the active site cavities than about the reactions in vivo. None of these unexpected properties had been predicted by modelling.
It seems likely that the activities of WtPKS1 in vitro cannot be understood without the knowledge of the subsequent reactions in vivo. Thus, the question is whether or not it is realistic to assume that a single enzyme (e.g. WtPKS1) carries out the initial condensation reaction as well as the second step expected in diarylheptanoid formation (Fig. 1). Formally, this would involve a thioesterase activity releasing the diketide, removal of the terminal carboxyl group for activation of the adjacent methyl group and condensation to a second different phenylpropanoid unit (Fig. 1) bound to the active site cysteine. The release of benzalacetone-type products after one condensation (at least with some substrates, see Results and Fig. 8) indicated that WtPKS1 could catalyse or at least facilitate thioesterase and decarboxylation reactions, as carried out by true BASs. A diarylheptanoid synthase activity, however, would postulate that the decarboxylated enzyme-bound product of the first condensation was not released, but served as chain extender for a new starter phenylpropanoid residue bound to the active site cysteine. This proposes essentially an ordered switch of chain extenders in sequential condensation reaction by the same protein. The experiments incubating NAC-diketides with phenylpropanoid-CoA esters intended to mimic the second condensation, but no product was detected. To the best of our knowledge, there is no example that type III PKSs carried out such ordered switch between chain extenders in sequential condensations. A comparable, though simpler, switch between malonyl-CoA and methylmalonyl-CoA had been discussed for the biosynthesis of methylated chalcones, but could not be demonstrated, and cooperation between several proteins had been proposed (Schröder et al. 1998).
It seems therefore likely that diarylheptanoid formation requires functional interactions of the PKS with another enzyme, and that might be another type III PKS with unusual properties or another type of condensing enzyme. Inter-protein interactions have been described for other type III PKSs. Cooperations of CHSs with reductases in the biosynthesis of 6′-deoxychalcones have been discussed, either on the level of enzyme-bound triketide or tetraketide intermediates (Welle et al. 1991) or, more likely, on the basis of new data, on the level of coumaroyl-triones prior to aromatization (Austin and Noel 2003; Bomati et al. 2005).
Similar interactions between STS and reductases must also be postulated in the biosynthesis of stilbenecarboxylic acids, and in this case it has been argued that the lack of the correct interaction partner was mainly responsible for not obtaining the expected end products (Eckermann et al. 2003). As discussed above, the products of WtPKS1 resulting from two condensations could therefore simply represent unphysiological activities due to lack of interaction with the appropriate other proteins, in particular as an even higher complexity of reactions appears likely. Not open-chain diarylheptanoids, but phenylphenalenones are the end products in W. thyrsiflora, and their formation requires additional reactions, e.g. a ketoreductase, a Diels–Alder cyclase and an aromatase.
In summary, the available evidence indicates that the biochemical properties of the recombinant WtPKS1 are compatible with the notion that it is a prime candidate for catalysing the initial step in diarylheptanoid biosynthesis. The availability of WtPKS1 will be an important key for the identification and characterization of the proteins responsible for the next steps.
Abbreviations
- BAS:
-
Benzalacetone synthase
- CHS:
-
Chalcone synthase
- IPTG:
-
Isopropyl-β-d-thiogalactopyranoside
- NAC:
-
N-acetylcysteamine
- PMSF:
-
Phenylmethanesulfonyl fluoride
- PKS:
-
Polyketide synthase
- 2PS:
-
2-Pyrone synthase
- STS:
-
Stilbene synthase
- WtPKS1:
-
Wachendorfia thyrsiflora polyketide synthase 1
References
Abe I, Sano Y, Takahashi Y, Noguchi H (2003) Site-directed mutagenesis of benzalacetone synthase. J Biol Chem 278:25218–25226
Austin MB, Noel JP (2003) The chalcone synthase superfamily of type III polyketide synthases. Nat Prod Rep 20:79–110
Austin MB, Bowman ME, Ferrer JL, Schröder J, Noel JP (2004a) An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chem Biol 11:1179–1194
Austin MB, Izumikawa M, Bowman M, Udwary D, Ferrer J-L, Moore B, Noel J (2004b) Crystal structure of a bacterial type III polyketide synthase and enzymatic control of reactive polyketide intermediates. J Biol Chem 279:45162–45174
Baranovsky A, Schmitt B, Fowler DJ, Schneider B (2003) Synthesis of new biosynthetically important diarylheptanoids and their oxa- and fluoro analogues by three different strategies. Synth Commun 33:1019–1045
Bazan AC, Edwards JM, Weiss U (1978) Synthesis of lachnanthocarpone [9-phenyl-2,6-dihydroxyphenalen-1(6)-one] by intramolecular Diels–Alder cyclization of a 1,7-diarylheptanoid orthoquinone; possible biosynthetic significance of Diels–Alder reactions. Tetrahedron 34:3005–3015
Beuerle T, Pichersky E (2002) Enzymatic synthesis and purification of aromatic coenzyme A esters. Arch Biochem Biophys 302:305–312
Bick IRC, Blackman AJ (1973) Haemodorin—a phenalenone pigment from Haemodorum distichophyllum. Aust J Chem 26:1377–1380
Bomati EK, Austin MB, Bowman E, Dixon RA, Noel JP (2005) Structural elucidation of chalcone reductase and implications for deoxychalcone biosynthesis. J Biol Chem 280:30496–30503
Borejsza-Wysocki W, Hrazdina G (1994) Biosynthesis of p-hydroxyphenylbutan-2-one in raspberry fruits and tissue cultures. Phytochemistry 35:623–628
Bradford MM (1976) Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal Biochem 72:248–254
Chattopadhyay I, Biswas K, Bandyopadhyay U, Banerjee RK (2004) Turmeric and curcumin: biological actions and medicinal applications. Curr Sci India 87:44–53
Dora GA (1991) Phytochemical study of some Haemodoraceous plants. PhD thesis, University of Connecticut
Eckermann S, Schröder G, Schmidt J, Strack D, Edrada RA, Helariutta Y, Elomaa P, Kotilainen M, Kilpelainen I, Proksch P, Teeri TH, Schröder J (1998) New pathway to polyketides in plants. Nature 396:387–390
Eckermann C, Schröder G, Eckermann S, Strack D, Schmidt J, Schneider B, Schröder J (2003) Stilbenecarboxylate biosynthesis: a new function in the family of chalcone synthase-related proteins. Phytochemistry 62:271–286
Edwards JM (1974) Phenylphenalenones from Wachendorfia species. Phytochemistry 13:90–291
Ferrer JL, Jez JM, Bowman ME, Dixon RA, Noel JP (1999) Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat Struct Biol 6:775–784
Funa N, Ohnishi Y, Ebizuka Y, Horinouchi S (2002) Properties and substrate specificity of RppA, a chalcone synthase-related polyketide synthase in Streptomyces griseus. J Biol Chem 277:4628–4635
Gilbert IH, Ginty M, O’Neill JA, Simpson TJ, Staunton J, Willis CL (1995) Synthesis of β-keto and α,β-unsaturated N-acetylcysteamine thioesters. Bioorg Med Chem Lett 5:1587–1590
Helariutta Y, Elomaa P, Kotilainen M, Griesbach RJ, Schröder J, Teeri TH (1995) Chalcone synthase-like genes active during corolla development are differentially expressed and encode enzymes with different catalytic properties in Gerbera hybrida (Asteraceae). Plant Mol Biol 28:47–60
Hölscher D (1996) Biosynthese und Strukturermittlung von Phenylphenalenonen aus Anigozanthos preissii Endl. PhD thesis, Martin Luther University Halle
Hölscher D, Schneider B (1995) A diarylheptanoid intermediate in the biosynthesis of phenylphenalenones in Anigozanthos preissii. J Chem Soc Chem Commun 525–526
Hölscher D, Schneider B (1996) A resveratrol dimer from Anigozanthos preissii and Musa Cavendish. Phytochemistry 43:471–473
Hölscher D, Schneider B (1997) Phenylphenalenones from root cultures of Anigozanthos preissii. Phytochemistry 45:87–91
Jez JM, Austin MB, Ferrer J, Bowman ME, Schröder J, Noel JP (2000a) Structural control of polyketide formation in plant-specific polyketide synthases. Chem Biol 7:919–930
Jez JM, Ferrer J-L, Bowman ME, Dixon RA, Noel JP (2000b) Dissection of malonyl-coenzyme A decarboxylation from polyketide formation in the reaction mechanism of a plant polyketide synthase. Biochemistry 39:890–902
Jez JM, Bowman ME, Noel JP (2002) Expanding the biosynthetic repertoire of plant type III polyketide synthases by altering starter molecule specificity. Proc Natl Acad Sci USA 99:5319–5324
Junghans H, Dalkin K, Dixon RA (1993) Stress responses in alfalfa (Medicago sativa L.). 15. Characterization and expression patterns of members of a subset of the chalcone synthase multigene family. Plant Mol Biol 22:239–253
Kamo T, Hirai N, Tsuda M, Fujioka D, Ohigashi H (2000) Changes in the content and biosynthesis of phytoalexins in banana fruit. Biosci Biotech Biochem 64:2089–2098
Lanz T, Tropf S, Marner F-J, Schröder J, Schröder G (1991) The role of cysteines in polyketide synthases: site-directed mutagenesis of resveratrol and chalcone synthases, two key enzymes in different plant-specific pathways. J Biol Chem. 266:9971–9976
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Opitz S (2002) Phenylphenalenones and related phenolic pigments of the Haemodoraceae: structure, biosynthesis and accumulation patterns in Xiphidium caeruleum and Wachendorfia thyrsiflora. PhD thesis, Friedrich Schiller University Jena
Opitz S, Schneider B (2003) Oxidative biosynthesis of phenylbenzoisochromenones from phenylphenalenones. Phytochemistry 62:307–312
Opitz S, Otálvaro F, Echeverri F, Quiñones W, Schneider B (2002) Isomeric oxabenzochrysenones from Musa acuminata and Wachendorfia thyrsiflora. Nat Prod Lett 16:335–338
Saitou N, Nei M (1987) The neighbour-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schmitt B, Schneider B (1999) Dihydrocinnamic acids are involved in the biosynthesis of phenylphenalenones in Anigozanthos preissii. Phytochemistry 52:45–53
Schröder J (1997) A family of plant-specific polyketide synthases: facts and predictions. Trends Plant Sci 2:373–378
Schröder J (2000) The family of chalcone synthase-related proteins: functional diversity and evolution. Recent Adv Phytochem 34:55–89
Schröder J, Raiber S, Berger T, Schmidt A, Schmidt J, Soares-Sello AM, Bardshiri E, Strack D, Simpson TJ, Veit M, Schröder G (1998) Plant polyketide synthases: a chalcone synthase-type enzyme which performs a condensation reaction with methylmalonyl-CoA in the biosynthesis of C-methylated chalcones. Biochemistry 37:8417–8425
Schüler G, Mithöfer A, Baldwin IT, Berger S, Ebel J, Santos JG, Herrmann G, Hölscher D, Kramell R, Kutchan TM, Maucher H, Schneider B, Stenzel I, Wasternack C, Boland W (2004) Coronalon: a powerful tool in plant stress physiology. FEBS Lett 563:17–22
Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31:3381–3385
Thomas R (2001) A biosynthetic classification of fungal and streptomycete fused-ring aromatic polyketides. ChemBioChem 2:612–627
Van de Peer Y, De Wachter R (1994) TREECON for Windows: a software package for the construction and drawing of evolutionary trees for the Microsoft Windows environment. Comput Appl Biosci 10:569–570
Wang CZ, Maier UH, Zenk MH (2000) Synthesis of 3,3′,5-trihydroxybiphenyl-2-carboxylic acid, a component of the bitterest natural product amarogentin and its coenzyme A and N-acetyl cysteamine thiol esters. J Nat Prod 63:371–374
Welle R, Schröder G, Schiltz E, Grisebach H, Schröder J (1991) Induced plant responses to pathogen attack: analysis and heterologous expression of the key enzyme in the biosynthesis of phytoalexins in soybean (Glycine max L. Merr. cv. Harosoy 63). Eur J Biochem 196:423–430
Zhu JP, Islas-Gonzales G, Bois-Choussy M (2000) Recent progress in isolation, bioactivity evaluation and total synthesis of diarylheptanoids. Org Prep Proced Int 32:505–546
Acknowledgements
The authors wish to thank Gudrun Schröder (Freiburg) for protein expression and practical advice and J.P. Noel (La Jolla) for providing the pHis8 vector. We are grateful to A. Schmidt and M. Reichelt (Jena) for assistance in radio-HPLC analysis and Bettina Raguschke (Jena) for DNA sequencing. This work was financially supported by the Deutsche Forschungsgemeinschaft (Schn 450–4).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Brand, S., Hölscher, D., Schierhorn, A. et al. A type III polyketide synthase from Wachendorfia thyrsiflora and its role in diarylheptanoid and phenylphenalenone biosynthesis. Planta 224, 413–428 (2006). https://doi.org/10.1007/s00425-006-0228-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-006-0228-x