
Abstract
At normal aging, the brain exhibits signs of compromised bioenergetic and increased levels of products of interaction between reactive oxygen/nitrogen species (ROS/RNS) and brain constituents. Under normal conditions, steady-state levels of ATP and ROS/RNS fluctuate in certain ranges providing basis for stable homeostasis. However, from time to time these parameters leave a “comfort zone,” and at adulthood, organisms are able to cope with these challenges efficiently, whereas at aging, efficiency of the systems maintaining homeostasis declines. That is very true for the brain due to high ATP demands which are mainly covered by mitochondrial oxidative phosphorylation. Such active oxidative metabolism gives rise to intensive ROS generation as side products. The situation is worsened by high brain level of polyunsaturated fatty acids which are substrates for ROS/RNS attack and production of lipid peroxides. In this review, organization of energetic metabolism in the brain with a focus on its interplay with ROS at aging is discussed. The working hypothesis on aging as a disbalance between oxidative stress and energy provision as a reason for brain aging is proposed. From this point of view, normal age-related physiological decline in the brain functions results from increased disbalance between decrease in capability of the brain to control constantly increased incapability to maintain ROS levels and produce ATP due to amplification of vicious cycles intensification of oxidative stress <----> impairment of energy provision.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Integrity of living organisms needs permanent supplementation of energy from environment according to the thermodynamic laws, and this energy is used to create and maintain their structure and functions. Especially it is true for mammalian brain which amounts for about 2% of body mass and consumes about 25% glucose and 20% oxygen utilized by the organism [37]. So, taking into account that carbohydrates are the main energy source here, the brain consumes about tenfold more glucose per mass unit than the rest mammalian body. Just a minor amount of energy for the brain demands under normal conditions is covered by ketone bodies (KB), free fatty acids, and certain amino acids. For example, recently, utilization of glutamate, the major excitatory neurotransmitter as a complementary energy contributor in the brain, was discovered: astrocytes could take up actively glutamate to use for energy production converting it to α-ketoglutarate by oxidative glutamate dehydrogenase [34]. Most universal energy equivalent ATP is used in the brain for ion transport systems to generate and maintain transmembrane ion gradients, axonal/dendritic transportation, biosynthetic processes, and operation of neurotransmitter systems [23, 46, 47, 74]. Some ATP amount is produced in glycolysis, but majority of ATP is generated by mitochondrial oxidative phosphorylation (OxPhos). At oxidative metabolism, reactive oxygen species (ROS) are generated as side products of operation of mitochondrial and other electron transport chains and some oxidases. Elimination of ROS is provided by antioxidants: high molecular mass ones include antioxidant enzymes such as superoxide dismutase and peroxidases, whereas low molecular mass antioxidants include vitamins A and C, glutathione, and carotenoids and anthocyanins. [23, 42]. Reactive carbonyl species such as methylglyoxal are also produced as side products of glycolysis and fatty acid oxidation. Since the role of reactive carbonyl species in the aging brain in this issue is covered by Dr. H. Semchyshyn, here I will focus on relationships between ATP production and ROS. Homeostasis of reactive nitrogen species (RNS) in extensive interplay with ROS and energy homeostasis is also affected during aging [23, 28] which will not be central but rather mentioned in this short review. It should be noted that the situation in the brain is “worsened” by the high amount of polyunsaturated fatty acids which easily enter ROS/RNS-induced oxidation called lipid peroxidation [23, 50].
Aging of the brain is caused by slow cooperative changes in number processes, and to date, one leading factor has not been identified due to multifactorial nature of the aging process. However, it is clear that at least two processes, namely energy and ROS/RNS homeostasis, are somehow associated with aging. But cause–effect relationships between these two groups of processes and aging have not been established finally to date. This causes disagreement in discussions on what is a primary reason for aging: corruption of energy homeostasis (balance between ATP generation and consumption) or poorly controlled ROS/RNS homeostasis (balance between ROS/RNS generation and elimination with maintaining of certain steady-state ROS/RNS levels) [23, 42]. I believe that they both together are responsible for the aging process, and their interplay during it leads to the formation number of vicious cycles. These vicious cycles of ROS/RNS production and disruption of energy-providing systems, along with an age-related increase in the levels of dysfunctional molecules and their aggregates consisting of proteins, lipids, and DNA (some of which became ROS generators), may be disrupted by the operation of certain rescue and repair mechanisms. One of such well-known and extensively studied mechanisms involves up-regulation of defense systems via stabilization of NF-E2-related factor 2 (Nrf2) [23, 42]. This transcription factor coordinates the expression of about 200 genes many of which encode proteins preventing ROS/RNS-induced damages, repair of these damages, or eliminate damaged molecules [23, 42, 47, 66].
In both above-mentioned blocks of processes, the interaction between them should be inspected at description of the processes of the aging: appearance or disappearance of certain compounds. This short review paper aims to cover reorganization of spatiotemporal architecture of energy and ROS homeostasis at the aging of the mammalian brain with aging-related amplification of vicious cycles intensification of oxidative stress ⬅➔ impairment of energy provision as a reason for the aging process.
Organization of energetic metabolism in the brain and its interplay with reactive oxygen species
Glucose level in the human blood plasma under normal conditions ranges from 5.5 to 7.8 mM, and in the brain it ranges from 0.82 to 2.4 mM [1, 15, 67]. At hyperglycemia, the glucose level in the plasma can enhance up to 15.2 mM, and extracellular glucose concentration in the brain can double from euglycemia [67]. These parameters clearly show that there is a permanent gradient of glucose concentration from the blood into the brain. This creates appropriate mechanistic conditions for facilitated diffusion of glucose from bloodstream into the brain provided by glucose transporters called GLUT [23, 46].
In this paper, I will concentrate mainly on three types of cerebral cells: endothelial, astrocytic, and neuronal ones (see Fig. 6 in [23]). Despite microglial cells are important for brain operation under normal life, pathologies, and aging, the main events in the brain related to energy homeostasis are associated with the operation of neurons and astrocytes [23, 46]. Leaving bloodstream via entherocytes in the brain, most glucose is taken by astrocytes, whereas neurons absorb directly only minor glucose amount consumed by the brain [46]. The gradient from the vessels into astrocytes and neurons is generated and maintained owing to permanent intracellular phosphorylation of glucose to glucose-6-phosphate by hexokinase or glucokinase for ATP expenses. Bioenergetic needs of the brain are mainly covered by hexokinase, whereas glucokinase, also known as hexokinase IV, serves as a glucose sensor [13, 54]. Glucokinase possesses a low affinity for glucose (KM ~ 10 mM) due to which its activity in physiological conditions is virtually linearly proportional to intracellular glucose levels which provides a mechanistic basis for serving as the glucose sensor [13]. In addition, glucokinase is not inhibited by glucose-6-phosphate, the product of the reaction catalyzed.
In the astrocytes, there are certain reserves of glucose units in the form of polysaccharide glycogen which can be included in active metabolism by phosphorylase to produce glucose-1-phopsphate further converted to glucose-6-phosphate [18, 60]. In the brain, the latter is used either in glycolysis or pentose phosphate pathway (PPP). Formed in glycolysis, pyruvate may enter tricarboxylic acid cycle (TCA cycle) also called the citric acid cycle or Krebs cycle. This cycle is a hub in metabolism where degradative pathways converge and precursors for anabolic pathways are produced. Some portion of pyruvate is used for energy production directly in astrocytes, whereas substantial amount of pyruvate in the astrocytes is reduced to lactate and exported from them. From intercellular space, lactate can be either absorbed by neurons or released from the brain into the blood stream [45, 61]. Cooperation in energy provision between astrocytes and neurons seems may protect neurons against carbonyl stress because most reactive carbonyl species resulting from glycolysis are produced in astrocytes, whereas utilization of lactate by neurons help them to minimize influence of glycolytic intermediates as carbonylating agents.
Formed in glycolysis pyruvate further can be used by mitochondria to produce ATP. Under normal conditions, a minor portion of energy demand can be covered by KB utilization and this portion may increase with aging and some pathologies [12]. Information on the utilization of fatty acids in the brain is controversial [49] but seems they also can be either used by astrocytes or converted to KB and provided for neurons [36].
The main portion of ATP produced in the brain is used to generate and maintain electrochemical transmembrane gradients of ions [23, 46]. Both, resting potential and action potential, require ATP for primary active transmembrane transport of ions against a concentration gradient. The basic functions of cerebral cells also need energy similarly to cells of other organs. But the specificity of the brain in energy homeostasis is that energy demands of “basic” processes occupy an only small part of total energy expenditures, whereas firing of neurons and signal transduction and processing use most energy produced in the brain.
General energy homeostasis includes the generation and utilization of ATP. The utilization of ATP in the mammalian brain is out of the scope of this paper, and interested readers may be recommended to read some review papers on the topic [21, 43]. The whole process of energy provision can be divided in three stages: (1) supply of energy substrates and co-substrates (carbohydrates, and lactate, fatty acids, amino acids, KB and oxygen), (2) production of ATP (glycolysis and OxPhos) and (3) elimination of the end products (mainly CO2). Therefore further I will analyze these stages from point of view of their modification by ROS and at aging.
Glucose is transported into the brain via GLUT1 membrane protein which provides facilitated transport through the blood–brain barrier from capillaries into astrocytes, whereas GLUT3 and GLUT4 transport glucose into neurons [23, 46, 47]. Monocarboxylate transporters (MCT) provide fast transport across the plasma membrane of diverse monocarboxylates such as lactate, pyruvate, and KB. In the mammalian brain, MCT1 is expressed in capillaries, MCT4 in astrocytes and MCT2 with MCT4 in neurons [23, 46, 47]. As mentioned above, astrocytes are well equipped with glycolytic machinery, whereas neurons possess high mitochondrial potential. Such specialization is well supported by specific transporters of carbohydrates and monocarboxylates highlighted above. All links of the energy-providing chain, namely transportation of substrates, glycolysis, TCA cycle, and OxPhos, are sensitive to ROS.
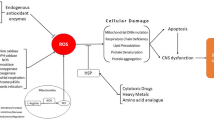
There are two principally different groups of ROS effects/responses on energy-generating machinery: short-term (acute) and long-term (chronic) ones (Fig. 1). Short-term effects usually do not involve effector-caused expression of specific genes and result from low specific oxidative modification of any cellular components which greatly varies in their sensitivity to ROS [6, 58]. More sensitive proteins/enzymes are links in the chain which provide ROS-sensitive regulation of the whole chain, that is, they are specific bottleneck providing interplay between energy and ROS/RNS homeostasis. If the information on oxidative inactivation of transporters by ROS in the brain is scarce, further steps of the ATP-producing chain got enough attention from this point of view. Among glycolytic enzymes glyceraldehyde-3-phosphate dehydrogenase, phosphofructokinase, and pyruvate kinase M2 are especially sensitive to ROS [6]. Inactivation of these enzymes may redirect glucose catabolism from glycolysis to PPP. That may be important for cell survival acute oxidative stress boots because NADPH produced in PPP can be used by the antioxidant system to combat ROS [42]. However, at a higher intensity of oxidative stress, key enzyme of PPP namely glucose-6-phosphate dehydrogenase may be also inactivated by ROS [56]. So ROS-promoted inactivation of the above-listed glycolytic enzymes seems to play an adaptive role as well. In TCA cycle, α-ketoglutarate dehydrogenase and aconitase are highly sensitive to ROS which may result in slowing operation of the cycle [71]. Finally, the electron transport chain (ETC) complexes I, III, IV, and V containing iron and copper ions are subjected to ROS-induced inactivation [69]. It should be mentioned that ETC complex V operates as ATP synthase and finalizes the whole process of ATP production in the mitochondria. Not all short-term ROS effects are negative. As mentioned above, inactivation of some targets (like phosphofructokinase and glyceraldehyde-3-phosphate dehydrogenase) may be important for cell survival oxidative stress and other stressful conditions [6].

Short-term and long-term effects of reactive oxygen species in the brain. Description in the text
Long-term ROS effects on the energy provision chain involve non-direct effects and provide an adaptive response via conditional (effector-caused) gene expression (Fig. 1). In the brain, there are at least two regulatory pathways that may provide interplay between ROS and ATP supply, namely transcription factors: hypoxia-inducible factor 1-alpha (HIF-1α) and nuclear factor erythroid 2-related factor 2 (Nrf2). Hypoxia-inducible factor 1-alpha is stabilized by ROS and up-regulates expression of number genes including one encoding GLUTs [35, 39] and glycolytic enzymes [9, 24] and providing in this manner increase in energy provision for the brain. Interestingly, in the brain, ROS may also induce translocation of GLUT1 from intracellular vesicles to cellular membrane like it takes place at an insulin-promoted increase of a number of glucose transporter molecules in cell membrane without affecting of gene expression [39]. Many adaptive responses to ROS are coordinated by transcription factor Nrf2 [42]. In response to increased ROS levels, it up-regulates expression of about 200 genes including ones encoding antioxidant enzymes, enzymes of glutathione biosynthesis, glucose-6-phosphate dehydrogenase; it also exerts anti-inflammatory effects and modulates mitochondrial function and biogenesis, proteostasis, and epigenetic mechanisms [4, 23, 42, 63]. Interestingly, up-regulation of glucose-6-phosphate dehydrogenase may increase the production of NADPH in PPP which has a dual effect: it is a cofactor used by the antioxidant system as mentioned above or co-substrate for ROS production by NADPH oxidase [31, 42]. However, the latter takes place preferentially in microglia and is out of the scope of this paper.
Energetic metabolism in the aged brain and reactive oxygen species involvement
It is recognized that in the healthy aging brain a significant decline in glucose metabolism either globally or regionally takes place [22, 30, 46, 47]. Positron emission tomography (PET) tracer in situ method is broadly used to characterize cerebral metabolic rate for glucose (CMRg) in the healthy aging brain and at pathologies (for review see [11]). Generalization of the experimental results does not conclude if there is age-related decline of glucose metabolic rate or no difference in the brain of normally aging people takes place [11]: ten of the cited papers indicated age-related decline, whereas eight of them did not demonstrate any difference. It is possible that some details in the studies led to such principal disagreement.
Since PET methods cannot respond all questions on glucose metabolic rate, other techniques should be applied also to characterize brain energy status at aging. It is especially important to differentiate energy metabolism in specific cerebral cells such as neurons or astrocytes. The topic of specialization of these cells was briefly covered above. Utilization of in vivo cultured astrocytes which are out of system control of the rest organism does not provide ideas on real functioning of cells in the organism but anyway may shed some light on their energetics [2, 26, 51]. It has been suggested that KB production by mammalian astrocytes followed by KB transportation into neurons may be responsible for the provision of nutrients for energy homeostasis in the brain [36]. Additional information was received with nuclear magnetic resonance (NMR) spectroscopy used to monitor the levels of glucose by 1H-NMR [33], glycogen by 31P-NMR [57], and ketones by 13C-NMR [48].
With 1H-magnetic resonance spectroscopy (MRS) it was shown that in the mouse frontal cortex lactate levels decreased with age [29]. Western blotting analysis of proteins from the frontal cortex indicated that the levels of proteins involved in lactate production, namely pyruvate dehydrogenase kinase (PDH 1), pyruvate dehydrogenase-E1α (PDH-E1α), lactate dehydrogenase A (LDHA), and pyruvate kinase M1 and M2 (PK M1, PK M2), declined with age. On the other hand, levels of lactate transporter proteins (MCT2 and MCT4) were significantly elevated at aging. Probably that is a compensatory adaptation to increase import of lactate in the frontal cortex in order to respond to decreased capability to convert glucose to lactate–pyruvate couple and normally feed mitochondria for ATP production. Interestingly, higher expression of the glycolytic enzymes positively correlated with better memory performance. On contrary, an inverse relationship was observed for lactate dehydrogenase B [29]. These results are in good agreement with previously received data by Duarte et al. [19], who found progressive decrease in lactate level in the cortex, whereas hippocampus did not show such pattern. Couple critical notes have to be provided here: (1) functional activities of the enzymes and transport proteins were not measured; (2) utilization of Western blotting analysis may not differentiate between active and non-active protein molecules; (3) operation of key glycolytic enzymes was not evaluated; and (4) inter-laboratory variation and specificity in mouse strain and their maintaining were not taken into account. Indeed, evaluation of levels of isoenzymes LDH-A and LDH-B is not very helpful here, because they catalyze a reversible reaction. Direction of the catalyzed reaction is mainly determined by the ratio between concentrations of substrates and cofactors and not only by the type isoenzyme and supposedly to a smaller extent by enzyme affinity to substrates and co-substrates. Standardization of protocols in the investigation and keeping of animals may help to get clear understanding here. Unfortunately, information on ROS involvement in the regulation of glycolytic enzymes in the aged brain in fact is not available.
Recently, using untargeted ultrahigh performance liquid chromatography-tandem mass spectroscopy (UPLC-MS/MS), Dong and Brewer [17] found that in the mouse brain, hippocampus glycolysis and the PPP metabolites declined with age in the reactions in which ATP was consumed (hexose phosphate intermediates) and increased in the reactions involved in ATP generation (tricabon glycolytic intermediates).
Again it is clear that just a combination of different techniques with standardization of approaches may shed light on the picture of real changes in glycolysis in the brain during its aging and especially on the ROS roles in these processes.
Mitochondria produce most energy in the cell using AcCoA in TCA cycle and OxPhos and, at the same time, they produce most cellular ROS [23, 27]. Due to this, mitochondria are constantly subjected to extensive ROS attacks. It is believed that enhanced ROS production by mitochondria at the brain aging is connected with ROS-promoted damage to cellular components of which damage to proteins, lipids, and DNA is critically important [23, 28, 41, 54]. Corruption of bioenergetic efficiency of glycolysis and especially OxPhos is of special interest because some glycolytic enzymes like glyceraldehyde-3-phosphate dehydrogenase [6] and iron–sulfur components and copper-containing clusters of mitochondrial electron transport complexes [23, 59] are rather sensitive to ROS and RNS. Most accepted theories of aging are related to ROS homeostasis and slow continuous accumulation of ROS-modified molecules [23, 27, 28].
At normal brain aging, in the mouse brain hippocampus, level of AcCoA increased but levels of citrate and cis-aconitate decreased [17]. This shows that potential inactivation of citrate synthase and aconitase by ROS [40] does not affect conversion the first steps of TCA cycle. But the levels of the rest intermediates of the cycle were found to be increased in the brain aged.
Two enzymes from the TCA cycle are supposed to be the main ROS targets: aconitase and α-ketoglutarate dehydrogenase [3, 40, 58]. Experiments with housefly thoracic flight muscles indicated directly that at aging aconitase is subjected to oxidative modification by ROS which was confirmed by measurement of both, oxidative inactivation, and enzymatic activity [73]. In the rat primary mesencephalic cultures, aconitase was found to be subjected to oxidative inactivation also [8]. The activity of α-ketoglutarate dehydrogenase in the brain of the aged mice was lower than that in young ones, and this positively correlated with glutathione level, whereas levels of ROS and malonic dialdehyde, a lipid peroxidation product, showed opposite age-related changes [75]. It should be noted here that the mitochondrial α-ketoglutarate dehydrogenase complex is not only sensitive to ROS but also itself can generate ROS [68].
Available information on OxPhos in the aged brain is rather limited and contradictory. Oxygen consumption or respiration with NADH-dependent substrates and complex I activity were lower in the mitochondria from the aged rats relatively to ones from young animals [10]. This might result in an age-promoted decrease of OxPhos. In the human aged brain, decreased activity of cytochrome oxidase could also reduce OxPhos efficiency [55]. In older Fischer 344 rat brains cytochrome oxidase activity was lower than that in younger ones [27, 70]. In mice, the activity of cytochrome c oxidase (complex IV) increased with age, whereas the activities of complexes I, II, and III showed no age-related changes [65]. Cytochrome oxidase activity in synaptic mitochondria from the cerebellar cortex of adult and old monkeys lowered with aging [52]. Generally, it can be concluded that in most studied cases, oxidative phosphorylation in the brain mitochondria is decreased with aging.
It is widely accepted that mitochondria from old mammals possess not only reduced capability to produce ATP but also they are morphologically altered [63]. Moreover, they produce more ROS [53]. That results in the decline of ATP production with age. Such events are generally supported experimentally. For example, in the hippocampus of aged (15-month old) rats, the level of ATP was 38% lower than that in their 3-month old counterparts without changes in ADP level [20]. Decreased by 35% ATP/ADP ratio in the older animals indicates reduced energy status of the aged brain. Unfortunately, the level of AMP was not evaluated due to which it is impossible to calculate energy charge (Atkinson charge), and one may suggest that AMP level could be relatively increased. That can be very important for the regulation of operation of the brain by AMP-activated protein kinase (AMPK), a master regulator of energy metabolism and related to number pathologies in the brain [7, 16, 23, 44].
Recently, to reveal metabolic changes mass spectrometry-based omic technologies were applied to the normally aged mouse brain [32]. Global, metabolomic, and proteomic analyses of different regions of the brain during adult life span did not find severe proteomic imbalance, whereas demonstrated an energy metabolic drift or significant imbalance in core metabolite levels in the aged animals. General energy homeostasis showed shifts at aging: levels of NAD decreased, whereas ratios AMP/ATP and purine/pyrimidine increased, and significant alteration of OxPhos was found [32]. It was suggested that energy metabolic drift was related to a failure of the cellular machinery to restore metabostasis in the aged brain [32]. Found changes could mirror decreased capability to respond properly to external stimuli leading to alterations in operation of signaling pathways like the above-mentioned possible AMPK activation to prevent age-related changes. The above-described findings may finally lead to perturbations in neuronal function and communication.
Finally, there is a reason to mention age-related damage to mitochondria which decreases the efficiency of ATP production. The defective mitochondria are characterized by increased ROS generation which is a reason for decreased ATP production [23]. Therefore living organisms developed a strategy to remove damaged mitochondria by autophagy and produce new ones efficient in coping with ROS and in ATP production. This strategy is realized via the mitochondria quality control process and the elimination of not efficient mitochondria. To do this, the cell identifies dysfunctional mitochondria because they have lower transmembrane potential, increased ROS generation and release toxic apoptotic mediators and apparently selectively can be removed by autophagy [62]. After identification of “problematic” mitochondria, they are fused with lysosomes leading further to the formation of the phagosome. In normal aging, more and more mitochondria are damaged, and the efficiency of autophagy is decreased due to which damaged molecules and organelles are accumulated which is a typical hallmark of aging [62] Corruption of both, mitochondria operation and autophagy, may lead to pathologic aging (reviewed in [14]). Unfortunately, the information on mitochondria–lysosome crosstalk is scarce, and that is an avenue for future studies because it may result in neurodegeneration.
Disbalance hypothesis between energy provision and oxidative stress at the brain aging
Energy generation in ATP form in living organisms is strictly associated with generation of reactive species of oxygen, nitrogen, and carbonyl ones [23, 25, 42, 69, 71]. At the first stage of carbohydrate catabolism glycolysis, several glycolytic intermediates are precursors for generation of reactive 1,2-dicarbonyl compounds such as methylglyoxal [64]. At the second stage of ATP production, namely mitochondrial TCA and OxPhos, some amount of electrons escape ETC and join molecular oxygen giving rise to ROS as side products which can be further combined with RNS [25, 66]. Therefore it is clear that increased demands in energy (ATP) may stimulate glycolysis and ETC operation which inevitably leads to coupled enhanced generation of side products such as reactive carbonyl, nitrogen, and oxygen species. Here I will concentrate mainly at ROS-related processes and their interplay with energy homeostasis.
Coordinated operation of homeostasis of ROS-related processes and energy-providing (bioenergetic) ones maintain ROS and ATP levels within certain ranges (Fig. 2). These fluctuations are rather well balanced at normal adulthood. Despite that, a slow increase in the level of end products of ROS/RNS-modified molecules takes place at this period of life. However, during normal aging and many pathologies, the “idyll” described above may be disturbed. In some cases, fluctuation of ROS levels may have enhanced amplitude or extended duration (Fig. 2). In other words, ROS level may leave the stationary (steady-state) corridor, i.e., escape regular steady-state (described in details in [42]). If such events take place at rare sporadic manner, the brain may cope with such disturbances without substantial functional consequences. It is well known that during life span the level of ROS/RNS-modified cellular components is slowly increased [23, 41, 72]. The disbalance between ROS production and elimination in favor of the first may result in slow increase of their steady-state level. If such challenges take place sporadically from time to time, it may not have serious consequences for living organisms, because they have high capacity to cope with them. At middle age, frequency of such events may increase, and some of challenges may not be counterbalanced by adequate compensatory or adaptive response of energy-producing and defense systems (Fig. 2). That may result in a decrease of the potential of energy-producing systems. Slow accumulation of ROS-modified molecules particularly such proteins takes place up to about half of life span, but later, it increases faster [38, 41]. Periodic increase in ROS level may corrupt antioxidant and energy-producing systems which results in both: (1) slow increase of steady-state ROS level and (2) decrease in the brain capacity to produce energy (Fig. 2). Therefore the range of fluctuation of ROS steady-state level is slowly going up, whereas the range of fluctuation of available energy (ATP)-producing capacity would be slowly going down. Moreover, vicious cycles are formed because ROS/RNS at enhanced levels inactivate energy-producing systems, whereas reduced efficiency of energy-producing systems may not provide enough energy to cover efficient operation of antioxidant defense systems. Due to this, both duration and amplitude of such fluctuations may increase during advanced aging and lead to disbalance causing the aging process.

Age-promoted divergence of two fluctuation zones in the brain: ROS-related and bioenergetic processes and amplification of vicious cycles formed by ROS-energy-providing systems. Description in the text
During life span, both corridors would separate, and formed between them, zone (Fig. 2 in gray) would characterize deviation of the system from normal operation. Divergence of two fluctuation zones (ROS/RNS-related and bioenergetic processes) makes the system unstable and more sensitive to diverse challenges. It may amplify vicious cycles formed by ROS/RNS production and disruption of energy-providing systems, increasing their amplitude and frequency. Age-related increase in the levels of ROS/RNS-modified molecules and their aggregates formed by modified proteins, lipids, and DNA (like lipofuscin) takes place. Some of these complexes can generate ROS being responsible for age-related increase in ROS production. Such a scenario results in further divergence of two zones making “gray zone” bigger that really reflects the aging process and can be its characteristic. Potentially the systems may return to some extent to initial levels due to disruption of vicious cycles (some sort of rejuvenation), but during a normal life span, the situation is worsened. At the least, the level of ROS-modified proteins starts to sharply increase at the last trimester of life [38, 41]. This clearly reflects processes taking place at advanced aging. So, at this age, frequency of boots and regular ROS levels become higher than those in previous stages of life, and such intensification of oxidative/nitrosative stress and decreased capacity to produce ATP are clearly characteristics of the aging brain [5, 25, 47].
The question which arises here is as follows: Why in different cases no clear signatures of intensification of oxidative stress and decrease in capacity to produce ATP in the aged brain have been found? This topic was partially covered by us earlier [23], and here I will briefly highlight the problem. To a big extent it is connected with the brain specificity concerning oxidative stress: (1) there is no reliable approach to evaluate accurately ROS/RNS level and, consequently, intensity of oxidative stress in the living organism; (2) markers used to evaluate oxidative stress intensity (intermediates or end products) have different spatiotemporal patterns; (3) it is not clear how defense systems operate to neutralize different ROS/RNS types in different cells and intracellular space; and, finally, (4) understanding of the interaction between different cell types in very complex cell specialization in the brain adds difficulties. The latter also adds complexity in the analysis of energy-providing processes in the brain. For example, the brain energy needs are mainly covered by glucose but to a minor extent also by KB and free fatty acids. Lactate and aspartate also can be taken by the brain and used to produce ATP. In addition, lactate operates as a signaling molecule. Both, cytosolic glycolysis and mitochondrial OxPhos are responsible for ATP production, and both pathways are inactivated by ROS/RNS, which is counterbalanced to a certain extent by de novo production to replace damaged components. Again, intracellular specialization adds complications.
Conclusions and perspectives
Energy in the ATP form is needed to create and maintain brain structure and provide its operation. High-intensity metabolism is accompanied by high production of ROS/RNS damaging any brain components. The situation in the brain is worsened by a high content of polyunsaturated fatty acids which are highly susceptible to ROS/RNS-initiated peroxidation [23]. So such brain peculiarities give rise to the question: which brain systems are so efficient in preventing age-related intensification of oxidative stress? Unfortunately, to date, such brain-specific systems have not been identified, and it is the avenue for future studies.
Many studies have demonstrated that brain aging is associated with the intensification of oxidative stress, but a less substantial amount of studies found no such intensification [23, 71, 73]. Similarly no clear situation takes place with energy-providing processes. The activity of certain glycolytic enzymes like glyceraldehyde-3-phosphate dehydrogenase may decrease at aging as well as operation of mitochondrial OxPhos does. It is well known that the production of ATP in mitochondria relies mainly on the operation of membrane electron gradient generated and utilized by operation of five iron–sulfur-rich protein megacomplexes located in the inner mitochondrial membrane. In young organisms, brain iron level is under strict control, but in advancing age, iron homeostasis shifts to enhance free, unbound iron which may be at least partially responsible for the age-promoted increase in ROS levels leading to intensification of oxidative stress [59]. But these data are controversial which to a big extent can be connected with models used and complex structural and functional architecture of the brain. Using different protocols of investigation adds complications.
The questions to be solved to clarify age-related changes in the aging brain with a focus on ROS-related processes are as follows: (1) Which mechanisms are responsible for keeping oxidative stress at low intensity? (2) Which regulatory mechanisms are responsible for the adjustment of antioxidant mechanisms to needs of the brain? (3) How different brain cell types cooperate to function in the environment highly susceptible to oxidative stress? Another group of perspective questions in the brain aging is related to energy (ATP) production. To my opinion, the most important key questions here are as follows: (1) Why do energy-producing mechanisms operate so efficiently at highly intensive generation of reactive species and (2) which regulatory mechanisms are responsible for the coordination of functioning of energy-producing mechanisms and high brain energy demands in combination with ROS-related processes? And finally, the most intriguing integrating question is as follows: Which rescue and repair mechanisms should be targeted to disrupt progress or even reverse (eternal theme of rejuvenation) aging via prevention or reduction of amplitude and frequency of vicious cycle oxidative stress ⬅➔ energy provision? One of the potential candidates for this was mentioned above—transcription regulator Nrf2—but, for sure, in cooperation with a number of other regulatory pathways and this warrants further investigation in the aging field.
Data Availability
Not applicable.
Code availability
Not applicable.
Abbreviations
- AcCoA:
-
Acetyl coenzyme A
- ETC:
-
Electron transport chain
- KB:
-
Ketone bodies
- LPO:
-
Lipid peroxidation
- NMR:
-
Nuclear magnetic resonance
- OxPhos:
-
Oxidative phosphorylation
- PPP:
-
Pentose phosphate pathway
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- TCA cycle:
-
Tricarboxylic acid cycle
References
Abi-Saab WM, Maggs DG, Jones T, Jacob R, Srihari V, Thompson J, Kerr D, Leone P, Krystal JH, Spencer DD, During MJ, Sherwin RS (2002) Striking differences in glucose and lactate levels between brain extracellular fluid and plasma in conscious human subjects: effects of hyperglycemia and hypoglycemia. J Cereb Blood Flow Metab 22(3):271–279. doi:https://doi.org/10.1097/00004647-200203000-00004
Auestad N, Korsak RA, Morrow JW, Edmond J (1991) Fatty acid oxidation and ketogenesis by astrocytes in primary culture. J Neurochem 56(4):1376–1386. https://doi.org/10.1111/j.1471-4159.1991.tb11435.x
Berndt N, Bulik S, Holzhütter H-G (2012) Kinetic modeling of the mitochondrial energy metabolism of neuronal cells: the impact of reduced α-ketoglutarate dehydrogenase activities on ATP production and generation of reactive oxygen species. Int J Cell Biol 2012:757594–757511. https://doi.org/10.1155/2012/757594
Brandes MS, Gray NE (2020) NRF2 as a therapeutic target in neurodegenerative diseases. ASN Neuro 12:1–23. https://doi.org/10.1177/1759091419899782
Butterfield DA (2020) Brain lipid peroxidation and alzheimer disease: synergy between the Butterfield and Mattson laboratories. Ageing Res Rev In press 101049:101049. https://doi.org/10.1016/j.arr.2020.101049
Butterfield DA, Hardas SS, Lange MLB (2010) Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer disease: many pathways to neurodegeneration. J Alzheimers Dis 20(2):369–393. https://doi.org/10.3233/JAD-2010-1375
Cai Z, Yan L, Li K et al (2012) Roles of AMP-activated protein kinase in Alzheimer's disease. Neuromol Med 14:1–14. https://doi.org/10.1007/s12017-012-8173-2
Cantu D, Schaack J, Patel M (2009) Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS One 4(9):e7095. https://doi.org/10.1371/journal.pone.0007095
Chen R, Lai UH, Zhu L, Singh A, Ahmed M, Forsyth NR (2018) Reactive oxygen species formation in the brain at different oxygen levels: the role of hypoxia inducible factors. Front Cell Dev Biol 6: 132. https://doi.org/10.3389/fcell.2018.00132
Cocco T, Sgobbo P, Clemente M, Lopriore B, Grattagliano I, Di PM, Villani G (2005) Tissue-specific changes of mitochondrial functions in aged rats: effect of a long-term dietary treatment with N-acetylcysteine. Free Radic Biol Med 38:796–805. https://doi.org/10.1016/j.freeradbiomed.2004.11.034
Cunnane S, Nugent S, Roy M (2011) Brain fuel metabolism, aging, and Alzheimer's disease. Nutrition 27(1):3–20. https://doi.org/10.1016/j.nut.2010.07.021
Cunnane SC, Courchesne-Loyer A, St-Pierre V, Vandenberghe C, Pierotti T, Fortier M, Croteau E, Castellano C-A (2016) Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer's disease. Ann N Y Acad Sci 1367(1):12–20. https://doi.org/10.1111/nyas.12999
De Backer I, Hussain SS, Bloom SR, Gardiner JV (2016) Insights into the role of neuronal glucokinase. Am J Physiol Endocrinol Metab 311(1):E42–E55. https://doi.org/10.1152/ajpendo.00034.2016
Deus CM, Yambire KF, Oliveira PJ, Raimundo N (2020) Mitochondria-lysosome crosstalk: from physiology to neurodegeneration. Trends Mol Med. 26(1):71–88. https://doi.org/10.1016/j.molmed.2019.10.009
De Vries MG, Arseneau LM, Lawson ME, Beverly JL (2003) Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes 52(11):2767–2773. https://doi.org/10.2337/diabetes.52.11.2767
Domise M, Didier S, Marinangeli C, Zhao H, Chandakkar P, Buée L, Viollet B, Davies P, Marambaud P, Vingtdeux V (2016) AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci Rep 6:26758. https://doi.org/10.1038/srep26758
Dong Y, Brewer GJ (2019) Global metabolic shifts in age and Alzheimer's disease mouse brains pivot at NAD+/NADH redox sites. J Alzheimers Dis 71(1):119–140. https://doi.org/10.3233/JAD-190408
Dringen R, Gebhardt R, Hamprecht B (1993) Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Res 623(2):208–214. https://doi.org/10.1016/0006-8993(93)91429-V
Duarte JM, Do KQ, Gruetter R (2014) Longitudinal neurochemical modifications in the aging mouse brain measured in vivo by 1H magnetic resonance spectroscopy. Neurobiol Aging 35(7):1660–1668. https://doi.org/10.1016/j.neurobiolaging.2014.01.135
El-Sawalhi MM, Darwish HA, Mausouf MN, Shaheen AA (2013) Modulation of age-related changes in oxidative stress markers and energy status in the rat heart and hippocampus: a significant role for ozone therapy. Cell Biochem Funct 31(6):518–525. https://doi.org/10.1002/cbf.2930
Erecinska M, Silver IA (1989) ATP and brain function. J Cereb Blood Flow Metab 9(1):2–19. https://doi.org/10.1038/jcbfm.1989.2
Forester BP, Berlow YA, Harper DG, Jensen JE, Lange N, Froimowitz MP, Ravichandran C, Iosifescu DV, Lukas SE, Renshaw PF, Cohen BM (2010) Age-related changes in brain energetic and phospholipid metabolism. NMR Biomed 23(3):242–250. https://doi.org/10.1002/nbm.1444
Garaschuk O, Semchyshyn HM, Lushchak VI (2018) Healthy brain aging: interplay between reactive species, inflammation and energy supply. Ageing Res Rev 43:26–45. https://doi.org/10.1016/j.arr.2018.02.003
Gaspar JM, Velloso LA (2018) Hypoxia inducible factor as a central regulator of metabolism – implications for the development of obesity. Front Neurosci 12:813. https://doi.org/10.3389/fnins.2018.00813
Grimm A, Eckert A (2017) Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem 143(4):418–431. https://doi.org/10.1111/jnc.14037
Guzmán M, Blázquez C (2004) Ketone body synthesis in the brain: possible neuroprotective effects. Prostaglandins Leukot Essent Fatty Acids 70(3):287–292. https://doi.org/10.1016/j.plefa.2003.05.001
Haripriya D, Devi MA, Kokilavani V, Sangeetha P, Panneerselvam C (2004) Age dependent alterations in mitochondrial enzymes in cortex, striatum and hippocampus of rat brain – potential role of L-carnitine. Biogerontology 5(5):355–364. https://doi.org/10.1007/s10522-004-2575-y
Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11(3):298–300
Harris RA, Tindale L, Lone A, Singh O, Macauley SL, Stanley M, Holtzman DM, Bartha R, Cumming RC (2016) Aerobic glycolysis in the frontal cortex correlates with memory performance in wild-type mice but not the APP/PS1 mouse model of cerebral amyloidosis. J Neurosci 36(6):1871–1878. https://doi.org/10.1523/JNEUROSCI.3131-15.2016
Hunsberger HC, Greenwood BP, Tolstikov V, Narain NR, Kiebish MA, Denny CA (2020) Divergence in the metabolome between natural aging and Alzheimer's disease. Sci Rep 10:12171. https://doi.org/10.1038/s41598-020-68739-z
Infanger DW, Sharma RV, Davisson RL (2006) NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal 8:9–10. https://doi.org/10.1089/ars.2006.8.1583
Ivanisevic J, Stauch KL, Petrascheck M, Benton HP, Epstein AA, Fang M, Gorantla S, Tran M, Hoang L, Kurczy ME, Boska MD, Gendelman HE, Fox HS, Siuzdak G (2016) Metabolic drift in the aging brain. Aging (Albany, NY) 8(5):1000–1020. https://doi.org/10.18632/aging.100961
Kaiser LG, Hirokazu K, Fukunaga M, Matson GB (2016) Detection of glucose in the human brain with 1H MRS at 7 Tesla. Magn Reson Med 76:1653–1660. https://doi.org/10.1002/mrm.26456
Karaca M, Frigerio F, Migrenne S, Martin-Levilain J, Skytt DM, Pajecka K, Martin-del-Rio R, Gruetter R, Tamarit-Rodriguez J, Waagepetersen HS, Magnan C, Maechler P (2015) GDH-dependent glutamate oxidation in the brain dictates peripheral energy substrate. Cell Rep 13(2):365–375. https://doi.org/10.1016/j.celrep.2015.09.003
Koepsell H (2020) Glucose transporters in brain in health and disease. Pflugers Arch - Eur J Physiol. 472:1299–1343. https://doi.org/10.1007/s00424-020-02441-x
Le Foll C, Levin BE (2016) Fatty acid-induced astrocyte ketone production and the control of food intake. Am J Physiol 310(11):R1186-R1192. https://doi.org/10.1152/ajpregu.00113.2016, 310
Lesnefsky EJ, Hoppel CL (2006) Oxidative phosphorylation and aging. Ageing Res Rev 5(4):402–433. https://doi.org/10.1016/j.arr.2006.04.001
Levine RL, Stadtman ER (2001) Oxidative modification of proteins during aging. Exp Gerontol 36(9:1495–1502. https://doi.org/10.1016/S0531-5565(01)00135-8
Liemburg-Apers DC, Willems PHGM, Koopman WJH, Grefte S (2015) Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch Toxicol 89:1209–1226. https://doi.org/10.1007/s00204-015-1520-y
Lushchak OV, Piroddi M, Galli F, Lushchak VI (2014) Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep 19(1):8–15. https://doi.org/10.1179/1351000213Y.0000000073
Lushchak VI (2007) Free radical oxidation of proteins and its relationship with functional state of organisms. Biochemistry (Mosc) 72(8):809–827. https://doi.org/10.1134/S0006297907080020
Lushchak VI (2014) Free radicals, reactive oxygen species, oxidative stress and its classification. Chem Biol Interact 224:164–175. https://doi.org/10.1016/j.cbi.2014.10.016
Magistretti PJ, Allaman I (2015) A cellular perspective on brain energy metabolism and functional imaging. Neuron. 86(4):883–901. https://doi.org/10.1016/j.neuron.2015.03.035
Martínez de Morentin PB, Urisarri A, Couce ML, López M (2016) Molecular mechanisms of appetite and obesity: a role for brain AMPK. Clin Sci (Lond) 130(19):1697–1709. https://doi.org/10.1042/CS20160048
Mason S (2017) Lactate shuttles in neuroenergetics—homeostasis, allostasis and beyond. Front Neurosci 11:43. https://doi.org/10.3389/fnins.2017.00043
Mattson MP (2012) Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab 16(6):706–722. https://doi.org/10.1016/j.cmet.2012.08.012
Mattson MP, Arumugam TV (2018) Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab 27(6):1176–1199. https://doi.org/10.1016/j.cmet.2018.05.011
McKenna MC, Scafidi S, Robertson CL (2015) Metabolic alterations in developing brain after injury: knowns and unknowns. Neurochem Res 40:2527–2543. https://doi.org/10.1007/s11064-015-1600-7
Melo HM, Santos LE, Ferreira ST (2019) Diet-derived fatty acids, brain inflammation, and mental health. Front Neurosci 13:265. https://doi.org/10.3389/fnins.2019.00265
Mueckler M, Thorens B (2013) The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med 34(2-3):121–138. https://doi.org/10.1016/j.mam.2012.07.001
Nakajima S, Kunugi H (2020) Lauric acid promotes neuronal maturation mediated by astrocytes in primary cortical cultures. Heliyon 6(5):e03892. https://doi.org/10.1016/j.heliyon.2020.e03892
Naudí A, Caro P, Jové M, Gómez J, Boada J, Ayala V, Portero-Otín M, Barja G, Pamplona R (2007) Methionine restriction decreases endogenous oxidative molecular damage and increases mitochondrial biogenesis and uncoupling protein in rat brain. Rejuvenation Res 10(4):473–484. https://doi.org/10.1089/rej.2007.0538
Nissanka N, Moraes CT (2018) Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett 592:728–742. https://doi.org/10.1002/1873-3468.12956
Ogunnowo-Bada EO, Heeley N, Brochard L, Evans ML (2014) Brain glucose sensing, glucokinase and neural control of metabolism and islet function. Diabetes Obes Metab 16(1):26–32. https://doi.org/10.1111/dom.12334
Ojaimi J, Masters CL, Opeskin K, McKelvie P, Byrnea E (1999) Mitochondrial respiratory chain activity in the human brain as a function of age. Mech Ageing Dev 111(1):39–47. https://doi.org/10.1016/S0047-6374(99)00071-8
Paolino N, Massimiliano D, Samuela C, Enrica B (2001) Rabbit brain glucose-6-phosphate dehydrogenase: biochemical properties and inactivation by free radicals and 4-hydroxy-2-nonenal. Neuroreport 12(18):4149–4153. https://doi.org/10.1097/00001756-200112210-00057
Phyu S, Tseng C, Fleming I et al (2016) Probing the PI3K/Akt/mTor pathway using 31P-NMR spectroscopy: routes to glycogen synthase kinase 3. Sci Rep 6:36544. https://doi.org/10.1038/srep36544
Quijano C, Trujillo M, Castro L, Trostchansky A (2016) Interplay between oxidant species and energy metabolism. Redox Biol 8:28–42. https://doi.org/10.1016/j.redox.2015.11.010
Raz N, Daugherty AM (2018) Pathways to brain aging and their modifiers: free-radical-induced energetic and neural decline in senescence (FRIENDS) model. Gerontology 64:49–57. https://doi.org/10.1159/000479508
Rich L, Brown AM (2016) Glycogen: multiple roles in the CNS. Neuroscientist 23(4):356–363. https://doi.org/10.1177/1073858416672622
Riske L, Thomas RK, Baker GB, Dursun SM (2017) Lactate in the brain: an update on its relevance to brain energy, neurons, glia and panic disorder. Ther Adv Psychopharmacol 7(2):85–89. doi:https://doi.org/10.1177/2045125316675579
Rubinsztein DC, Mariño G, Kroemer G (2011) Autophagy and aging. Cell. 146(5):682–695. https://doi.org/10.1016/j.cell.2011.07.030
Salman M, Tabassum H, Parvez S (2020) Nrf2/HO-1 mediates the neuroprotective effects of pramipexole by attenuating oxidative damage and mitochondrial perturbation after traumatic brain injury in rats. Dis Model Mech 13(8):dmm045021. https://doi.org/10.1242/dmm.045021
Semchyshyn HM (2014) Reactive carbonyl species in vivo: generation and dual biological effects. Sci World J 2014:417842–417810. https://doi.org/10.1155/2014/417842
Sharman EH, Bondy SC (2001) Effects of age and dietary antioxidants on cerebral electron transport chain activity. Neurobiol Aging 22(4):629–634. https://doi.org/10.1016/S0197-4580(01)00226-3
Sies H, Jones DP (2020) Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 21:363–383. https://doi.org/10.1038/s41580-020-0230-3
Silver IA, Erecinska M (1994) Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci 14(8):5068–5076. https://doi.org/10.1523/JNEUROSCI.14-08-05068.1994
Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF (2004) Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. Neurosci. 24(36):7779–7788. https://doi.org/10.1523/JNEUROSCI.1899-04.2004
Stefanatos R, Sanz A (2018) The role of mitochondrial ROS in the aging brain. FEBS Lett 592:743–758. https://doi.org/10.1002/1873-3468.12902
Tian L, Cai Q, Wei H (1998) Alterations of antioxidant enzymes and oxidative damage to macromolecules in different organs of rats during aging. Free Radic Biol Med 24(9):1477–1484. https://doi.org/10.1016/S0891-5849(98)00025-2
Tretter L, Adam-Vizi V (2005) Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Phil Trans R Soc B 360:2335–2345. https://doi.org/10.1098/rstb.2005.1764
Walsh ME, Shi Y, Van Remmen H (2014) The effects of dietary restriction on oxidative stress in rodents. Free Radic Biol Med 66:88–99. https://doi.org/10.1016/j.freeradbiomed.2013.05.037
Yan L-J, Levine RL, Sohal RS (1997) Oxidative damage during aging targets mitochondrial aconitase. Proc Natl Acad Sci U S A 94(21):11168–11172. https://doi.org/10.1073/pnas.94.21.11168
Yin F, Sancheti H, Patil I, Cadenas E (2016) Energy metabolism and inflammation in brain aging and Alzheimer's disease. Free Radic Biol Med 100:108–122. https://doi.org/10.1016/j.freeradbiomed.2016.04.200
Zhao Z, Yu Z, Hou Y, Zhang L, Fu A (2020) Improvement of cognitive and motor performance with mitotherapy in aged mice. Int J Biol Sci 16(5):849–858. https://doi.org/10.7150/ijbs.40886
Acknowledgments
The author would like to thank Drs. H. Semchyshyn and M. Bayliak for critical reading of the manuscript and two anonymous reviewers for their careful reading of the manuscript and their many constructive comments with suggestions that resulted in better presentation of the material.
Funding
This work was partially supported by a grant #90233 from the Volkswagen Foundation (VolkswagenStiftung, Germany) and a grant #0118U003477 from the Ministry of Education and Science of Ukraine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Conflict of interest
The author declares no conflict of interest.
Consent to participate
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue on Aging Brain in Pflügers Archiv—European Journal of Physiology
Rights and permissions
About this article

Cite this article
Lushchak, V.I. Interplay between bioenergetics and oxidative stress at normal brain aging. Aging as a result of increasing disbalance in the system oxidative stress–energy provision. Pflugers Arch - Eur J Physiol 473, 713–722 (2021). https://doi.org/10.1007/s00424-021-02531-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-021-02531-4