
Abstract
The activated disease resistance (ADR) 1 gene encodes a protein that possesses an N-terminal coiled-coil (CC) motif, nucleotide-binding site (NBS) and C-terminal leucine-rich repeat (LRR) domains. ADR1 belongs to a small, atypical Arabidopsis thaliana sub-class containing four CC–NBS–LRR genes. The NBS region of most NBS–LRR proteins possesses numerous conserved motifs. In contrast, the LRR domain, which is subject to positive selection, is highly variable. Surprisingly, sequence analysis revealed that the LRR domain of the ADR1 sub-class was more conserved than the NBS region. Sequence analysis identified two novel conserved motifs, termed TVS and PKAE, specific for this CC–NBS–LRR sub-class. The TVS motif is adjacent to the P-loop, whereas the PKAE motif corresponded to the inter-domain region termed the NBS–LRR linker, which was conserved within the different CC–NBS–LRR classes but varied among classes. These ADR1-specific motifs were employed to identify putative ADR1 homologs in phylogenetically distant and agronomically important plant species. Putative ADR1 homologs were identified in 11 species including rice and in 3 further Poaceae species. The ADR1 sub-class of CC–NBS–LRR proteins is therefore conserved in both monocotyledonous and dicotyledonous plant species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pathogen recognition is thought to result from the direct or indirect interaction between a product of a microbial avirulence (avr) gene and the corresponding plant disease resistance (R) gene (Staskawicz et al. 1995). The most prevalent class of R genes encodes proteins that contain a nucleotide-binding site (NBS) and C-terminal leucine-rich repeat (LRR) domains (Dangl and Jones 2001). NBS–LRR proteins were divided into two sub-families based on the presence or absence of an N-terminal region homologous to the toll/interleukin-1 receptor (TIR) domain (Pan et al. 2000). Most of the non-TIR NBS–LRR proteins exhibited a coiled-coil (CC) domain in the N-terminal region (Meyers et al. 1999, 2003).
Comparative analysis of a complete set of Arabidopsis NBS–LRR genes revealed a greater structural and sequence diversity among CC–NBS–LRR (CNL) proteins compared to TIR–NBS–LRR (TNL) proteins suggesting that CNL proteins may be more ancient than TNLs (Meyers et al. 2003). Four CNL sub-groups, designated as CNL clade A, B, C and D, were defined in Arabidopsis (Meyers et al. 2003). These four sub-groups were identified in several plant species and named clade N4, N3, N2 and N1, respectively (Cannon et al. 2002; Zhu et al. 2002; Ashfield et al. 2004).
The activated disease resistance (ADR) 1 gene encodes a CNL protein that belongs to the smallest and least characterised Arabidopsis clade CNL-A, which is also referred to as non-TIR N4 in different plant species (Cannon et al. 2002; Baumgarten et al. 2003; Meyers et al. 2003). The N-terminus of ADR1 also possesses a number of sub-domains typically found in protein kinases (Grant et al. 2003). An activation tagged allele of adr1, which resulted in ADR1 over-expression, produced constitutive activation of salicylic acid-dependent defence genes and conveyed broad-spectrum disease resistance (Grant et al. 2003). Intriguingly, this mutant line also exhibited enhanced drought tolerance suggesting significant overlap between biotic and abiotic stress signalling networks (Chini et al. 2004).
Materials and methods
Comparisons and alignments of protein sequences were performed by ClustalW (http://www.ebi.ac.uk/clustalw/index.html) (Chenna et al. 2003). The programme multiple expectation maximisation for motif elicitation (MEME) was employed to analyse the sequence alignment to search for conserved motifs (P-value lower than 5e-20) (Bailey and Elkan 1995; http://meme.sdsc.edu/meme/website/intro.html).
In order to identify ADR1 homologs in different plant species, similarity searches were performed using sequences available, during the period from April 2004 to September 2004, on 37 plant EST databases from The Institute for Genomic Research (TIGR; http://www.tigr.org/) and Munich Information Centre for Protein Sequences (MIPS; http://mips.gsf.de/). Consensus sequences were derived by Consensus (Bork et al. 1996; http://www.bork.embl-heidelberg.de/Alignment/consensus.html). The amino acid sequences of the NBS region were analysed and a phenogram representation of the neighbour-joining tree was generated using Phylodendron ClustalW (Felsenstein 1989; http://www.es.embnet.org/Doc/phylodendron/clustal-form.html). The tree was rooted using the sequence of the human Apf-1 protein. Bootstrapping provided an estimate of the confidence for each branch point.
Results and discussion
ADR1 exhibits high sequence identity to three other ADR1-like (ADR1-L) proteins within the CNL-A clade: At4g33300 (ADR1-L1), At5g04720 (ADR1-L2) and At5g47280 (ADR1-L3), which are 68, 65 and 66% identical to ADR1 respectively (Grant et al. 2003). Two additional proteins, At5g66900 and At5g66910, are also found within the clade CNL-A (Meyers et al. 2003). However, At5g66900 and At5g66910 exhibit only 31 and 30% sequence identity in ADR1 respectively, and comprehensive sequence analyses classified them into a different sub-group within the clade CNL-A (Mondragon-Palomino et al. 2002; Meyer et al. 2003).
In order to search for conserved motifs, the programme MEME (P-value lower than 5e-20) (Bailey and Elkan 1995) was employed to analyse the sequence alignment of the four proteins that comprise the ADR1 family. This analysis did not identify conserved motifs within the N-terminal CC domain (from ADR1 M1 to L187). Seven motifs were identified by MEME analysis of the NBS domain (from ADR1 F188 to N472). Six of these corresponded to conserved motifs previously described for ADR1: P-loop, kinase-2, RNBS-A, GLPL, RNBS-D and MHDV (Meyers et al. 1999; Cannon et al. 2002; Grant et al. 2003). These six motifs from the ADR1 protein family were compared with those present in the complete set of Arabidopsis NBS–LRR proteins. The RNBS-D and MHDV motifs alone showed sequence specificities unique for the ADR1 family (Figs. 1, 2). Only this family and the fellow CNL-A clade members At5g66900 and At5g66910 contained a glutamine (Q) instead of a methionine (M) as the third residue of the MHDV motif. This methionine is conserved among all the remaining 156 Arabidopsis NBS–LRR proteins and was integral to the core sequence (MHDV) defining the motif name (Meyers et al. 2003). The corresponding motif will therefore be referred to as QHDV in the ADR1 protein family.

Full-length alignment of the four ADR1 family proteins generated by ClustalW (Chenna et al. 2003). Amino acids boxed in black are invariant, whereas residues shaded in grey are conserved in >75% of the sequences. The six previously reported conserved motifs of the NBS domain (P-loop, kinase2, RNBS-A, GLPL, RNBS-D and MHDV) are boxed in black lines (Meyers et al. 1999; Cannon et al. 2002; Grant et al. 2003). Blue boxes designate novel conserved motifs (TVS, LMP and PKAE). Individual LRR are indicated by red lines

Multiple sequence alignment of motif regions specific for the Arabidopsis ADR1 protein family and homologous plant sequences. Accession numbers of individual plant sequences shown are: Glycine max TC195419, Gossypium hirsutum CD486153, Lotus japonicus AV417020, Lycopersicon esculentum AW039749, Medicago truncatula TC87505, Oryza sativa TC149181, Solanum tuberosum TC62931, Sorghum bicolor TC57671, Triticum aestivum TC107913, Vitis vinifera CA32EN0005 and Zea mays BG836496. a Alignment of all available plant sequences containing the novel TVS motif by ClustalW (Chenna et al. 2003). Amino acids boxed in black are invariant, whereas residues shaded in dark grey are conserved in more than 85% of the sequences. Light grey shaded amino acids are conserved in more than 70% of the sequences. The ADR1 consensus sequence shown beneath the alignment was derived by Consensus (Bork et al. 1996). Notations for variable amino acid are as follows: “a”, aromatic (F, Y, W, H); “l”, aliphatic (I, V, L); “h”, hydrophobic (aromatic, aliphatic, A, G, M, C, K, R, T); “+”, positive (H, K, R); “-“, negative (D, E); “p”, polar (positive, negative, Q, N, S, T, C); “u”, tiny (G, A, S); “s”, small (tiny, V, T, D, N, P, C); “.”, any. b Sequence alignment of all available plant RNBS-D motif regions by ClustalW. The ADR1 consensus sequence was derived as described above. c Sequence of the ADR1 family-specific QHDV motif region from all available sequences aligned by ClustalW. A consensus sequence was derived; amino acids notations are as described above. d Sequence alignment of the ADR1 family-specific PKAE motif, which corresponded to the clade-specific NBS-LRR linker region, created by ClustalW. Again, a consensus sequence was derived and is shown below the alignment
The presence of an additional ADR-specific motif (from ADR1 L221 to L234), designated TVS according to its sequence (Figs. 1, 2), was also uncovered. The conservation rate of the TVS motif was compared to that of conserved NBS motifs: 71% of the amino acid positions within the TVS motif were conserved throughout the four ADR1 proteins, greater than other conserved NBS motifs, with the exception of the P-loop and kinase-2. Among the complete set of Arabidopsis NBS–LRR proteins, the novel TVS motif was found only within the ADR1 protein family. The other CNL-A clade members At5g66900 and At5g66910 lacked the TVS motif. Furthermore, the TVS motif spanned a similar, but more defined sequence designated as the NBS-22 motif (Meyers et al. 2003).
The NBS domain conventionally terminates with the MHDV motif (Meyers et al. 2003; Ashfield et al. 2004), while the LRR domain starts approximately 40–65 amino acids C-terminal of this motif. This inter-domain region is termed the NBS–LRR (NL) linker (Meyers et al. 2003). The NL sequence is conserved within the different CNL classes but varies among classes (Meyers et al. 2003). The NL linker of the ADR1 family is 78 residues long (from ADR1 R473 to S551); and one motif, designated PKAE according to its sequence, was identified by MEME analysis within the ADR1 NL (Figs. 1, 2). Moreover, the ADR1 linker region was the most conserved NL among the four clades. The putative PKAE motif showed 71% position identity among the four ADR1 protein family members. Consequently, this novel motif exhibited greater conservation than the other six conserved NBS motifs, with the exception of the P-loop and kinase-2. Again, the other CNL-A clade members At5g66900 and At5g66910 lacked this motif.
The LRR domain in R proteins is thought to recognise, directly or indirectly, pathogen molecules (Jia et al. 2000; Dangl and Jones 2001). Structurally, LRRs are thought to form repeats of β-strand-loop and α-helix-loop units, with non-leucine residues responsible for protein recognition (Thomas et al. 1996; Michelmore and Meyers 1998; Kobe and Kajava 2001). Comparative analysis of NBS–LRR genes from tomato, lettuce, rice and Arabidopsis revealed that the non-leucine residues of the LRR are hypervariable and subject to positive selection (Parniske et al. 1997; Meyers et al. 1998; Wang et al. 1998; Noel et al. 1999; Ellis et al. 2000). Comprehensive analysis of Arabidopsis NBS–LRR protein sequences showed that the NBS domain was significantly more conserved than the variable LRR region (Meyers et al. 1999, 2003; Pan et al. 2000).
Comparative analysis of the LRR domain (from ADR1 R552 to L734) showed 57% position identity (83% homology) among the four ADR1 proteins. Furthermore, the conserved residues were uniformly distributed throughout the LRR domain (Fig. 1). Residues identical among the four ADR1 proteins not only corresponded to the amino acids expected to form the structural backbone of each LRR unit but also represented amino acids predicted to form β sheets responsible for ligand binding. In contrast, the NBS domain (from ADR1 F188 to N472), which represents the most conserved domain of all NBS–LRR proteins, exhibited 50% position identity (75% homology) among the four proteins of the ADR1 family. These identical amino acids were predominantly located within the conserved motifs of the NBS domain (Fig. 1). The ADR1 LRR domain was therefore more highly conserved than the NBS domain. To our knowledge, this is the first report describing this feature in an Arabidopsis R gene family. These conclusions were consistent with a previous analysis of the lettuce RGC2 gene family that reported an alternating pattern of conservation and hyper-variability within the LRR region (Meyers et al. 1998).
In order to identify ADR1 homologs in different plant species, similarity searches specifically using the highly conserved sequence fragment surrounding the TVS, RNBS-D, QHDV and PKEA motifs were performed on 37 plant EST databases (from TIGR and MIPS). To minimize errors, only sequences encoding a minimum of 200 amino acids were selected for further analyses. These searches identified putative ADR1 homologues from 12 plant species, whereas searches of the remaining 25 plant databases failed to identify sequences significantly homologous to ADR1. These results were robust, since the same sequences were also found in a database search employing the full-length ADR1 protein sequence. The accession numbers for these ESTs are reported in Fig. 3. Surprisingly, only one sequence showing high homology to ADR1 was identified within the complete rice genome.

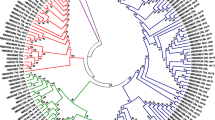
Phenogram representation of the neighbour-joining tree for the Arabidopsis ADR1 protein family and homologous plant sequences, which was constructed according to the method of Saitou and Nei (1987). The amino acidic sequences of the NBS region were analysed and a tree was generated using Phylodendron ClustalW (Felsenstein 1989). The NBS sequence from the human Apf1 protein rooted as an out-group. The number adjacent to each node indicates the percentage of 1,000 bootstrap replicates that support it; bootstrap results were not reported for support <50%. Branch lengths are proportional to the estimated evolutionary distance
To assess whether these ESTs represent ADR1 homologs, the four ADR1 Arabidopsis proteins were aligned with these 12 amino acid sequences. The difference in length of the 12 ESTs precluded a comprehensive analysis of the full-length protein. Therefore only the TVS, RNBS-D, QHDV and PKAE motif regions were analysed. Multiple alignments are shown in Fig. 2. Only the Oryza sativa and Medicago truncatula sequences homologous to ADR1 were sufficiently extended to include the TVS motif region, which is located at the N-terminus of the NBS domain. The RNBS-D, QHDV and PKAE motifs were highly conserved, even among several phylogenetically distant plant species. Moreover, all sequences exhibited the characteristic glutamine (Q) residue in the third position of the QHDV motif region, which seems to be a unique feature of the ADR1 protein family (Fig. 2c). The novel PKAE motif, which corresponds to the ADR1 clade-specific NL linker domain, was also highly conserved (Fig. 2d). These results suggest that the plant sequences reported here are likely to belong to the ADR1 protein family.
To further investigate this, phylogenetic analysis of the NBS domains from the ADR1 protein family together with the NBS domains from the putative protein homologs was undertaken. The CNL-A clade members At5g66900 and At5g66910 were included as controls. RMP1, RPP8 and RPS2 were included as representative proteins of the CNL-B, CNL-C and CNL-D clades respectively, whereas RPP1 served as controls from the TNL family. The phylogenetic analysis revealed that the NBS sequences derived from the 11 identified ESTs robustly grouped with the four members of the ADR1 protein family. In contrast, all control sequences grouped in distant branches of the tree (Fig. 3). These results suggest that at least 11 of the 12 plant sequences reported here are likely homologs of the ADR1 protein family.
The presence of the CNL-A clade (alternatively termed non-TIR NBS clade N4), which contains the ADR1 protein family, has been confirmed in many plant species (Cannon et al. 2002; Zhu et al. 2002). The previous failure to identify any sequences belonging to this clade from several Poaceae families suggested that it might be absent from the monocotyledonous plant lineage (Cannon et al. 2002). Our analysis, based on recently available data, has identified sequences with significant homology to the ADR1 gene family, which are members of the CNL-A clade, in four Poaceae species (Oryza sativa, Sorghum bicolor, Triticum aestivum and Zea mays). Thus, suggesting the CNL-A clade of NBS–LRR proteins is in fact present in monocotyledonous plants.
References
Ashfield T, Ong LE, Nobuta K, Schneider CM, Innes RW (2004) Convergent evolution of disease resistance gene specificity in two flowering plant families. Plant Cell 16:309–318
Bailey T L, Elkan C (1995) The value of prior knowledge in discovering motifs with MEME. Proc Int Conf Intell Syst Mol Biol 3:21–29
Baumgarten A, Cannon S, Spangler R, May G (2003) Genome-level evolution of resistance genes in Arabidopsis thaliana. Genetics 165:309–319
Bork P, Brown NP, Hegyi H, Schultz J (1996) The protein phosphatase 2C (PP2C) superfamily: detection of bacterial homologues. Protein Sci 5:1421–1425
Cannon SB, Zhu H, Baumgarten AM, Spangler R, May G, Cook DR, Young ND (2002) Diversity, distribution, and ancient taxonomic relationships within the TIR and non-TIR NBS–LRR resistance gene subfamilies. J Mol Evol 54:548–562
Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31:3497–3500
Chini A, Grant JJ, Seki M, Shinozaki K, Loake GJ (2004) Drought tolerance established by enhanced expression of the CC-NBS-LRR gene, ADR1, requires salicylic acid, EDS1 and ABI1. Plant J 38:810–822
Dangl JL, Jones JD (2001) Plant pathogens and integrated defence responses to infection. Nature 411:826–833
Ellis RP, Forster BP, Robinson D, Handley LL, Gordon DC, Russell JR, Powell W (2000) Wild barley: a source of genes for crop improvement in the 21st century? J Exp Bot 51:9–17
Felsenstein J (1989) PHYLIP—phylogeny inference package, version 3.2. Cladistics 5:164–166
Grant JJ, Chini A, Basu D, Loake GJ (2003) Targeted activation tagging of the Arabidopsis NBS-LRR gene, ADR1, conveys resistance to virulent pathogens. Mol Plant Microbe Interact 16:669–680
Jia Y, McAdams SA, Bryan GT, Hershey HP, Valent B (2000) Direct interaction of resistance gene and avirulence gene products confers rice blast resistance. Embo J 19:4004–4014
Kobe B, Kajava AV (2001) The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol 11:725–732
Meyers BC, Chin DB, Shen KA, Sivaramakrishnan S, Lavelle DO, Zhang Z, Michelmore RW (1998) The major resistance gene cluster in lettuce is highly duplicated and spans several megabases. Plant Cell 10:1817–1832
Meyers BC, Dickerman AW, Michelmore RW, Sivaramakrishnan S, Sobral BW, Young ND (1999) Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J 20:317–332
Meyers BC, Kozik A, Griego A, Kuang H, Michelmore RW (2003) Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15:809–834
Michelmore RW, Meyers BC (1998) Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res 8:1113–1130
Mondragon-Palomino M, Meyers BC, Michelmore RW, Gaut BS (2002) Patterns of positive selection in the complete NBS-LRR gene family of Arabidopsis thaliana. Genome Res 12:1305–1315
Noel L, Moores TL, van Der Biezen EA, Parniske M, Daniels MJ, Parker JE, Jones JD (1999) Pronounced intraspecific haplotype divergence at the RPP5 complex disease resistance locus of Arabidopsis. Plant Cell 11:2099–2112
Pan Q, Wendel J, Fluhr R (2000) Divergent evolution of plant NBS-LRR resistance gene homologues in dicot and cereal genomes. J Mol Evol 50:203–213
Parniske M, Hammond-Kosack KE, Golstein C, Thomas CM, Jones DA, Harrison K, Wulff BB, Jones JD (1997) Novel disease resistance specificities result from sequence exchange between tandemly repeated genes at the Cf-4/9 locus of tomato. Cell 91:821–832
Staskawicz BJ, Ausubel FM, Baker BJ, Ellis JG, Jones JDG (1995) Molecular genetics of plant disease resitance. Science 248:189–194
Thomas D, Rozell TG, Liu X, Segaloff DL (1996) Mutational analyses of the extracellular domain of the full-length lutropin/choriogonadotropin receptor suggest leucine-rich repeats 1-6 are involved in hormone binding. Mol Endocrinol 10:760–768
Wang GL, Ruan DL, Song WY, Sideris S, Chen L, Pi LY, Zhang S, Zhang Z, Fauquet C, Gaut BS et al. (1998) Xa21D encodes a receptor-like molecule with a leucine-rich repeat domain that determines race-specific recognition and is subject to adaptive evolution. Plant Cell 10:765–779
Zhu H, Cannon SB, Young ND, Cook DR (2002) Phylogeny and genomic organization of the TIR and non-tIR NBS-LRR resistance gene family in Medicago truncatula. Mol Plant Microbe Interact 15:529–39
Acknowledgements
We would like to thank Dietlind Gerloff for critically reading this manuscript. AC was the recipient of a Darwin Trust Studentship. This work was supported by BBSRC grant P16595 to GJL.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chini, A., Loake, G.J. Motifs specific for the ADR1 NBS–LRR protein family in Arabidopsis are conserved among NBS–LRR sequences from both dicotyledonous and monocotyledonous plants. Planta 221, 597–601 (2005). https://doi.org/10.1007/s00425-005-1499-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-005-1499-3