
Abstract
While the vast majority of the genome is transcribed into RNA, only a small fraction of these transcripts have protein-coding potential. A large fraction of the transcribed RNA belongs to the class known as long non-coding RNAs (lncRNAs). Several recent studies have shown that at least some of these lncRNA transcripts represent functional RNA molecules. LncRNAs can utilize a wide range of mechanisms to regulate the RNA and/or the protein content of a cell on the transcriptional and the post-transcriptional levels. So far, many studies have identified differentially expressed lncRNAs in various physiological contexts, genetic disorders and human diseases. A steadily increasing number of studies could establish functional roles for some of these lncRNAs in developmental processes, cancer and tissue homeostasis. Taken together, these functions provide an additional layer of gene regulation and contribute to the high complexity of physiological and disease-related phenotypes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The advent of high-resolution microarrays and unbiased transcriptome analysis using massive parallel sequencing technologies has provided the opportunity to identify and characterize most, if not all, RNA species in humans and various animal models, including mice. The finding that ~70 % of the human genome is actively transcribed, while only 2 % of the total genome is represented in the ~20,000 protein-coding transcripts, has revolutionized our view of genome organization and content [17]. The vast amount of transcripts that lack protein-coding potential are collectively referred to as non-coding RNAs (ncRNAs). Many of these ncRNAs are well-characterized, constitutively expressed “housekeeping” RNAs, such as transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs) involved in translation, small nuclear RNAs (snRNAs) involved in splicing regulation, and small nucleolar RNAs (snoRNAs) involved in ribosome biogenesis [18]. In recent years, it has become apparent that many ncRNAs are involved in regulating the RNA and protein layout of cells. The classes of ncRNAs that have currently received the most attention are microRNAs (miRNAs), small interfering RNAs (siRNAs), or PIWI-interacting RNAs (piRNAs), collectively referred to as short ncRNAs (18–31 nt) [19]. The role of miRNAs in post-transcriptional gene regulation by targeting mRNAs and thus repressing translation has been extensively described. There is also an abundance of studies demonstrating the role of miRNAs in cancer [88], cardiovascular [86], neurological and other diseases [76]. Based on these studies, miRNAs have been proposed as promising prognostic, diagnostic, and treatment targets.
Although the current body of scientific literature is dominated by studies featuring short ncRNAs, the majority of mammalian non-coding transcripts encompass RNAs longer than 200 nt; currently referred to as long non-coding RNAs (lncRNAs) [59]. LncRNAs were first described during the large-scale sequencing of full-length cDNA libraries in the mouse [12, 87]. Given the important functional roles uncovered for small ncRNAs, these lncRNAs have received increasing attention in recent years with respect to their possible functions.
Current genome annotation efforts estimate the number of lncRNA genes to be around 16,000 in the human genome (GENCODE 24) and around 9000 in the mouse genome (GENCODE M8). Because the annotation process is ongoing, these numbers are likely to increase. In addition, since one gene can provide a template for several different splice variants of a lncRNA, the number of lncRNA transcripts exceeds the number of genes. It is still a matter of debate whether these transcripts represent transcriptional “noise” and are simply an outcome of transcriptional regulatory processes, or if the transcripts themselves have a specific function [2]. Although the vast majority of long non-coding RNAs have yet to be characterized thoroughly, several studies have clearly demonstrated that some lncRNAs can indeed be functional. They exhibit developmental and tissue-specific expression patterns, localize to specific subcellular compartments, and participate in various mechanisms of gene regulation during development, cell differentiation [16], imprinting [49], apoptosis and cell cycle [84], and metabolism and aging [68]. In addition, deregulated expression of lncRNAs has been associated with a variety of diseases, including cancer. While these studies have indeed shown that lncRNAs serve biological roles, the majority were performed in vitro. Therefore, the precise in vivo biological roles and mechanisms for lncRNAs in normal cellular processes during development, differentiation, and disease progression remains, for the most part, less clear.
In this review, we present an overview of the different types of lncRNAs, their general characteristics and their mechanisms-of-action as transcriptional and post-transcriptional regulators. We highlight the emerging role of lncRNAs in both normal physiology and disease states. We have mainly focused on studies that have used either in vivo models, patient samples, or ex vivo models to provide evidence for the functional relevance of lncRNAs in development, cell differentiation, cancer, genetic disorders, and other diseases (Table 1). Additionally, we address some critical points that should be taken into consideration for the functional analysis of novel lncRNAs. The discovery and study of lncRNAs is relevant to furthering our understanding of human biology and diseases, since this heterogeneous and largely unexplored class of transcripts represents new molecules with functional mechanisms that could aid in the explanation of organismal and disease complexity.
Characteristics of lncRNA transcripts
The class of lncRNAs is a very heterogeneous collection of RNA transcripts, which is solely defined as any RNA having an obvious lack of open reading frame (ORF) and a minimum length of 200 nucleotides. LncRNA genes are mainly transcribed by RNA polymerase II, and are processed similarly to mRNAs, including 5′-end capping, 3′-end polyadenylation, splicing of introns, and intracellular transport. Although polyadenylation occurs, it is not the rule, as there are examples of lncRNAs such as the antisense asOct4-pg5, or the brain-associated BC200, which are functional, but not polyadenylated [13]. In general, lncRNAs contain a lesser number of exons than mRNAs and are generally shorter, with median length of 592 nt, compared to 2453 nt for mRNAs [12, 29]. Their predicted ORFs have poor start codon and ORF contexts; hence translation of lncRNAs is less probable but not completely unlikely [32]. The functions of lncRNAs are mainly ascribed to their sequence homology/complementarity with other nucleic acids, and by structure; forming molecular frames and scaffolds for assembly of macromolecular complexes. Some lncRNA transcripts are processed into smaller RNAs, and each mature transcript can display a distinct function [22]. Their sequences are less conserved than mRNAs among related species, but it seems that their positions in relation to neighboring protein-coding genes and secondary structures are often maintained [13].
LncRNAs fold into double and single stranded stems, loops and bulges, which can fold further into three-dimensional structures, resulting in highly structured macromolecules with a variety of shapes. Novel methods like SHAPE-MaP (Selective 2′-Hydroxyl Acylation analyzed by Primer Extension and Mutational Profiling) allow the determination of secondary structures in living cells on a genome-wide scale [73]. Many lncRNAs are localized to the nucleus, but there are also lncRNAs enriched in the cytoplasm. Their localization within a cell can be used as an indication of their function. The lncRNAs examined to date also show more tissue-specific expression patterns compared to protein-coding genes which, in combination with their lower expression levels across different tissues, raises the question as to whether there is a low abundance of lncRNA in all cells of a tissue, or rather a specific subpopulation of cells in the tissue preferentially expresses the lncRNA [13].
Genomic organization of lncRNA genes
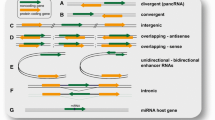
As far as the genomic architecture is concerned, lncRNA genes are positioned in various genomic regions with respect to mRNA coding genes. Generally, the genomic localization of lncRNA genes can be classified into three large groups according to their proximity to protein-coding genes (group I), to gene-regulatory regions (group II) and to specific chromosomal regions (group III) (Fig. 1). The first group is sub-classified into (1) exonic lncRNAs, when the lncRNA overlaps one or more exons of a protein-coding gene and can be transcribed in the sense or antisense direction relative to the mRNA (these antisense lncRNAs are also called natural antisense transcripts, NATs), (2) bidirectional lncRNAs when the transcription of the lncRNA and a neighboring protein-coding gene on the opposite strand is initiated in close genomic proximity, (3) intronic lncRNAs, when the lncRNA is transcribed from an intron of a protein-coding gene, and (4) intergenic lncRNAs, when the lncRNA is transcribed completely within an intergenic genomic region between protein-coding genes [9]. The second group is sub-classified into (1) 3′-UTR-associated lncRNAs, also called uaRNAs, when the lncRNA is transcribed from the 3′-untranslated region of a protein-coding gene [60], (2) promoter-associated lncRNAs, also called pRNAs or plncRNAs, when the lncRNA is transcribed from the promoter domain upstream of a protein-coding gene [37] [56] (3) enhancer lncRNAs, when the lncRNA is transcribed from an enhancer, termed eRNAs if they are transcribed into both directions or elncRNAs if they are transcribed unidirectionally [56, 64]. The third group includes, for example, the telomeric repeat-containing lncRNAs referred to as TERRA [1]. Analyzing the genomic context of lncRNAs can provide indications as to their functional roles, as lncRNAs often regulate genes located in cis, even though they act via a trans mechanism (see below).

Genomic organization of lncRNA coding genes according to their proximity to protein-coding genes, regulatory elements, and chromosomal regions. LncRNAs are depicted in red, protein-coding genes in grey, enhancers in black, and promoters in blue. The arrows indicate the direction of transcription
The histone methylation pattern at lncRNA loci is similar to that at protein-coding genes. These include histone marks at regulatory elements of lncRNAs genes, such as H3K4me2/3 (histone 3 lysine 4 di-/tri-methylation), H3K9ac (histone 3 lysine 9 acetylation), and H3K27ac at the upstream regulatory elements and promoters, and H3K36me3 over the actively transcribed region. They are, hence, subject to common regulatory machinery, however, lncRNAs which are transcribed from enhancer regions harbor histone marks found typically at enhancers [56].
Molecular functions of lncRNA transcripts
LncRNAs are able to regulate the expression of target genes through transcriptional and post-transcriptional mechanisms (Fig. 2). They influence histone modifications, chromatin remodeling (nucleosome positioning), and transcription factor binding to influence gene expression on the transcriptional level. When lncRNAs physically associate with regulatory protein complexes, they can guide them, stabilize them, or evict them from specific genomic regions to establish and maintain the active or inactive chromatin state for target gene expression. They are able to bind and target components of the Polycomb (PcG) repressive group, which is responsible for tri-methylation of histone H3 at lysine 27 (H3K27me3), or components of the Mixed-lineage Leukemia/Trithorax activating group (Mll/TrxG), which is responsible for the tr i-methylation of histone H3 at lysine 4 (H3K4me3), to specific loci. It has been also shown that lncRNAs can recruit DNA methyltransferases (DNMTs) and the LSD1/REST/coREST complex in order to silence specific loci [79]. They can also coordinate the actions of these chromatin influencing complexes by acting as molecular scaffolds for different histone modifying complexes, for example, interacting both with histone methylases and demethylases [79]. Targeting of these histone modifying complexes to specific loci is under tight spatial and temporal control during development, and it is unclear how they are able to precisely locate their target genes, since most do not possess sequence-specific DNA-binding subunits [27]. Therefore, the involvement of lncRNAs able to both bind and, via other functional domains, target these complexes to specific loci, could provide specificity for the activity of these histone modifying complexes. The ability to provide single locus specificity distinguishes lncRNAs as key components of regulatory networks in comparison to transcription factors and small ncRNAs (see below).

Summary of lncRNA function at transcriptional level (1–7) and post-transcriptional level (8–12). DNMT DNA methyltransferases, Ac acetylated chromatin
LncRNAs can also regulate gene expression directly through binding to transcription factors and/or polymerase II, and thereby either titrating them away or guiding them to target loci [29]. They can also indirectly influence gene expression by regulating the localization of proteins involved in transcription, for example by facilitating or preventing their nuclear localization [50, 72, 89]. Conclusively, the collaboration between locus specific lncRNAs, transcription factors and chromatin modifiers is essential for spatial and temporal regulation of gene expression. However, in addition to acting as key regulators of gene expression, lncRNAs can also function as key structural components by forming complexes with other nucleic acids, e.g., RNA:RNA hybridization, RNA:DNA hybridization, and RNA:dsDNA triplex formation.
With respect to post-transcriptional regulation, lncRNAs can again act in a variety of ways. They have been implicated in pre-mRNA splicing, by influencing the distribution of splicing regulatory proteins and directing alternative splicing in a cell type-specific manner [78, 92] and in mRNA decay, by influencing stabilization and degradation of the mRNA. They are able to hybridize with target mRNA, and hence prevent decay [20], and have also been reported to function jointly with STAU1 to promote the decay of Alu-containing mRNAs [46] [26]. LncRNAs have the potential to either repress or activate translation via interaction with partially complementary mRNAs and recruitment of translation repressors, or ribosomes to enhance translation [10, 82, 93]. In other cases, lncRNAs regulate post-transcriptional processes by functional association with miRNAs wherein they can “sponge” away microRNAs or compete for the mRNA target to increase translation, or even give rise to microRNAs and hence decrease mRNA translation [11, 21, 43].
Given the great variety of functional mechanisms for lncRNAs, they are able to act in cis, in trans, and in some cases even in both ways. Cis-acting lncRNAs are restricted to the site of synthesis and directly act on one or several neighboring genes on the same chromosome from which it is transcribed. Moreover, the transcription of the lncRNA rather than the transcript itself might account for the function. Trans-acting lncRNAs diffuse from the site of synthesis and can act directly on many genes on different chromosomes. Therefore, trans-acting lncRNAs are more likely to be involved in large gene-regulatory networks. Gene specific targeting seems to happen naturally for cis-acting lncRNAs because they are tethered to the site of transcription, while for trans-acting lncRNAs the formation of RNA:DNA triplex via Hoogsteen base-pairing is one possible mechanism [31].
LncRNAs involved in dosage compensation and imprinting
The first functions that were ascribed to lncRNAs were dosage compensation (Xist) and genomic imprinting (H19), and these have been extensively described and reviewed elsewhere [49]. Briefly, X-inactive-specific transcript (XIST/Xist) is expressed only from the inactive X chromosome (Xi) and acts in cis to promote the silencing and dosage compensation processing of the X chromosome [67]. Xist binds the PRC2 complex and targets it to X chromosome by interacting with YY1. Two other lncRNAs control Xist action, Tsix (negatively) and Jpx (positively). Male mice lacking Xist are unaffected whereas females die during the first half of embryonic development.
The first imprinted lncRNA identified was H19. H19 expression ensures that only one allele of the paternal Igf2 (Insulin-like growth factor 2) will be expressed [3]. Upon H19 deletion, Igf2 expression, if maternally inherited, is increased in accordance with body weight. Deletion of one copy of the Igf2 gene rescues the phenotype [51]. H19 knockout mice are viable and fertile but have reduced muscle regeneration capacity mediated by loss of two miRNAs that are encoded within the H19 transcript [14].
LncRNAs involved in development and cell differentiation
Embryo development and cell differentiation require strict spatiotemporal regulation of gene expression and lncRNAs are essential regulators of gene-regulatory networks in these processes. A remarkable example of how lncRNAs can coordinate the function of opposing chromatin modification complexes on target genes in order to keep their expression level balanced is Fendrr (fetal-lethal non-coding developmental regulatory RNA, alternative: Foxf1 adjacent non-coding developmental regulatory RNA). It is divergently expressed from the Foxf1 promoter in the lateral plate mesoderm of the early developing embryo. Surprisingly, Fendrr interacts with both PRC2 and WDR5 (a member of the TrxG/MLL complex) to establish the chromatin landscape for genes controlling differentiation of the ventral body wall and heart, both tissues derived from the lateral plate mesoderm. Consistently, disruption of Fendrr expression in developing embryos is lethal, in part as a consequence of heart failure [28]. At later developmental stages, Fendrr is expressed in lungs and gut, where it might contribute to additional detrimental defects [71].
The embryonic ventral forebrain 2, Evf-2 lncRNA, expressed from an ultra-conserved distal enhancer region in the medial ganglionic eminence of the E13.5 mouse embryo is an example of a lncRNA that binds a transcription factor, affects its activity, and in turn regulates gene expression. Particularly, Evf-2 associates with the transcription factor DLX2 (distal-less homeobox 2) to control the expression of Dlx5/6 via an enhancer. A stable complex of Evf-2 with DLX1/2 proteins binds to the regulatory elements in the Dlx5/6 locus in vivo, most likely preventing repressive enhancer DNA methylation [23, 39]. Deletion of Evf-2 leads to a reduction of GABAergic interneurons in adult mice due to a decrease in the number of neurons specified for this lineage during embryonic development [5].
Another lncRNA involved in nervous system development is the retinal non-coding RNA4 (RNACR4). It is expressed in the postnatal developing retina and is part of a regulatory network including the RNA helicase DDX3X and the miR-183/96/182, which are essential for the organization of retinal architecture. Deletion of RNACR4 impairs the uniformity of the different layers of the retina, with the photoreceptor layer being the most affected. When present, RNACR4 indirectly activates pre-miR-183/96/182 processing by inhibiting the processing repressor DDX3X. RNACR4 and the DDX3X-miR-183/96/182 network is an interesting example of how lncRNAs are able to regulate gene expression indirectly [47].
The lncRNA Neat1 was identified as an essential architectural component of paraspeckle nuclear bodies, but a physiological role had not been linked to this lncRNA until recently, when two independent groups investigated Neat1 knockout mice in detail. The first group observed that Neat1 deletion resulted in impaired mammary gland branching morphogenesis, lobular-alveolar development, and lactation [74]. The second group observed that, although deletion of Neat1 still resulted in viable and fertile mice, knockout females had about 50 % reduced fertility due to corpus luteum dysfunction and reduced progesterone production. Unilateral transplantation of wild-type ovaries or the administration of progesterone partially rescued the phenotype, suggesting the importance of Neat1 for corpus luteum formation and function [63]. However, the molecular mechanism of Neat1 function in these two physiological processes remains unknown.
Playrr (Pitx2 locus asymmetric regulated RNA) was identified as a lncRNA transcribed from a known enhancer region in the right side of the dorsal mesentery (DM). Asymmetric cell behavior in the DM requires Pitx2 expression on the left to direct gut looping. The expression of Pitx2 and Playrr are mutually downregulated through a mechanism that involves changes in chromatin conformation and promoter-enhancer looping [85]. Cells expressing Playrr-Pitx2 are proximal in the left DM, where Pitx2 is preferentially expressed, but positioned further apart in the right DM where Playrr is expressed and Pitx2 is not. Deletion of Pitx2 using CRISPR/Cas9 genome editing systems, leads to Playrr expression on both sides and deletion of Playrr increases Pitx2 expression.
During early stages of murine myoblast differentiation in vitro, a lncRNA named linc-MD1 was identified as being able to induce myogenic differentiation. Linc-MD1 provides an example of how the interplay between lncRNAs and miRNAs regulates gene expression. Linc-MD1 originates from the activation of the DIST promoter upon induction of differentiation, and accumulates in the cytoplasm where it binds two miRNAs, miR-133 and miR-135 [11]. Binding to the miRNAs alleviates the repression of the miRNA target genes MAML1 and MEF2, key myogenic factors, allowing for their expression and progression of differentiation. Linc-MD1 downregulation is associated with the pathogenesis of Duchenne muscular dystrophy, as the myoblasts show a reduced ability to undergo terminal differentiation.
The terminal differentiation-induced lncRNA (TINCR) is a lncRNA involved in somatic tissue differentiation. Using organotypic human epidermal tissue it was found that TINCR is expressed during epidermal differentiation and increases the stability of mRNAs that induce skin differentiation [46]. Depletion of TINCR leads to epidermis lacking terminal differentiation ultrastructure, including keratohyalin granules and intact lamellar bodies. TINCR binds directly to mRNAs with a TINCR box motif, and promotes their stability by recruiting Staufen 1 protein (STAU1), acting at a post-transcriptional level to regulate gene expression.
LncRNAs can also play important roles in immune cell differentiation. The lnc-DC, was exclusively identified in human dendritic cells (DCs), and knockdown experiments showed its important role in promoting DC differentiation from human monocytes in vitro and from mouse bone marrow cells in vivo. The derived DCs had an impaired capacity to activate T cells. Lnc-DC mediates its function by binding to signal transducer and activator of transcription 3 (STAT3), promoting STAT3 phosphorylation and thus activation [83]. Additionally, lncRNAs have been shown to be involved in immune regulation and susceptibility to infectious disease. Specifically, the lncRNA NeST (Nettoie Salmonella pas Theiler’s [cleanup Salmonella not Theiler’s]) is required for inducible IFN-γ. NeST controls the chromatin state at the interferon gamma (IFN-γ) locus by binding to trithorax group protein WDR5 and altering histone 3 methylation marks. Thus, NeST regulates INF-γ expression and susceptibility to a viral and a bacterial pathogen [25].
LncRNAs associated with genetic disorders
Genome-wide association studies have revealed that only 7 % of disease or trait-associated mutations or single nucleotide polymorphisms (SNPs) reside in protein-coding exons, while 43 % are found outside of protein-coding genes [36]. Considering the wide range of roles that lncRNAs play in cellular networks, it is not surprising that non-coding RNAs have been implicated in genetic diseases and disorders.
FSHD (facioscapulohumeral muscular dystrophy) is an autosomal-dominant disease, the third most common myopathy, and is caused predominantly by a reduction in the copy number of D4Z4 repeats at chromosomal region 4q35 [8]. In healthy subjects, D4Z4 repeats recruit Polycomb complexes to promote the formation of a repressive chromatin state that inhibits the expression of genes at 4q35. In contrast, in muscle biopsies and primary muscle cells from FSHD patients, the reduction of D4Z4 leads the transcription of a novel lncRNA named DBE-T, which coordinates the establishment of active chromatin state and de-repression of genes from 4q35 [7]. DBE-T acts in cis by recruiting the Trithorax group protein ASH1L to the FSHD locus, driving histone H3 lysine 36 demethylation, chromatin remodeling, and finally 4q35 gene expression.
ACD/MPV (Alveolar capillary dysplasia with misalignment of pulmonary veins) is a lethal lung developmental disorder associated with dysregulation of the FOXF1 gene, a key regulator of lung development. Apart from deletions or inactivating mutations in the FOXF1 locus, deletions in an upstream region that contains a distal FOXF1 enhancer have been also identified as disease-causing deletions [75]. This region also contains two lncRNAs expressed specifically in the lung, which raise the hypothesis that these lncRNAs establish a chromatin loop that brings the enhancer in close proximity to FOXF1 [77].
The HELLP syndrome is a life-threatening pregnancy-associated disease that occurs in the third trimester in the mother and includes hemolysis, elevated liver enzymes, and low platelets. However, the origin of the disease can be traced back to the first trimester fetal placenta. Genome-wide linkage analysis of families with HELLP syndrome identified a region on chromosome 12q23 between PMCH and IGF1 to be associated with the disease [15]. The region contains a lncRNA more than 205 kb in length which is expressed in early placenta extravillous trophoblasts. Although the precise function of this lncRNA has yet to be established, knockdown of this lncRNA revealed a role in trophoblast transition from G2 to mitosis and cell invasion. Interestingly, injection of morpholino oligonucleotides complementary to the mutation site in extravillous trophoblast cells boosted and reversed the expression levels of HELLP lncRNA and cell invasion defects. This was the first study that linked a lncRNA to a Mendelian disorder with autosomal-recessive inheritance.
Examination of two families with BDE (Brachydactyly type E) a disease with translocations on chromosome 12q, illustrated that the disease state involved regulation of expression by a lncRNA. The identified translocations disrupt a regulatory region referred to as CISTR-ACT that interacts in cis with parathyroid hormone-like hormone (PTHLH) gene and in trans with the sex-determining region Y-box 9 (SOX9) gene on chromosome 17q, and leads to downregulation of both of these genes [55]. Interestingly, a lncRNA (DA125942) was also found to be transcribed from this region and to interact with both loci, regulating PTHLH and SOX9 expression. The translocations disrupted the higher order chromatin organization and cis-regulatory elements, resulting in low lncRNA occupancy on the target loci and downregulation of the target genes.
LncRNAs with roles in cancer
Several studies have transcriptionally profiled numerous primary tumor samples, metastatic biopsies, and cancer cell lines which has led to the identification of thousands of differentially expressed lncRNAs with implications in cancer. One of the most comprehensive studies so far encompasses poly A+ RNA-seq data from more than 7000 samples, including tumors, normal samples and tumor cell lines. The expression of 8000 of the estimated 58,000 lncRNAs was restricted to only one cancer type, highlighting the specific association of some lncRNAs to cancers of different tissue origin [57]. These findings open up the possibility of using lncRNA expression for accurate identification of tumor subclasses or response to a given therapy. Although numerous in vitro cell-based assays have linked lncRNAs to both oncogenic and tumor-suppressive pathways, only a handful of animal models have been developed to examine the association lncRNAs with cancer in vivo.
The first lncRNA described to have a fundamental role in cancer invasiveness and metastasis was HOX antisense intergenic RNA (HOTAIR). HOTAIR is transcribed from the antisense strand of HOXC gene cluster and mediates silencing of the HOXD locus through direct interaction with the PRC2 complex and guiding to target genes [69]. This feature of HOTAIR mediates cancer invasiveness and metastasis, as HOTAIR overexpression leads to re-targeting of PRC2, altered histone H3 lysine 27 methylation, and abnormal gene expression in different types of cancer. In breast cancer metastasis, HOTAIR is expressed hundreds to thousands of fold higher than in noncancerous tissue, being a powerful predictor of metastasis and death. The altered PRC2 occupancy led to cancer cells resembling embryonic fibroblasts, explaining the increased lung metastatic ability in a xenograft model for breast cancer, where HOTAIR was overexpressed [30]. Moreover, in a pancreatic cancer xenograft model, loss of HOTAIR reduced tumor growth and in hepatocellular carcinoma tissue HOTAIR overexpression increased the risk of recurrence after hepatectomy and lymph node metastasis, and in colorectal cancer loss correlated with liver metastasis and poor prognosis [24, 44].
Another cancer-associated lncRNA is MALAT1 (metastasis-associated lung adenocarcinoma transcript 1). MALAT1 was originally identified as a prognostic marker for metastasis and patient survival in non-small cell lung cancer. It is extremely abundant in many human cell types and highly conserved over its full length among mammals [40]. The contribution of MALAT1 to cancer progression was proven using a xenograft mouse model of human lung cancer, where implanted MALAT1 knockout human lung cancer cells were impaired in migration and formed fewer and smaller tumor nodules [45]. The same study also showed that loss of MALAT1 downregulated expression of several metastasis-associated genes, hence uncovering MALAT1’s role in lung cancer as an active regulator of metastasis.
Interestingly, it has been shown that Xist is not only important for X chromosome dosage compensation, but also plays a role in suppressing hematologic cancer in vivo. Deletion of Xist in the blood compartment of mice led to the development of a highly aggressive myeloproliferative neoplasm and myelodysplastic syndrome (mixed MPN/MDS) in mutant females with 100 % penetrance. In addition, Xist-deficient hematopoietic stem cells show aberrant maturation and age-dependent loss. In this context, X-reactivation causes DNA replication and mitotic anomalies, genome instability, and dysregulation of the hematopoiesis pathway [91].
As a special case, the non-coding pseudogene BRAFP1 has been found to be mutated and aberrantly expressed in cancers, similarly to its coding homologue BRAF. Mice overexpressing Braf-rs1 (the murine homologue of BRAFP1) displayed malignancies resembling human diffuse large B cell lymphoma, which regressed when the transgene was knocked down. Expression of Braf-rs1 causes upregulation of Braf by acting as a ceRNA (competing endogenous RNA) and “sponging” miRNAs that would otherwise target Braf, [42].
LncRNAs with roles in other diseases
The antisense lncRNA BACE1-AS has been strongly correlated with Alzheimer’s disease (AD) severity, since elevated BACE1-AS levels have been detected in subjects with AD and in amyloid precursor protein transgenic mice. BACE1-AS is transcribed from the strand opposite to BACE1, an aspartyl protease that cleaves APP in amyloid precursor protein transgenic mice. Accumulation of the Aβ neuropeptide and increased BACE1 protein levels have been implicated in the pathogenesis of the disease. BACE1-AS regulates BACE1 expression at the post-transcriptional level, as BACE1-AS and BACE1 directly associate to form a duplex which stabilizes BACE1 mRNA. Stabilization of BACE1 mRNA leads to elevated BACE1 protein levels, higher APP cleavage, and toxic accumulation of Aβ plaques maintaining the deleterious feed-forward cycle of disease progression [20]. BACE1-AS is an excellent example of how dysregulated levels of a lncRNA can play a significant role in the pathogenesis of a disease.
Cardiovascular diseases (CVDs) have also been associated with various lncRNAs. Hundreds of lncRNAs have been found to be dysregulated after induced myocardial infarction in mice through transaortic constriction [65, 80, 94]. Analysis of the myosin heavy-chain-associated RNA transcript (Mhrt), transcribed divergently to Myh6 and overlapping with Myh7 in an antisense orientation, revealed a lncRNA with cardioprotective effects. Mhrt is down regulated after induction of myocardial infarction, and ectopic expression of Mhrt in mice following the operation resulted in much less severe symptoms during recovery as compared to control mice [34]. Mhrt binds to the helicase domain of BRG1, an ATPase subunit of the BAF chromatin-remodeling complex that is activated by stress to induce cardiac hypertrophy. The binding prevents BRG1 from recognizing its genomic targets, including the promoters of α-myosin and β-myosin heavy chain (Myh6 and Myh7), critical genes in cardiac physiology but also in pathological hypertrophy. BRG1, in turn, downregulates Mhrt, and thus escapes tethering to promote cardiac hypertrophy under stress conditions. Human MHRT also originates from the MYH7 locus, has conserved sequence and secondary structure, and is repressed in various types of cardiomyopathies [33].
An examination of patients with coronary artery disease (CAD) by genome-wide association studies has identified SNPs in both non-coding regions and genomic regions with no coding potential which are associated with the disease. A case-control study of over 4000 CAD cases confirmed that the high-risk CAD haplotype overlaps with exons 13–19 of an antisense non-coding RNA in the INK4 (CDKN2A) locus (ANRIL) on chromosome 9q21. ANRIL recruits CBX7 (and thus PRC1) to its locus via a POLII-dependent mechanism in order to repress the surrounding genes CDKN2A and CDKN2B, and promotes cell cycle activity [90]. Further characterization of the identified polymorphisms in the ANRIL locus showed that SNPs could disrupt ANRIL splicing, resulting in a circular transcript that affects its function. ANRIL is expressed in atheromatous human vessels and cell types such as vascular endothelial cells, monocyte-derived macrophages, and coronary smooth muscle cells, all involved in atherosclerosis. In another study, deletion of a 70 kb region including a large portion of the ANRIL locus led to dysregulation of gene expression in cis, and increased mortality in mice, especially after high fat diet [81]. A smaller scale case-control study of 188 myocardial infarction patients and 752 control patients significantly linked 6 SNPs in the myocardial infarction associated transcript (MIAT) on chromosome 22q12.1 with susceptibility for myocardial infarction [38].
LncRNAs in diagnostics and therapies
Current and future analysis of lncRNAs and their role as regulatory molecules will broaden our understanding of human diseases, and opens up opportunities for new prognostic, diagnostic, and therapeutic strategies. Some specific features of lncRNAs make them good candidates for developing diagnostics and therapies. Another strategy takes advantage of the highly tissue- and cell type-specific expression patterns of lncRNAs to identify cell populations to target, such as different subclasses of tumors, or to even predict treatment responses in these populations; hence lncRNAs could be useful as biomarkers [6, 35]. Currently, the lncRNA prostate cancer-associated 3 (PCA3, also called DD3) is used as a specific and sensitive marker for prostate cancer in patient urine samples, and the hepatocellular carcinoma (HCC) upregulated lncRNA, HULC, which is highly expressed in HCC patients, can be detected in the blood by conventional PCR methods [6, 66]. MALAT1 was originally identified as a prognostic marker for metastasis and patient survival in non-small cell lung cancer [40] and LIPCAR (long intergenic non-coding RNA predicting cardiac remodeling) was found in plasma from myocardial infarction patients to significantly correlate with mortality and could thus be considered as a useful biomarker for CVD [48]. Beyond these, the highly specific expression of lncRNAs has proven advantageous in the delivery of treatments to specific cell populations, mitigating the risk of affecting normal tissues. The only clinically developed strategy thus far uses a plasmid that carries the gene for the A subunit of diphtheria toxin under the regulation of the H19 promoter. Intratumoral injection of the plasmid induces the expression of high levels of diphtheria toxin specifically in the tumor, resulting in a reduction of tumor size in human trials for a broad range of carcinomas [62].
Other therapeutic strategies are based on manipulating lncRNA expression or function. As previously discussed, a characteristic of lncRNAs is that they bind proteins and provide gene locus specificity to the protein’s activity. Therefore, protein activity can be indirectly targeted by modulating the interaction with the lncRNA, and hence affecting expression of specific genes. Due to this increased specificity, such a strategy could be result in less off-target effects than conventional protein-targeting drugs. Many drug development companies focus on making small molecules or synthetic oligonucleotide antagonists that specifically block the effects of proteins. For example, in the case of HOTAIR, inhibition of the interaction with PRC2 and LSD1 complexes may limit the metastatic potential of breast cancer. Knockdown strategies could be also applied using antisense oligonucleotides (ASOs) that knockdown the lncRNA. To this effect, an ASO has recently been used in mice for the treatment of Angelman syndrome. The ASO blocks the lncRNA ubiquitin protein ligase E3a gene antisense lncRNA (Ube3a-ats), leading to the expression of the imprinted Ube3a and, hence, the Angelman phenotype is reversed [53]. In another study, MALAT1 expression was targeted in established human xenograft tumors in mice with free-uptake ASOs. Targeted MALAT1 expression dramatically reduced lung cancer metastasis formation [45]. In contrast to knockdown strategies, overexpression of lncRNAs can also be used for therapeutics, as for transgenic expression of the human XIST which has been applied to silence one of the triplicated chromosome 21 in cells derived from a person with Down syndrome [41]. Major challenges of these strategies include the delivery of the oligonucleotides or transgenes, both to the tissue of interest and to the nucleus in case of nuclear lncRNAs, and maintaining long-lasting effects.
Considerations when analyzing lncRNAs
LncRNAs have emerged as a new class of key regulatory molecules in every level of cell physiology and are associated with multiple human diseases and disorders. However, even lncRNAs that have been known for quite some time and that have been investigated in great detail, like H19 and Xist, have proven elusive with regards to fully identifying their functional significance and molecular mechanisms. This makes loss-of-function in vivo experiments crucial in the understanding of the roles of novel lncRNAs. A variety of approaches, such as locus deletion, promoter deletion, inversions, transcriptional termination, and RNAi-mediated knockdown have been used so far to disrupt lncRNA function. A quick approach, which also allows for a small to medium sized primary screen, is the use of knockdown experiments employing siRNAs. However, siRNAs are less efficient for nuclear localized lncRNAs. Therefore, it is favorable to use an approach exploiting LNA modified antisense RNAs, which have been shown to work significantly better [54]. In addition, such lncRNA inhibitory molecules can be injected into mice to evaluate potential in vivo defects caused by loss of the lncRNA function post-development [61]. The gold standard, however, remains the mouse knockout model [52], since in some cases the observed phenotype of knockdowns and knockouts differ significantly. For example, in the case of the Evf-2 and lincRNA-p21 lncRNAs the two strategies showed contradictory effects on neighboring gene expression and mechanism-of-action [57]. Therefore, strategies such as introduction of a transcriptional terminator that cause minimal interference to the surrounding genomic DNA sequence are recommended. However, in this case care should also be taken to control for changing the spacing between regulatory elements. In addition, considerations should be made with regards to elucidating the functional role of the RNA, as opposed to the process of transcription itself [4]. Despite the robust phenotypes identified by the few lncRNA knockout models examined to date, some others, such as Malat1 and Neat1, have been found to display no overt or subtle phenotypes, which may be the result of compensatory mechanisms [70]. It also remains possible that the gene-regulatory functions of some lncRNAs result in mild and specific phenotypic effects that are only detectable following thorough analyses.
Concluding remarks
An unanticipated and tremendous amount of the non-coding sequence of the human genome is transcribed, and lncRNAs constitute the largest fraction of the non-protein-coding transcripts. Mediating gene expression regulation at all levels, lncRNAs have emerged as a new and bright field in human genetics which has broadened our understanding of the complexity of disease and of gene regulation. Mice and humans, perhaps surprisingly, share a highly conserved set of protein-coding genes that compose the basic instructions for body plan and organ formation, but the diversity and complexity at the organismal level hinges on the variety in non-coding genes. The higher the complexity of an organism, the bigger the non-coding regulatory repertoire [58]. LncRNAs are an important component of the non-coding regulatory repertoire involved in fine-tuning the gene-regulatory networks, and they are associated with specific cell types, growth stage, physiology, and disease. Despite their importance, many questions remain to be addressed in the remarkably diverse field of lncRNAs. First, the full range of lncRNA mechanisms has to be determined. In addition, while the catalog of identified lncRNAs is rapidly expanding, the in vivo function of the majority remains unknown. Furthermore, a more concerted effort should be made to elucidate the structure-function relationship, as this currently lacking and will greatly contribute to our understanding of lncRNAs and realizing their therapeutic potential.
References
Azzalin CM, Reichenbach P (2007) Telomeric repeat-containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318(80):798–801
van Bakel H, Nislow C, Blencowe BJ, Hughes TR (2010) Most “dark matter” transcripts are associated with known genes. PLoS Biol 8:e1000371. doi:10.1371/journal.pbio.1000371
Bartolomei MS, Zemel STS (1991) Parental imprinting of the mouse H19 gene. Nature 9:153–155
Bassett AR, Akhtar A, Barlow DP, Bird AP, Brockdorff N, Duboule D, Ephrussi A, Ferguson-Smith AC, Gingeras TR, Haerty W, Higgs DR, Miska EA, Ponting CP (2014) Considerations when investigating lncRNA function in vivo. Elife 3:1–14. doi:10.7554/eLife.03058
Bond AM, Vangompel MJ, Sametsky EA, Clark MF, Savage JC, Disterhoft JFKJ (2009) Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat Neurosci 12:1020–1027
Bussemakers MJ, van Bokhoven A, Verhaegh GW, Smit FP, Karthaus HF, Schalken JA, Debruyne FM, Ru NIW (1999) D3: a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res 59:5975–5979
Cabianca DS, Casa V, Bodega B, Xynos A, Ginelli E, Tanaka Y, Gabellini D (2012) A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 149:819–831. doi:10.1016/j.cell.2012.03.035
Cabianca DS, Gabellini D (2010) The cell biology of disease: FSHD—copy number variations on the theme of muscular dystrophy. J Cell Biol 191:1049–1060. doi: 10.1083/jcb.201007028
Cabili CT, Loyal G, Koziol M, Barbara AR, Tazon-Vega LR (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25:1915–1927. doi:10.1101/gad.17446611
Carrieri C, Cimatti L, Biagioli M, Beugnet A, Zucchelli S, Fedele S, Pesce E, Ferrer I, Collavin L, Santoro C, Forrest ARR, Carninci P, Biffo S, Stupka E, Gustincich S (2012) Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 491:454–457. doi:10.1038/nature11508
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I (2011) A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147:358–369. doi:10.1016/j.cell.2011.09.028
Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, Sementchenko V, Piccolboni A, Bekiranov S, Bailey DK, Ganesh M, Ghosh S, Bell I, Gerhard DS, Gingeras TR (2005) Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308(80):1149–1154. doi:10.1126/science.1108625
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigó R (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22:1775–1789. doi:10.1101/gr.132159.111
Dey BK, Pfeifer K, Dutta A (2014) The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev 28:491–501. doi:10.1101/gad.234419.113
van Dijk M, Thulluru HK, Mulders J, Michel OJ, Poutsma A, Windhorst S, Kleiverda G, Sie D, Lachmeijer AMA, Oudejans CBM (2012) HELLP babies link a novel lincRNA to the trophoblast cell cycle. J Clin Invest 122:4003–4011. doi:10.1172/JCI65171
Dinger M, Amaral P, Mercer T (2008) Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res 18:1433–1445. doi:10.1101/gr.078378.108.7
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Röder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See L-H, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigó R, Gingeras TR (2012) Landscape of transcription in human cells. Nature 489:101–108. doi:10.1038/nature11233
Eddy SR, Hughes H (2001) Non-coding RNA genes and the modern RNA world. Nat Rev Genet 2:919–929. doi:10.1038/35103511
Esteller M (2011) Non-coding RNAs in human disease. Nat Rev Genet 12:861–874. doi:10.1038/nrg3074
Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G, Kenny PJ, Wahlestedt C (2008) Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med 14:723–730. doi:10.1038/nm1784
Faghihi MA, Zhang M, Huang J et al. (2010) Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol 11
Fejes-Toth K, Sotirova V, Sachidanandam R, Assaf G, Hannon GJ, Kapranov P, Foissac S, Willingham AT, Duttagupta R, Dumais E, Gingeras TR (2009) Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature 457:1028–1032. doi:10.1038/nature07759
Feng J, Bi C, Clark BS, Mady R, Shah P, Kohtz JD (2006) The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev 20:1470–1484. doi:10.1101/gad.1416106
Geng YJ, Xie SL, Li Q, Ma J, Wang GY (2011) Large intervening non-coding RNA HOTAIR is associated with hepatocellular carcinoma progression. J Int Med Res 39:2119–2128. doi:10.1177/147323001103900608
Gomez JA, Wapinski OL, Yang YW, Bureau J, Monack DM, Chang HY, Brahic M, Kirkegaard K (2013) NeST, a long noncoding RNA, controls microbial susceptibility and epigenetic activation of the Ifng locus. Cell 152:743–754. doi:10.1016/j.cell.2013.01.015.NeST
Gong C, Maquat LE (2011) lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 470:284–288. doi:10.1038/nature09701
Greer Eric L, Yang S (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 13:343–357
Grote P, Wittler L, Hendrix D, Koch F, Währisch S, Beisaw A, Macura K, Bläss G, Kellis M, Werber M, Herrmann BG (2013) The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell. doi:10.1016/j.devcel.2012.12.012
Guo X, Gao L, Wang Y et al. (2015) Advances in long noncoding RNAs: identification, structure prediction and function annotation. Brief Funct Genomics 1–9. doi: 10.1093/bfgp/elv022
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai M-C, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464:1071–1076. doi:10.1038/nature08975
Guttman M, Rinn JL (2012) Modular regulatory principles of large non-coding RNAs. Nature 482:339–346. doi:10.1038/nature10887
Guttman M, Russell P, Ingolia NT, Weissman JS, Lander ES (2013) Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell 154:240–251. doi:10.1016/j.cell.2013.06.009
Han P, Li W, Lin C-H, Yang J, Shang C, Nurnberg ST, Jin KK, Xu W, Lin C-Y, Lin C-J, Xiong Y, Chien H-C, Zhou B, Ashley E, Bernstein D, Chen P-S, Chen H-SV, Quertermous T, Chang C-P (2014) A long noncoding RNA protects the heart from pathological hypertrophy. Nature 514:102–106. doi:10.1038/nature13596
Hang CT, Yang J, Han P, Cheng H-L, Shang C, Ashley E, Zhou B, Chang C-P (2010) Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 466:62–67. doi:10.1038/nature10222
Hessels D, Klein Gunnewiek JMT, Van Oort I, Karthaus HFM, Van Leenders GJL, Van Balken B, Kiemeney LA, Witjes JA, Schalken JA, Culig Z (2003) DD3PCA3-based molecular urine analysis for the diagnosis of prostate cancer. Eur Urol 44:8–16. doi:10.1016/S0302-2838(03)00201-X
Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 106:9362–9367. doi:10.1073/pnas.0903103106
Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, Wang Y, Kong B, Langerød A, Børresen-Dale A-L, Kim SK, van de Vijver M, Sukumar S, Whitfield ML, Kellis M, Xiong Y, Wong DJ, Chang HY (2011) Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet 43:621–629. doi:10.1038/ng.848
Ishii N, Ozaki K, Sato H, Mizuno H, Saito S, Takahashi A, Miyamoto Y, Ikegawa S, Kamatani N, Hori M, Saito S, Nakamura Y, Tanaka T (2006) Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet 51:1087–1099. doi:10.1007/s10038-006-0070-9
Cajigas I, Leib DE, Cochrane J, Luo H, Swyter KR, Chen S, Clark BS, Thompson J, Yates JR III, Robert E, Kingston JDK (2015) Evf2 lncRNA/BRG1/DLX1 interactions reveal RNA-dependent inhibition of chromatin remodeling. Development 142:2641–2652
Ji P, Diederichs S, Wang W, Böing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, Thomas M, Berdel WE, Serve H, Müller-Tidow C (2003) MALAT-1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 22:8031–8041. doi:10.1038/sj.onc.1206928
Jiang J, Jing Y, Cost GJ, Chiang J-C, Kolpa HJ, Cotton AM, Carone DM, Carone BR, Shivak DA, Guschin DY, Pearl JR, Rebar EJ, Byron M, Gregory PD, Brown CJ, Urnov FD, Hall LL, Lawrence JB (2013) Translating dosage compensation to trisomy 21. Nature 500:296–300. doi:10.1038/nature12394
Karreth FA, Reschke M, Ruocco A, Ng C, Chapuy B, Léopold V, Sjoberg M, Keane TM, Verma A, Ala U, Tay Y, Wu D, Seitzer N, Velasco-Herrera MDC, Bothmer A, Fung J, Langellotto F, Rodig SJ, Elemento O, Shipp MA, Adams DJ, Chiarle R, Pandolfi PP (2015) The BRAF pseudogene functions as a competitive endogenous RNA and induces lymphoma in vivo. Cell 161:319–332. doi:10.1016/j.cell.2015.02.043
Keniry A, Oxley D, Monnier P, Kyba M, Dandolo L, Smits G, Reik W (2012) The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell Biol 14:659–665. doi:10.1038/ncb2521
Kim K, Jutooru I, Chadalapaka G, Johnson G, Frank J, Burghardt R, SK and SS (2013) HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 32:1616–1625
Koebnick C, Smith N, Black MH, Porter AH, Richie BA, Hudson S, Gililland D, Steven J, Longstreth GF, Baldwin P, Medical P, Park B (2013) The non-coding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res 73:1180–1189. doi:10.1097/MPG.0b013e31824d256f.Pediatric
Kretz M, Siprashvili Z, Chu C, Webster DE, Zehnder A, Qu K, Lee CS, Flockhart RJ, Groff AF, Chow J, Johnston D, Kim GE, Spitale RC, Flynn RA, Zheng GX, Aiyer S, Raj A, Rinn JL, Chang HYKP (2013) Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 493:231–235. doi:10.1038/nature11661.Control
Krol J, Krol I, Alvarez CPP, Fiscella M, Hierlemann A, Roska B, Filipowicz W (2015) A network comprising short and long noncoding RNAs and RNA helicase controls mouse retina architecture. Nat Commun 6:7305. doi:10.1038/ncomms8305
Kumarswamy R, Bauters C, Volkmann I, Maury F, Fetisch J, Holzmann A, Lemesle G, de Groote P, Pinet F, Thum T (2014) Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ Res 114:1569–1575. doi:10.1161/CIRCRESAHA.114.303915
Lee J (2012) Epigenetic regulation by long noncoding RNAs. Science 338(80):1435–1439. doi:10.1126/science.1231776
Lee JT (2009) Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev 23:1831–1842. doi:10.1101/gad.1811209
Leighton PA, Ingram RS, Eggenschwiler J, Efstratiadis ATS (1995) Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature 4:34–39
Li L, Chang HY (2014) Physiological roles of long noncoding RNAs: insight from knockout mice. Trends Cell Biol 24:594–602. doi:10.1016/j.tcb.2014.06.003
Meng L, Ward AJ, Seung C, Bennett CF, Arthur L, Beaudet FR (2015) Towards a therapy for Angelman syndrome by reduction of a long non-coding RNA. Nature 518:409–412. doi:10.1038/nature13975.Towards
Maamar H, Cabili MN, Rinn J, Raj A (2013) linc-HOXA1 is a noncoding RNA that represses HoXa1 transcription in cis. Genes Dev 27:1260–1271. doi:10.1101/gad.217018.113
Maass PG, Rump A, Schulz H, Stricker S, Schulze L, Platzer K, Aydin A, Tinschert S, Goldring MB, Luft FC, Bahring S, Bähring S (2012) A misplaced IncRNA causes brachydactyly in humans. J Clin Invest 122:3990–4002. doi:10.1172/JCI65508
Marques AC, Hughes J, Graham B, Kowalczyk MS, Higgs DR, Ponting CP (2013) Chromatin signatures at transcriptional start sites separate two equally populated yet distinct classes of intergenic long noncoding RNAs. Genome Biol 14:R131. doi:10.1186/gb-2013-14-11-r131
Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, Poliakov A, Cao X, Dhanasekaran SM, Wu Y-M, Robinson DR, David GB, AMC (2015) The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 47:199–208. doi:10.1016/j.ygyno.2014.12.035.Pharmacologic
Mattick JS (2001) Non-coding RNAs: the architects of eukaryotic complexity. EMBO Rep 2:986–991. doi:10.1093/embo-reports/kve230
Mercer TR, Dinger ME, Mattick JS (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10:155–159. doi:10.1038/nrg2521
Mercer TR, Wilhelm D, Dinger ME, Solda G, Korbie DJ, Glazov EA, Truong V, Schwenke M, Simons C, Matthaei KI, Saint R, Koopman P, Mattick JS (2011) Expression of distinct RNAs from 3′ untranslated regions. Nucleic Acids Res 39:2393–2403. doi:10.1093/nar/gkq1158
Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T, John D, Ponomareva Y, Chen W, Uchida S, Boon RA, Dimmeler S (2014) Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res 114:1389–1397. doi:10.1161/CIRCRESAHA.114.303265
Mizrahi A, Czerniak A, Levy T, Amiur S, Gallula J, Matouk I, Abu-lail R, Sorin V, Birman T, de Groot N, Hochberg A, Ohana P (2009) Development of targeted therapy for ovarian cancer mediated by a plasmid expressing diphtheria toxin under the control of H19 regulatory sequences. J Transl Med 7:69. doi:10.1186/1479-5876-7-69
Nakagawa S, Shimada M, Yanaka K, Mito M, Arai T, Takahashi E, Fujita Y, Fujimori T, Standaert L, Marine J-C, Hirose T (2014) The lncRNA Neat1 is required for corpus luteum formation and the establishment of pregnancy in a subpopulation of mice. Development 141:4618–4627. doi:10.1242/dev.110544
Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R (2010) Long noncoding RNAs with enhancer-like function in human cells. Cell 143:46–58. doi:10.1016/j.cell.2010.09.001.Long
Ounzain S, Micheletti R, Beckmann T, Schroen B, Alexanian M, Pezzuto I, Crippa S, Nemir M, Sarre A, Johnson R, Dauvillier J, Burdet F, Ibberson M, Guigo R, Xenarios I, Heymans S, Pedrazzini T (2015) Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur Heart J 36:353–368. doi:10.1093/eurheartj/ehu180
Panzitt K, Tschernatsch MMO, Guelly C, Moustafa T, Stradner M, Strohmaier HM, Buck CR, Denk H, Schroeder R, Trauner M, Zatloukal K (2007) Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 132:330–342. doi:10.1053/j.gastro.2006.08.026
Penny GD, Kay GF, Sheardown SA, Rastan SBN (1996) Requirement for Xist in X chromosome inactivation. Nature 379:131–137
Rando TA, Chang HY (2012) Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell 148:46–57. doi:10.1016/j.cell.2012.01.003
John R, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough H, Helms JA, Farnham PJ, Segal E, HYC (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by non-coding RNAs. Cell 129:1311–1323. doi:10.1021/ja8019214.Optimization
Rossi A, Kontarakis Z, Gerri C, Nolte H, Hölper S, Krüger M, Stainier DYR (2015) Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 13:230–233. doi:10.1038/nature14580
Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, Sanchez-Gomez DB, Hacisuleyman E, Li E, Spence M, Liapis SC, Mallard W, Morse M, Swerdel MR, D’Ecclessis MF, Moore JC, Lai V, Gong G, Yancopoulos GD, Frendewey D, Kellis M, Hart RP, Valenzuela DM, Arlotta P, Rinn JL (2013) Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2:e01749. doi:10.7554/eLife.01749
Schmitz K-M, Mayer C, Postepska A, Grummt I (2010) Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev 24:2264–2269. doi:10.1101/gad.590910
Siegfried NA, Busan S, Rice GM, Nelson JAE, Weeks KM (2014) RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat Methods 11:959–965. doi:10.1038/nmeth.3029
Standaert L, Adriaens C, Radaelli E, Standaert L, Adriaens C, Radaelli E, Keymeulen AVAN, Blanpain C, Hirose T, Nakagawa S, Marine J (2014) The long noncoding RNA Neat1 is required for mammary gland development and lactation. RNA 20:1–6. doi:10.1261/rna.047332.114
Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, Ou Z, Wiszniewska J, Driscoll DJ, Maisenbacher MK, Bolivar J, Bauer M, Zackai EH, McDonald-McGinn D, Nowaczyk MMJ, Murray M, Hustead V, Mascotti K, Schultz R, Hallam L, McRae D, Nicholson AG, Newbury R, Durham-O’Donnell J, Knight G, Kini U, Shaikh TH, Martin V, Tyreman M, Simonic I, Willatt L, Paterson J, Mehta S, Rajan D, Fitzgerald T, Gribble S, Prigmore E, Patel A, Shaffer LG, Carter NP, Cheung SW, Langston C, Shaw-Smith C (2009) Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet 84:780–791. doi:10.1016/j.ajhg.2009.05.005
Sun SB and T (2011) Functions of noncoding RNAs in neural development and neurological diseases. Mol Neurobiol 44:359–373. doi:10.1016/j.micinf.2011.07.011.Innate
Szafranski P, Dharmadhikari AV, Brosens E, Gurha P, Kolodziejska KE, Zhishuo O, Dittwald P, Majewski T, Mohan KN, Chen B, Person RE, Tibboel D, de Klein A, Pinner J, Chopra M, Malcolm G, Peters G, Arbuckle S, Guiang SF, Hustead VA, Jessurun J, Hirsch R, Witte DP, Maystadt I, Sebire N, Fisher R, Langston C, Sen P, Stankiewicz P (2013) Small noncoding differentially methylated copy-number variants, including lncRNA genes, cause a lethal lung developmental disorder. Genome Res 23:23–33. doi:10.1101/gr.141887.112
Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, Blencowe BJ, Prasanth SG, Prasanth KV (2010) The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 39:925–938. doi:10.1016/j.molcel.2010.08.011
Tsai M, Manor O, Wan Y, Mosammaparast N, Wang JK, Shi Y, Segal E, Chang HY (2010) Long noncoding RNA as modular scaffold of histone modification complexes. Science 329(80):689–693. doi:10.1126/science.1192002.Long
Vausort M, Wagner DR, Devaux Y (2014) Long noncoding RNAs in patients with acute myocardial infarction. Circ Res 115:668–677. doi:10.1161/circresaha.115.303836
Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, Blow MJ, Cohen JC, Rubin EM, Pennacchio A (2010) Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 464:409–412. doi:10.1038/nature08801
Wang H, Iacoangeli A, Lin D, Williams K, Denman RB, Hellen CUT, Tiedge H (2005) Dendritic BC1 RNA in translational control mechanisms. J Cell Biol 171:811–821. doi:10.1083/jcb.200506006
Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, Jiang Z, Xu J, Liu Q, Cao X (2014) The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science 344:310–313. doi:10.1126/science.1251456
Wapinski O, Chang HY (2011) Long noncoding RNAs and human disease. Trends Cell Biol 21:354–361. doi:10.1016/j.tcb.2011.04.001
Welsh IC, Kwak H, Chen FL, Werner M, Shopland LS, Danko CG, Lis JT, Zhang M, Martin JF, Kurpios NA (2015) Chromatin architecture of the Pitx2 locus requires CTCF- and Pitx2-dependent asymmetry that mirrors embryonic gut laterality. Cell Rep 13:337–349. doi:10.1016/j.celrep.2015.08.075
Wu P, Zuo X, Ji A (2012) Stroke-induced microRNAs: the potential therapeutic role for stroke. Exp Ther Med 3:571–576. doi:10.3892/etm.2012.452
Wu Q, Kim YC, Lu J, Xuan Z, Chen J, Zheng Y, Zhou T, Zhang MQ, Wu C-I, Wang SM (2008) Poly A-transcripts expressed in HeLa cells. PLoS One 3:e2803. doi:10.1371/journal.pone.0002803
Liua X, Liua L, Xua Q, Wua P, XZ & AJ (2012) MicroRNA as a novel drug target for cancer therapy. Expert Opin Biol Ther 12:573–580
Yang Y, Wen L, Zhu H (2015) Unveiling the hidden function of long non-coding RNA by identifying its major partner-protein. Cell Biosci 5:59. doi:10.1186/s13578-015-0050-x
Yap KL, Li S, Muñoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou M-M (2010) Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell 38:662–674. doi:10.1016/j.molcel.2010.03.021
Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, Lee JT (2013) Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 152:727–742. doi:10.1016/j.cell.2013.01.034
Yin QF, Yang L, Zhang Y, Xiang JF, Wu YW, Carmichael GG, Chen LL (2012) Long noncoding RNAs with snoRNA ends. Mol Cell 48:219–230. doi:10.1016/j.molcel.2012.07.033
Yoon JH, Abdelmohsen K, Srikantan S, Yang X, Martindale JL, De S, Huarte M, Zhan M, Becker KG, Gorospe M (2012) LincRNA-p21 suppresses target mRNA translation. Mol Cell 47:648–655. doi:10.1016/j.molcel.2012.06.027
Zangrando J, Zhang L, Vausort M, Maskali F, Marie P-Y, Wagner DR, Devaux Y (2014) Identification of candidate long non-coding RNAs in response to myocardial infarction. BMC Genomics 15:460. doi:10.1186/1471-2164-15-460
Acknowledgments
We thank Tracie Pennimpede and Reinier Boon for comments on the manuscript. We apologize to those colleagues whose work is not mentioned in this review due to space restrictions. The authors’ work is funded by the LOEWE-CGT (M.T.M.) and the DFG (German Research Foundation) Excellence Cluster Cardio-Pulmonary System (Exc147-2) (P.G.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Melissari, MT., Grote, P. Roles for long non-coding RNAs in physiology and disease. Pflugers Arch - Eur J Physiol 468, 945–958 (2016). https://doi.org/10.1007/s00424-016-1804-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-016-1804-y