
Abstract
Exchange proteins directly activated by cyclic AMP (Epac) were discovered 10 years ago as new sensors for the second messenger cyclic AMP (cAMP). Epac family, including Epac1 and Epac2, are guanine nucleotide exchange factors for the Ras-like small GTPases Rap1 and Rap2 and function independently of protein kinase A. Given the importance of cAMP in the cardiovascular system, numerous molecular and cellular studies using specific Epac agonists have analyzed the role and the regulation of Epac proteins in cardiovascular physiology and pathophysiology. The specific functions of Epac proteins may depend upon their microcellular environments as well as their expression and localization. This review discusses recent data showing the involvement of Epac in vascular cell migration, endothelial permeability, and inflammation through specific signaling pathways. In addition, we present evidence that Epac regulates the activity of various cellular compartments of the cardiac myocyte and influences calcium handling and excitation–contraction coupling. The potential role of Epac in cardiovascular disorders such as cardiac hypertrophy and remodeling is also discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cyclic adenosine 3′,5′-monophosphate (cAMP) is a universal second messenger which is produced from ATP by adenylyl cyclase upon activation of Gs-protein-coupled receptor (GPCR) [6]. cAMP plays central roles in cardiovascular regulation influencing gene expression, cell morphology, and function. In the vasculature, cAMP influences contraction/relaxation of blood vessel smooth muscle cells as well as their movement and the permeability of vascular–endothelial cells (VECs) [4, 29]. In the heart, cAMP regulates many physiological processes such as contractility, relaxation, and automaticity [7]. Besides these physiological roles, various actions of cAMP can be altered in cardiovascular diseases such as hypertrophy and heart failure [65].
Although protein kinase A (PKA) is generally recognized as the primary effector of cAMP signaling [53], other effectors are known to transduce cyclic nucleotide-encoded information. They encompass a class of cyclic nucleotide gated (CNG) cation channels and phosphodiesterases (PDEs) [9, 33, 40, 59]. The role of CNG channels appears to be specific to certain cell types such as pacemaker cells where distinct ion fluxes are regulated. More recently, a family of proteins directly activated by cAMP was discovered adding another complexity in the cAMP-mediated signaling mechanism [25, 46]. These proteins, named exchange proteins directly activated by cAMP (Epac), are activated by cAMP and function in a PKA-independent manner and therefore represent a novel mechanism for governing signaling specificity within the cAMP cascade. Specifically, they are guanine nucleotide exchange factors (GEFs) for the Ras family of small GTPases, Rap1 and Rap2 [16, 82]. Small G proteins, including Rap, act as molecular switches, cycling between an active GTP-bound state and an inactive GDP-bound state [17] (Fig. 1). cAMP-dependent activation of Epac promotes the exchange of GDP for GTP, hence switching on the Rap GTPases. The functions of cAMP-regulated GEFs in various cellular contexts are currently being unraveled, but a large body of evidence indicates that Epac is involved in multiple biological actions of cAMP such as insulin secretion and memory formation [15, 38, 70, 82, 103]. In this review, we describe the current understanding of Epac signaling and its roles in cardiovascular physiology and pathophysiology.

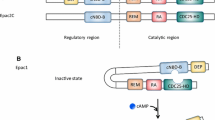
Structure and mode of action of Epac proteins. a Schematic representation of Epac proteins. Epac1 and Epac2 are multidomain proteins encoded by two distinct genes and are characterized by a N-terminal regulatory region that directly binds cAMP with high affinity (cAMP-B). Epac2A possesses a second lower affinity cAMP-binding domain whose role is unknown (cAMP-A). The catalytic domain of Epac is composed of a CDC25 homology catalytic site stabilized by a REM. Epac proteins also possess a dishevelled, Egl-10, pleckstrin domain (DEP) responsible for membrane association. Epac2B is a splice variant of Epac2 and lacks the first cAMP-A-binding domain. Epac2B is expressed in the mouse adrenal gland. b cAMP is produced by adenylyl cyclase (AC) in response to GPCR stimulation and activates its classical downstream effector, protein kinase A (PKA). PDE degrade cAMP and thereby regulate its availability for diffusion. The identification of Epac proteins as novel sensors for cAMP has broken the dogma surrounding cAMP and PKA. Epac proteins are nucleotide exchange factors for the Ras-like small GTPases, Rap1 and Rap2, that function independently of PKA. Upon binding of cAMP, Epac undergoes conformational changes that release the C-terminal catalytic region responsible for Rap activation. 8-pCPT-2′-O-Me-cAMP is a synthetic cAMP analog that specifically activates Epac
Epac protein structure and mechanisms of action
So far, two isoforms of Epac named Epac1 and Epac2 have been identified (Fig. 1). They are encoded by two distinct genes, RAPGEF3 and RAPGEF4 [25, 46]. RAPGEF3 codes for Epac1, whereas RAPGEF4 generates a long and a short variant named Epac2A and Epac2B, respectively [67]. Epac1 mRNA is expressed ubiquitously with a high expression in the heart, kidneys, ovaries, and thyroid glands, whereas Epac2 mRNA is detectable most notably in the brain, pituitary, and adrenal gland [46, 67]. Based on quantitative RT-PCR and immunoblot blot analysis, Epac1 has been shown to be more abundant than Epac2 in the myocardium [62, 101].
Epac are multi domain proteins characterized by a regulatory region and a catalytic domain. The regulatory region of Epac1 and Epac2B contains two recognized domains, a dishevelled–Egl-10–Pleckstrin (DEP) domain and a high-affinity cAMP-binding domain (cAMP-B). Epac2A shares this organization, but possesses an additional cyclic nucleotide-binding domain, which has low affinity for cAMP and uncertain biological function [24, 79]. The DEP domain of Epac proteins is responsible for membrane association and is required for the translocation of Epac1 to the plasma membrane [24, 72, 75]. The catalytic region of Epac isoforms is constructed from a Ras exchange motif (REM) and a CDC25 homology domain. The crystal structure of the inactive state of the Epac2 protein revealed that the cyclic mononucleotide-binding domain sterically blocks the access of Rap to the catalytic site in the CDC25 homology domain, which is responsible for mediating the exchange activity [79, 80]. Binding of cAMP to Epac induces large conformational changes within the protein and releases the auto-inhibitory effect of the N-terminal region, leading to Rap activation [80]. Epac isoforms can be activated by GPCRs that are positively linked to adenylyl cyclase, including serotonin 5-HT4 receptors and β-adrenergic receptors (β-AR) [58, 62, 76, 84]. Also of interest, the light chain 1 of microtubule-associated protein 1B (LC1) has been shown to act as a molecular chaperone of Epac1, increasing the binding of cAMP to this GEF and consequently increasing Epac1 signaling in rat pheochromocyta (PC12) cells [14].
Epac proteins are observed at many locations in the cell, including the cytosol, the nucleus as well as the nuclear and plasma membranes [41, 62, 72, 75]. Depending on their cellular localization and molecular partners, Epac proteins activate different downstream effectors [14, 94]. Thus, the distinction between the coupling of Epac to a specific signaling pathway is determined by its localization to micro sub-cellular compartments, explaining the different biological effects of Epac inside a given cell. In this line, muscle-specific A-kinase anchoring protein (mAKAP) plays a key role in compartmentalization of Epac-dependent signaling [20]. It is a scaffold protein that has been shown to localize cAMP-hydrolyzing PDEs, regulatory cAMP-binding subunits of PKA, ERK5, protein phosphatase 2A as well as Epac into specific intracellular compartments, thereby controlling the cellular actions of cAMP [60]. In addition, as for PKA, there is now strong evidence that PDEs contribute to the specificity of Epac signaling in a spatial and temporal manner [41, 60, 77]. For instance, PDE4B activity specifically controls the ability of nuclear Epac1 to drive nuclear export of DNA-dependent protein kinase (DNA-PK), an enzyme that provides a key part of the DNA repair systems, while cytosolic PDE4D regulates PKA-mediated nuclear import of DNA-PK [41]. But PKA and Epac may also be interconnected to regulate cellular processes [49]. In that way, a PDE4 inhibitor roflumilast has been shown to protect cardiac myocytes against NO-induced apoptosis via both cAMP–PKA and Epac-dependent pathways [49]. Therefore, by virtue of the membrane targeting and their ability to interact with other signaling proteins, Epac directs cAMP signaling to specific cellular sites.
Epac-selective cAMP analogs
The major part of available data regarding Epac-dependent functions in the cardiovascular system has been obtained by the use both in vitro and in vivo of Epac activators, which do not discriminate between Epac1 and Epac2. These Epac-selective cAMP analogs incorporate a 2′-O-methyl substitution on the ribose ring of cAMP, a modification that impairs their ability to activate PKA [30, 37, 93]. Among them, 8-pCPT-2′-O-Me-cAMP (8-(4-chloro-phenylthio)-2'-O-methyladenosine-3',5'-cyclic monophosphate) is the most commonly used Epac agonist and exhibits high affinity for Epac (K d 2.2 µM for Epac1) as well as reduced affinity for PKA (K d 200–300 µM) [30] (Fig. 1). However, like all pharmacological agents, Epac-selective cAMP analogs may be metabolized (i.e., by PDEs) into bioactive products, which may have non-specific effects [31, 73]. Therefore, it is important to mention that the involvement of Epac in a particular function is firmly established only when pharmacological data are supplemented by molecular analyses.
Epac functions in vascular cells
VECs and vascular smooth muscle cells (VSMCs) constitute the bulk of blood vessel cells and cooperate to regulate blood vessel structure and function [78]. VECs and VSMCs of healthy undamaged arteries function primarily to regulate blood vessel lumen diameter by coordinating contractility and relaxation. In contrast, during cardiovascular stresses, VSMCs and VECs undergo a process of phenotypic modulation that results in the development of cells with migratory and proliferative phenotypes, which can contribute to severe vasculopathies, such as atherosclerosis or restenosis after angioplasty [99]. Because cAMP influences many facets of the biology of VSMCs (relaxation–contraction coupling, proliferation, migration, and cellular metabolism) and VECs (proliferation, migration, cellular metabolism, and permeability), it is obviously important to determine how Epac may influence these cellular processes (Fig. 2).

Role of Epac in vascular smooth cells (VSMCs) and vascular–endothelial cells (VECs). Epac promotes (probably via Rap1) the migration of various types of VSMCs and facilitates the development of neointima thickening, a process involved in vascular restenosis. It also contributes to the proliferation of airway SMCs induced by β2-adrenergic receptors. In VECs, Epac activation increases endothelial barrier function by increasing junctional molecules such as vascular–endothelial (VE) cadherin at cell–cell contacts, by inducing cortical actin rearrangement and enhancing microtubule growth. In addition, Epac mediates activation of integrins involved in adhesion of VECs to the basal membrane. The anti-inflammatory effect of Epac involves the induction of suppressor of cytokine signaling-3 (SOCS-3), a negative regulator of IL-6 receptor signaling. Epac/Rap activation induces expression of thrombospondin-1, a key anti-angiogenic molecule. On the other hand, Epac activation stimulates VECS proliferation, a process known to be involved in angiogenesis
Epac and VSMC migration
The functional roles of Epac proteins in VSMCs are just beginning to be defined. In contrast with PKA, direct activation of Epac promotes the migration of various types of VSMCs and facilitates the development of neointima thickening, a process involved in vascular restenosis [99, 100] (Fig. 2). The pharmacological approach with the Epac-specific agonist, 8-pCPT-2′-O-Me-cAMP was verified by siRNA-mediated knockdown of Epac1, which decreased serum-mediated SMC migration [100]. The exact molecular mechanisms that lead to Epac activation and increased migration in VSMCs are unknown. The activation of Rap1, Akt, and/or ERK may be involved, as demonstrated in the regulation of cell adhesion and migration [34, 61, 76, 101]. The role of Epac in the regulation of cell migration is not restricted to VSMCs since several reports have shown that this GEF may influence this cellular process in various cell types including tumor cell migration [3, 51, 57, 101]. Interestingly, Epac contributes to the proliferation of airway SMCs induced by β2-adrenergic receptors indicating that this cAMP-GEF controls different aspects of SMC fates and may also be implicated in the pathological remodeling seen in asthmatic airways [45].
Role of Epac in VECs
Regulation of vascular–endothelial barrier function by Epac
Endothelial barrier function restricts the passage of plasma proteins and circulating cells across endothelial cells, and its dysfunction may result in an increase in vascular permeability, thereby causing edema, inflammatory, or metastatic cell infiltration [91]. Vascular permeability is induced by various pro-inflammatory mediators including thrombin and histamine. Thus, the selective regulation of vascular permeability is critical for maintaining vascular integrity in homeostasis and disease. Several stimuli such as prostaglandins that are linked to cAMP production inhibit vascular permeability of endothelial cell monolayers [34]. Some studies tend to demonstrate that Epac mediates this effect independently of PKA and may represent a new therapeutic target for modulating vascular permeability. Indeed, using pharmacological and molecular approaches in a monolayer of a human umbilical vein endothelial cell line (HUVEC), it has been reported that increased cAMP levels trigger Epac–Rap1 signaling to reduce vascular permeability by augmentation of junctional molecules such as vascular–endothelial cadherin at cell–cell contacts [22, 34, 48]. Epac1-specific siRNA showed that Rap1 activation by Epac1, but not by Epac2, was responsible for the increase in endothelial adherens junctions [48]. Inversely, Epac signaling antagonizes thrombin-induced cell junction disruption and strongly attenuates the platelet activating factor-induced permeability in HUVECs and intact rat microvessels, respectively [1, 22, 34, 48].
Since the endothelial barrier function is largely dependent on the dynamic of the actin cytoskeleton [12], several groups investigated the involvement of Epac in this process. Epac activation indeed induces reorganization of cortical actin, which supports junctional adhesion molecules thereby contributing to stabilize endothelial barrier function [11, 22, 34, 48]. The effect of Epac on actin remodeling involves the activation of the Ras-like GTPase, Rap, which can then modulate indirectly the activity of the Rho GTPase family such as Rho and Rac. In turn, Rho proteins interact with downstream cytoskeletal and cell adhesion effectors and promote cytoskeletal remodeling [10, 32]. It is suggested that the connection between the Ras and Rho GTPase families probably occurs through the recruitment of specific GEFs of the Rho family (e.g., Tiam, Vav) [10, 11, 22]. Of note, such a cross talk between Rap and RhoGTPases regulated by cAMP and Epac exists in other cellular systems including primary neurons [58, 81, 102]. Finally, besides actin remodeling, Sehrawat and colleagues [85] reported that Epac regulates microtubule dynamics in HUVECs. According to these authors, the effect of Epac activation on microtubule dynamics might have physiological consequences for barrier function as activating Epac could reverse vascular permeability increased by TGFβ and TNF-α, which are cytokines that promote barrier dysfunction by destabilizing microtubule.
Epac and inflammation
Uncontrolled and excessive migration of leukocytes from the peripheral blood into tissues is a hallmark of chronic inflammation. Importantly, it has been reported that Epac inhibits endothelial leukocyte transmigration [95]. These data suggest that the regulation of the endothelial barrier function by Epac–Rap may have some important biological consequences under conditions in which an exaggerated inflammatory response leads to vascular disease as observed in atherosclerosis [95]. In contrast, Cullere et al. [22] did not observe any blocking effect on neutrophil migration during endothelial Epac1 stimulation. This discrepancy is currently unclear. Various aspects of transendothelial leukocyte migration such as leukocyte adhesion (primarily through the activation of β1 integrins) and migration can also be influenced by Epac [5, 35, 55, 56].
VECs represent a major cellular target for many pro- and anti-inflammatory cytokines. Activated VECs are capable of producing interleukin (IL)-6, and accumulation of IL-6 has been noted within arterial lesions in several models of atherosclerosis [89]. Epac has been identified as a modulator of IL-6 signaling in HUVECs, making this cAMP sensor as a potential anti-inflammatory protein in VECs [83] (Fig. 2). It is demonstrated that the anti-inflammatory effect of Epac is PKA independent and is associated with the induction of suppressor of cytokine signaling 3 (SOCS-3), a bona fide inhibitor in vivo of gp130, the signal-transducing component of the IL-6 receptor complex (Fig. 2) [83]. Targeting the cAMP-Epac–Rap1–SOCS-3 pathway might therefore prove to be a useful strategy for combating pathologies associated with chronic vascular inflammation [15]. Interestingly, the induction of SOCS-3 expression in response to Epac activation occurs at the transcriptional level and involves the binding of the CCAAT/enhancer-binding proteins (C/EBPs) to the promoter of SOC-3 gene [97]. Although it remains to be demonstrated in VECs, PKCα is a critical requirement for efficient ERK- and C/EBP-dependent SOCS-3 induction by Epac in COS cells [13]. It should be noted that the involvement of Epac in inflammatory processes is not limited to VECs since this cAMP-GEF has been reported to display pro- and anti-inflammatory actions in various cell types including dendritic cells and macrophage cell lines [2, 15, 42, 88, 96].
Others roles of Epac in the vasculature
Obviously, other important functions of Epac in the vascular system need to be explored. For instance, a recent observation points out a role of Epac in the regulation of angiogenesis [28]. Angiogenesis is a tightly regulated process that is important in development as well as pathologic disease states such as tumor growth and metastasis [19]. In their study, Doebele and colleagues [28] show that pharmacologic activation of Epac with the cAMP analog, 8-pCPT-2′-O-Me-cAMP, inhibits vascular–endothelial growth factor (VEGF)-induced angiogenesis in vivo in a matrigel mouse model as well as in a severe combined immunodeficient mouse model of human angiogenesis. It is proposed that Epac/Rap activation induces expression of thrombospondin-1, a key anti-angiogenic molecule, which counteracts VEGF signaling via MEK5/ERK5 (Fig. 2) [28]. However, it should be noted that Epac activation stimulates in vitro VECS proliferation, a process known to be involved in angiogenesis [66]. Interestingly, 8-pCPT-2′-O-Me-cAMP has also been shown to decrease the in vitro ability of aggressive MUN-2B melanoma cells to engage in vasculogenic mimicry, a process by which a vessel is formed from tumor cells [54]. It is therefore possible that Epac may be involved in the regulation of tumor progression though various effects on new blood vessel formation.
Epac functions in the myocardium
Cellular contraction is initiated by a transient elevation in intracellular calcium ([Ca2+]i transients). During an action potential, Ca2+ influx induced by activation of voltage-dependent L-type Ca2+ channels upon membrane depolarization triggers the release of Ca2+ via intracellular Ca2+ release channels (ryanodine receptors (RyRs)) of sarcoplasmic reticulum (SR) through a Ca2+-induced Ca2+ release (CICR) mechanism [7]. Relaxation follows the decrease in [Ca2+]i mainly by the Ca2+ uptake into the SR through the SR Ca2+ ATPase and Ca2+ extrusion through the Na+/Ca2+ exchanger (Fig. 3).

Epac regulates the activity of various cellular compartments of the cardiomyocyte. Upon binding to cAMP, Epac stimulates PLC, which hydrolyzes PIP2 to produce DAG and IP3 leading to PKC and CaMKII activation. Both kinases are involved in the regulation of contractile protein phosphorylation and myofilament Ca2+ sensitivity. Epac-Rap1 signaling cAMP induces Cx43 accumulation in cell–cell contacts and enhances gating function. Epac contributes to the hypertrophic effect of β-adrenergic stimulation through the activation of the small GTPase Ras, Ca2+-sensitive proteins (CaMKII and calcineurin) and downstream transcription factors such as NFAT. This cAMP-binding guanine nucleotide exchange factor also stimulates RyR2 phosphorylation via CaMKII, and subsequent Ca2+ leak may contribute to CaMKII and calcineurin activation and arrhythmia. Epac has been reported to phosphorylate phospholamban in a CaMKII-dependent manner. AC adenylyl cyclase, CaMKII Ca2+/calmodulin-dependent protein kinase II, CN calcineurin, DAG diacylglycerol, IP3 inositol triphosphate, NFAT nuclear factor of activated T cell, PLB phospholamban, PIP2 phosphatidylinositol bisphosphate, PLC phospholipase C, RyR ryanodine receptors, SERCA SR Ca2+ ATPase, SR sarcoplasmic reticulum
cAMP, acting through PKA, influences the activity of several key cardiac proteins involved in excitation–contraction coupling, such as L-type Ca2+ channels, phospholamban, RyR, and troponin I [8]. These effects produce PKA-dependent increases in Ca2+ current (I Ca), SR Ca2+ release and uptake, as well as a desensitization of the myofilaments to Ca2+. The net result is the characteristic positive inotropic (contractile force) and lusitropic (relaxation) effects of β-AR activation in cardiac myocytes. In addition, binding of cAMP to the hyperpolarization-activated cyclic nucleotide-gated channels (HCN) that carry the pacemaker current, I f, helps to increase heart rate in response to a sympathetic stimulation (chronotropic effect). The discovery of the existence of Epac in cardiac myocytes has raised the possibility of alternative, PKA-independent, cAMP-dependent mechanisms of action involved in Ca2+ handling.
Epac in excitation–contraction coupling
The Epac1 isoform is highly expressed in the heart and shows sarcolemmal and perinuclear linear localization in neonatal and adult rat ventricular myocytes [27, 62]. Initially, Epac activation was found to stimulate CICR in pancreatic β-cells confirming a role of this cAMP-GEF in Ca2+ handling [44]. Our subsequent analyses in adult rat cardiomyocytes showed that the activity of the cardiac RyR during diastole, measured as Ca2+ sparks, was rapidly increased by Epac activation due to Ca2+/calmodulin-dependent protein kinase II (CaMKII) phosphorylation of the RyR, confirming a role of Epac in cardiac Ca2+ handling [71]. However, published reports on the amplitude of the [Ca2+]i transient are not unanimous. In this sense, the [Ca2+]i transient amplitude has been shown to be reduced by acute Epac activation due to a decrease in the SR Ca2+ load subsequent to the enhanced diastolic Ca2+ leak through the phosphorylated RyRs in rat cardiomyocytes [21, 71]. Conversely, Oestreich and colleagues [69] showed that acute treatment of single mouse ventricular cardiac myocytes with 8-pCPT-2′-O-Me-cAMP increased [Ca2+]i transient amplitude in field-stimulated cells. This process was dependent on Rap1 and phospholipase C-ε, which was also shown to be a downstream effector of Epac in neuronal cells [47, 82, 84]. The reasons for the discrepancies between the two studies are unclear and may involve species differences and/or methodological approaches such as the frequency of cardiac myocyte electrical stimulation.
Regarding the functional impact of Epac activation in cardiac myocytes, one could think that because contraction is activated by [Ca2+]i transient, which is decreased by Epac in rat cells, contraction would be weaker. However, this is not the case since we recently found that despite a decrease in the amplitude of [Ca2+]i transient, Epac shows a positive inotropic effect [21]. Specific activation of Epac or overexpression of a constitutively active form of Epac increases myofilament Ca2+ sensitivity in permeabilized rat ventricular cardiac myocytes in a PKA-independent manner. This is correlated with an increase in phosphorylation of sarcomeric proteins such as cardiac myosin-binding protein-C and troponin I [21]. Epac-dependent effects on myofilament proteins involve both protein kinase C (PKC) and CaMKII [21].
Thus, until now there are some collected pieces of evidence showing that Epac exerts a role in cardiac Ca2+ handling and excitation–contraction coupling. The involved signaling pathway is beginning to be unraveled: phospholipase C, CaMKII, and PKC have been identified so far as Epac effectors [21, 68, 69, 71]. However, more analyses are still needed to further understand Epac actions on EC coupling in physiology and to elucidate its possible implication in Ca2+ handling alterations during cardiac pathologies.
Roles of Epac in cardiac electrophysiology
Very little is known concerning the possible role of Epac on cardiac electrophysiology. To our knowledge, only I Ca, main trigger of CICR, has been analyzed. The results collected so far in rat and in mice cardiomyocytes failed to show any significant effect of 8-pCPT-2′-O-Me-cAMP on I Ca [68, 71]. However, other ionic currents in cardiac myocytes must be analyzed before excluding any effect of Epac on cardiac electrical activity. For instance in other cell types, Epac has been shown to affect ionic channels function such as the Ca2+-sensitive potassium currents in cerebellar neurons [87] and the ATP-sensitive potassium current (I K,ATP) in chromaffin and in vascular cells [43, 74]. Whether Epac effects in these ionic currents are direct or mediated by intracellular Ca2+ mobilization remains unclear. Among the Epac modulated ionic currents cited above, the I K,ATP has an important role in the heart. I K,ATP is normally inactivated by the intracellular ATP concentration present in cardiac cell. In pathological situations such as ischemia, ATP levels fall and I K,ATP is activated, promoting an outward potassium current that hyperpolarizes the cell. Membrane hyperpolarization lowers cell excitability and thus protects the cardiomyocyte in these situations. As indicated below Epac has been shown to decrease I K,ATP in non-cardiac cells [43, 74]. Therefore, one could hypothesize that, if Epac also decreases I K,ATP in cardiac myocytes, this could have deleterious actions and eventually increase excitability and promote arrhythmias. In this sense, Epac has been shown to favor arrhythmias in mice hearts [39]. However, this effect seems to be independent of ionic currents activated during the action potential (AP) since Epac did not alter AP duration and refractory periods. Proarrhythmic Epac effects would be instead dependent on Ca2+ handling alterations through CaMKII [39].
Epac and intercellular communication
In the heart, Epac may also modulate intercellular communication at the level of channels called gap junctions (GJ). GJ are formed by two docking connexins constituted by six connexins (Cx). The most predominantly expressed Cx isoform in cardiomyocytes is Cx-43 although Cx-40 and Cx-45 are also present [98]. Few studies have evaluated a possible role of Epac on connexins and GJ in the heart. Prolonged (24 h) treatment of cultured cells with the cAMP analog db-cAMP was shown to increase the expression of Cx-43 and Cx-45 but the molecular mechanisms appeared as disparate [23]. These mechanisms were more recently evaluated by Somekawa et al. [86] who demonstrated that, after 12-h treatment with a cAMP analog and/or specific activators or inhibitors of PKA or Epac, GJ neoformation was both PKA and Epac-dependent with independent mechanisms. This was associated with an increased intercellular communication. A possible role of an acute stimulation of Epac on intercellular communication in the heart has not been studied yet. Preliminary studies from our laboratory indicated that Epac-induced PKCε stimulation increased Cx-43 phosphorylation, a known target of PKC. It is interesting to note that RhoA GTPase activity was recently shown to increase channels permeability of Cx-43 in rat cardiomyocytes with a modulation by the actin cytoskeleton [26]. Since Rap, which is activated by Epac, can modulate indirectly the activity of the Rho GTPase family members, it is likely that Epac may increase intercellular permeability by this means but this needs to be verified.
Epac in cardiac hypertrophy and remodeling
Epac in cardiac hypertrophy
With respect to Ca2+ handling, the role of Epac in cardiac myocytes is not limited to the regulation of the contractile machinery. Indeed, recent research has implicated Epac as a new actor in the regulation of cardiac myocyte hypertrophy through the activation of specific Ca2+-dependent signaling pathway, suggesting that this cAMP-GEF regulates distinct pools of calcium involved in contractility and remodeling [62–64].
Adult myocyte hypertrophy is the compensatory response of the heart to stress and is characterized by non-mitotic growth, addition of new sarcomeres, fetal gene expression, and specific changes in ion channel properties. Maladaptive cardiac hypertrophy can progress to heart failure, a leading cause of morbidity and mortality in industrialized countries [36, 50]. A role of Epac1 in myocardial growth is strongly suggested by its upregulation in various models of cardiac hypertrophy such as chronic isoprenaline infusion and pressure overload induced by thoracic aortic constriction [62, 90]. A more direct evidence of its role in the regulation of cardiac growth came from the observation that Epac1 activation led to morphological changes associated with an increase in cell surface area, protein synthesis, and the expression of cardiac hypertrophic markers such as the atrial natriuretic factor [62, 64]. Consistent with its role in cardiac myocyte hypertrophy, Epac-specific activation triggers signaling pathway involving the Ca2+/calmodulin dependant phosphatase, calcineurin, and the small GTPases, Rac, and Ras in rat cardiac myocytes [52, 62, 64]. Similarly, the hypertrophic effects of Epac are dependent on both calcineurin and CaMKII in adult rat cardiac myocytes [62]. These two well-known calcium-sensitive proteins activate a hypertrophic gene program through the activation of transcriptions factors such as NFAT.
In response to long term β-adrenergic receptor stimulation, Epac induces adult cardiomyocyte hypertrophy in a cAMP-dependent but PKA-independent manner. Knockdown of Epac1 using ShRNA decreases cardiomyocyte hypertrophy induced by β-adrenergic receptor activation in neonatal cardiac myocytes [62]. Altogether, these data combined with the observation that the sympathetic activity is highly increased in heart failure indicate that Epac contributes to the progression of pathological cardiac growth, and its activation may be exacerbated in heart failure, and this contributes to the maladaptive remodeling of the heart [63].These data contrast with the observation that Epac1 inhibits the hypertrophic extracellular signal-regulated kinase 5 (ERK5) pathway by a mechanism involving Rap1 in neonatal cardiomyocytes [27]. As mentioned above, this apparent discrepancy suggests the existence of spatiotemporal dynamics of Epac signaling, which determines its coupling to different effectors and hence functional effect in a given cell type.
Epac in cardiac fibrosis
Recent data pointed to the role of Epac in the regulation of cardiac fibrosis [92, 101]. Cardiac fibroblasts synthesize and secrete components of the extracellular matrix such as fibrillar collagens [18]. In pathological conditions such as those seen in heart failure, excessive deposition of collagen occurs throughout the myocardium (i.e., interstitial fibrosis). Excess fibrosis can lead to impaired diastolic and systolic functions [18]. In their study, Yokoyama and colleagues [101] show that the profibrotic agonist, transforming growth factor β1 (TGFβ1), inhibits Epac1 expression in fibroblasts from multiple tissues. Inversely, overexpression of Epac1 inhibits TGFβ1-induced collagen synthesis in cardiac fibroblast, implying that a decrease in Epac expression is required for profibrotic response. In line with these data, the effect of adenosine agonists to inhibit angiotensin II-stimulated collagen synthesis occurs via a cAMP-Epac pathway in cardiac fibroblasts [92]. The observation that Epac interacts with matrix metalloproteinases [81] suggests that Epac might play a key role in other aspects of tissue remodeling such as the shedding of certain growth factors.
Conclusions and future directions
Besides the well-known cAMP substrate PKA, Epac is now recognized as an incontrovertible factor leading to complex and diversified cAMP signaling pathways. The evidence reviewed here demonstrate that this cAMP-GEF is involved in the control of various cardiovascular functions such as vascular cell adhesion, migration, endothelial barrier permeability, and excitation–contraction coupling. However, some studies are controversial, and it is just the beginning of the functional characterization of Epac, and obviously the list of its cellular function is growing very fast. Depending of their relative abundance, distribution, partners, and localization, Epac and PKA may act independently, cooperate, or oppose to each other in regulating a specific cellular function. The understanding of Epac and PKA connection represents therefore a real research area of interest but it is also crucial to study the individual contribution of Epac (as an isolated protein or integrated in a macromolecular complex) within the overall cAMP signaling. Further studies will presumably unravel new Epac regulators and effectors as well as specific function for Epac isoforms within the cell. For instance the contribution of Rap proteins in the function of Epac is not always clear as observed in cardiac myocytes. Data are also missing concerning the ability of hormones or neurotransmitters to activate Epac isoforms.
Importantly, Epac interferes with the regulation of cellular mechanisms intimately involved in the manifestation of diseases, such as atherosclerosis, cardiac hypertrophy, and tumor invasion. Thus, Epac proteins are not only at the crossroads of different physiological processes but also may represent attractive therapeutic targets for the treatment of various cardiovascular disorders. An in-depth analysis of the structure, the spatiotemporal regulation, and the mechanism of action of Epac will allow the development of new pharmacological compounds targeting specifically this cAMP sensor. To date, the pharmacology of Epac proteins is poor, and current studies on Epac functional effects clearly suffer from the lack of Epac-selective pharmacological inhibitors. In addition, the development of Epac knock-out mice will help to understand the pathophysiological role of Epac in the cardiovascular system and other tissues.
References
Adamson RH, Ly JC, Sarai RK, Lenz JF, Altangerel A, Drenckhahn D, Curry FE (2008) Epac/Rap1 pathway regulates microvascular hyperpermeability induced by PAF in rat mesentery. Am J Physiol Heart Circ Physiol 294:H1188–H1196
Aronoff DM, Carstens JK, Chen GH, Toews GB, Peters-Golden M (2006) Short communication: differences between macrophages and dendritic cells in the cyclic AMP-dependent regulation of lipopolysaccharide-induced cytokine and chemokine synthesis. J Interferon Cytokine Res 26:827–833
Baljinnyam E, Iwatsubo K, Kurotani R, Wang X, Ulucan C, Iwatsubo M, Lagunoff D, Ishikawa Y (2009) Epac increases melanoma migration by a heparan sulfate-related mechanism. Am J Physiol Cell Physiol 297:C802–C813
Barlow CA, Rose P, Pulver-Kaste RA, Lounsbury KM (2006) Excitation-transcription coupling in smooth muscle. J Physiol 570:59–64
Basoni C, Nobles M, Grimshaw A, Desgranges C, Davies D, Perretti M, Kramer IM, Genot E (2005) Inhibitory control of TGF-beta1 on the activation of Rap1, CD11b, and transendothelial migration of leukocytes. FASEB J 19:822–824
Beavo JA, Brunton LL (2002) Cyclic nucleotide research—still expanding after half a century. Nat Rev Mol Cell Biol 3:710–718
Bers DM (2002) Cardiac excitation–contraction coupling. Nature 415:198–205
Bers DM (2008) Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70:23–49
Biel M, Michalakis S (2009) Cyclic nucleotide-gated channels. Handb Exp Pharmacol 191:111–136
Birukova AA, Zagranichnaya T, Alekseeva E, Bokoch GM, Birukov KG (2008) Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J Cell Physiol 215:715–724
Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, Birukov KG (2007) Prostaglandins PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res 313:2504–2520
Bogatcheva NV, Verin AD (2008) The role of cytoskeleton in the regulation of vascular endothelial barrier function. Microvasc Res 76:202–207
Borland G, Bird RJ, Palmer TM, Yarwood SJ (2009) Activation of protein kinase Calpha by EPAC1 is required for the ERK- and CCAAT/enhancer-binding protein beta-dependent induction of the SOCS-3 gene by cyclic AMP in COS1 cells. J Biol Chem 284:17391–17403
Borland G, Gupta M, Magiera MM, Rundell CJ, Fuld S, Yarwood SJ (2006) Microtubule-associated protein 1B-light chain 1 enhances activation of Rap1 by exchange protein activated by cyclic AMP but not intracellular targeting. Mol Pharmacol 69:374–384
Borland G, Smith BO, Yarwood SJ (2009) EPAC proteins transduce diverse cellular actions of cAMP. Br J Pharmacol 158:70–86
Bos JL (2006) Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci 31:680–686
Bos JL, Rehmann H, Wittinghofer A (2007) GEFs and GAPs: critical elements in the control of small G proteins. Cell 129:865–877
Bujak M, Frangogiannis NG (2007) The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 74:184–195
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438:932–936
Carnegie GK, Means CK, Scott JD (2009) A-kinase anchoring proteins: from protein complexes to physiology and disease. IUBMB Life 61:394–406
Cazorla O, Lucas A, Poirier F, Lacampagne A, Lezoualc'h F (2009) The cAMP binding protein Epac regulates cardiac myofilament function. Proc Natl Acad Sci U S A 106:14144–14149
Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN (2005) Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 105:1950–1955
Darrow BJ, Fast VG, Kleber AG, Beyer EC, Saffitz JE (1996) Functional and structural assessment of intercellular communication. Increased conduction velocity and enhanced connexin expression in dibutyryl cAMP-treated cultured cardiac myocytes. Circ Res 79:174–183
de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL (2000) Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem 275:20829–20836
de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396:474–477
Derangeon M, Bourmeyster N, Plaisance I, Pinet-Charvet C, Chen Q, Duthe F, Popoff MR, Sarrouilhe D, Herve JC (2008) RhoA GTPase and F-actin dynamically regulate the permeability of Cx43-made channels in rat cardiac myocytes. J Biol Chem 283:30754–30765
Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD (2005) The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437:574–578
Doebele RC, Schulze-Hoepfner FT, Hong J, Chlenski A, Zeitlin BD, Goel K, Gomes S, Liu Y, Abe MK, Nor JE, Lingen MW, Rosner MR (2009) A novel interplay between Epac/Rap1 and MEK5/ERK5 regulates thrombospondin to control angiogenesis. Blood. PMID: 19710505 (in press)
Dudek SM, Garcia JG (2001) Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 91:1487–1500
Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Doskeland SO, Blank JL, Bos JL (2002) A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol 4:901–906
Enyeart JA, Enyeart JJ (2009) Metabolites of an Epac-selective cAMP analog induce cortisol synthesis by adrenocortical cells through a cAMP-independent pathway. PLoS One 4:e6088
Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420:629–635
Fischmeister R, Castro LR, Abi-Gerges A, Rochais F, Jurevicius J, Leroy J, Vandecasteele G (2006) Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res 99:816–828
Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N (2005) Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol 25:136–146
Goichberg P, Kalinkovich A, Borodovsky N, Tesio M, Petit I, Nagler A, Hardan I, Lapidot T (2006) cAMP-induced PKCzeta activation increases functional CXCR4 expression on human CD34+ hematopoietic progenitors. Blood 107:870–879
Ho KK, Pinsky JL, Kannel WB, Levy D (1993) The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol 22:6A–13A
Holz GG, Chepurny OG, Schwede F (2008) Epac-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell Signal 20:10–20
Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG (2006) Cell physiology of cAMP sensor Epac. J Physiol 577:5–15
Hothi SS, Gurung IS, Heathcote JC, Zhang Y, Booth SW, Skepper JN, Grace AA, Huang CL (2008) Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflugers Arch 457:253–270
Houslay MD, Baillie GS, Maurice DH (2007) cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling. Circ Res 100:950–966
Huston E, Lynch MJ, Mohamed A, Collins DM, Hill EV, MacLeod R, Krause E, Baillie GS, Houslay MD (2008) EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. Proc Natl Acad Sci U S A 105:12791–12796
Jing H, Yen JH, Ganea D (2004) A novel signaling pathway mediates the inhibition of CCL3/4 expression by prostaglandin E2. J Biol Chem 279:55176–55186
Kang G, Chepurny OG, Malester B, Rindler MJ, Rehmann H, Bos JL, Schwede F, Coetzee WA, Holz GG (2006) cAMP sensor Epac as a determinant of ATP-sensitive potassium channel activity in human pancreatic beta cells and rat INS-1 cells. J Physiol 573:595–609
Kang G, Joseph JW, Chepurny OG, Monaco M, Wheeler MB, Bos JL, Schwede F, Genieser HG, Holz GG (2003) Epac-selective cAMP analog 8-pCPT-2'-O-Me-cAMP as a stimulus for Ca2 + -induced Ca2+ release and exocytosis in pancreatic beta-cells. J Biol Chem 278:8279–8285
Kassel KM, Wyatt TA, Panettieri RA Jr, Toews ML (2008) Inhibition of human airway smooth muscle cell proliferation by beta 2-adrenergic receptors and cAMP is PKA independent: evidence for EPAC involvement. Am J Physiol Lung Cell Mol Physiol 294:L131–L138
Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282:2275–2279
Keiper M, Stope MB, Szatkowski D, Bohm A, Tysack K, Vom Dorp F, Saur O, Oude Weernink PA, Evellin S, Jakobs KH, Schmidt M (2004) Epac- and Ca2+-controlled activation of Ras and extracellular signal-regulated kinases by Gs-coupled receptors. J Biol Chem 279:46497–46508
Kooistra MR, Corada M, Dejana E, Bos JL (2005) Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett 579:4966–4972
Kwak HJ, Park KM, Choi HE, Chung KS, Lim HJ, Park HY (2008) PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways. Cell Signal 20:803–814
Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP (1990) Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 322:1561–1566
Lezoualc'h F (2009) Epac in melanoma: a contributor to tumor cell physiology? Am J Physiol Cell Physiol 297:797–799
Lezoualc'h F, Metrich M, Hmitou I, Duquesnes N, Morel E (2008) Small GTP-binding proteins and their regulators in cardiac hypertrophy. J Mol Cell Cardiol 44:623–632
Lincoln TM, Komalavilas P, Boerth NJ, MacMillan-Crow LA, Cornwell TL (1995) cGMP signaling through cAMP- and cGMP-dependent protein kinases. Adv Pharmacol 34:305–322
Lissitzky JC, Parriaux D, Ristorcelli E, Verine A, Lombardo D, Verrando P (2009) Cyclic AMP signaling as a mediator of vasculogenic mimicry in aggressive human melanoma cells in vitro. Cancer Res 69:802–809
Lorenowicz MJ, Fernandez-Borja M, Hordijk PL (2007) cAMP signaling in leukocyte transendothelial migration. Arterioscler Thromb Vasc Biol 27:1014–1022
Lorenowicz MJ, van Gils J, de Boer M, Hordijk PL, Fernandez-Borja M (2006) Epac1-Rap1 signaling regulates monocyte adhesion and chemotaxis. J Leukoc Biol 80:1542–1552
Lyle KS, Raaijmakers JH, Bruinsma W, Bos JL, de Rooij J (2008) cAMP-induced Epac-Rap activation inhibits epithelial cell migration by modulating focal adhesion and leading edge dynamics. Cell Signal 20:1104–1116
Maillet M, Robert SJ, Cacquevel M, Gastineau M, Vivien D, Bertoglio J, Zugaza JL, Fischmeister R, Lezoualc'h F (2003) Crosstalk between Rap1 and Rac regulates secretion of sAPPalpha. Nat Cell Biol 5:633–639
Maurice DH, Palmer D, Tilley DG, Dunkerley HA, Netherton SJ, Raymond DR, Elbatarny HS, Jimmo SL (2003) Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol Pharmacol 64:533–546
McConnachie G, Langeberg LK, Scott JD (2006) AKAP signaling complexes: getting to the heart of the matter. Trends Mol Med 12:317–323
Mei FC, Qiao J, Tsygankova OM, Meinkoth JL, Quilliam LA, Cheng X (2002) Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J Biol Chem 277:11497–11504
Métrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, Lezoualc'h F (2008) Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ Res 102:959–965
Métrich M, Morel E, Berthouze M, Pereira L, Charron P, Gomez AM, Lezoualc'h F (2009) Functional characterization of the cAMP-binding proteins Epac in cardiac myocytes. Pharmacol Rep 61:146–153
Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompre AM, Vandecasteele G, Lezoualc'h F (2005) cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ Res 97:1296–1304
Movsesian MA, Bristow MR (2005) Alterations in cAMP-mediated signaling and their role in the pathophysiology of dilated cardiomyopathy. Curr Top Dev Biol 68:25–48
Namkoong S, Kim CK, Cho YL, Kim JH, Lee H, Ha KS, Choe J, Kim PH, Won MH, Kwon YG, Shim EB, Kim YM (2009) Forskolin increases angiogenesis through the coordinated cross-talk of PKA-dependent VEGF expression and Epac-mediated PI3K/Akt/eNOS signaling. Cell Signal 21:906–915
Niimura M, Miki T, Shibasaki T, Fujimoto W, Iwanaga T, Seino S (2009) Critical role of the N-terminal cyclic AMP-binding domain of Epac2 in its subcellular localization and function. J Cell Physiol 219:652–658
Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV (2009) Epac and phospholipase Cepsilon regulate Ca2+ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. J Biol Chem 284:1514–1522
Oestreich EA, Wang H, Malik S, Kaproth-Joslin KA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV (2007) EPAC-mediated activation of phospholipase Cepsilon plays a critical role in beta -adrenergic receptor dependent enhancement of Ca2+ mobilization in cardiac myocytes. J Biol Chem 282:5488–5495
Ostroveanu A, van der Zee EA, Eisel UL, Schmidt M, Nijholt IM (2009) Exchange protein activated by cyclic AMP 2 (Epac2) plays a specific and time-limited role in memory retrieval. Hippocampus (in press)
Pereira L, Métrich M, Fernandez-Velasco M, Lucas A, Leroy J, Perrier R, Morel E, Fischmeister R, Richard S, Benitah JP, Lezoualc'h F, Gomez AM (2007) The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol 583:685–694
Ponsioen B, Gloerich M, Ritsma L, Rehmann H, Bos JL, Jalink K (2009) Direct spatial control of Epac1 by cyclic AMP. Mol Cell Biol 29:2521–2531
Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, Beavo JA, Butt E (2008) Cyclic nucleotide analogs as probes of signaling pathways. Nat Methods 5:277–278
Purves GI, Kamishima T, Davies LM, Quayle JM, Dart C (2009) Exchange protein activated by cAMP (Epac) mediates cAMP-dependent but protein kinase A-insensitive modulation of vascular ATP-sensitive potassium channels. J Physiol 587:3639–3650
Qiao J, Mei FC, Popov VL, Vergara LA, Cheng X (2002) Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J Biol Chem 277:26581–26586
Rangarajan S, Enserink JM, Kuiperij HB, de Rooij J, Price LS, Schwede F, Bos JL (2003) Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the beta 2-adrenergic receptor. J Cell Biol 160:487–493
Raymond DR, Wilson LS, Carter RL, Maurice DH (2007) Numerous distinct PKA-, or EPAC-based, signalling complexes allow selective phosphodiesterase 3 and phosphodiesterase 4 coordination of cell adhesion. Cell Signal 19:2507–2518
Regan CP, Adam PJ, Madsen CS, Owens GK (2000) Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest 106:1139–1147
Rehmann H, Arias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL (2008) Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature 455:124–127
Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL (2006) Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature 439:625–628
Robert S, Maillet M, Morel E, Launay JM, Fischmeister R, Mercken L, Lezoualc'h F (2005) Regulation of the amyloid precursor protein ectodomain shedding by the 5-HT4 receptor and Epac. FEBS Lett 579:1136–1142
Roscioni SS, Elzinga CR, Schmidt M (2008) Epac: effectors and biological functions. Naunyn Schmiedebergs Arch Pharmacol 377:345–357
Sands WA, Woolson HD, Milne GR, Rutherford C, Palmer TM (2006) Exchange protein activated by cyclic AMP (Epac)-mediated induction of suppressor of cytokine signaling 3 (SOCS-3) in vascular endothelial cells. Mol Cell Biol 26:6333–6346
Schmidt M, Evellin S, Weernink PA, von Dorp F, Rehmann H, Lomasney JW, Jakobs KH (2001) A new phospholipase-C-calcium signalling pathway mediated by cyclic AMP and a Rap GTPase. Nat Cell Biol 3:1020–1024
Sehrawat S, Cullere X, Patel S, Italiano J Jr, Mayadas TN (2008) Role of Epac1, an exchange factor for Rap GTPases, in endothelial microtubule dynamics and barrier function. Mol Biol Cell 19:1261–1270
Somekawa S, Fukuhara S, Nakaoka Y, Fujita H, Saito Y, Mochizuki N (2005) Enhanced functional gap junction neoformation by protein kinase A-dependent and Epac-dependent signals downstream of cAMP in cardiac myocytes. Circ Res 97:655–662
Ster J, De Bock F, Guerineau NC, Janossy A, Barrere-Lemaire S, Bos JL, Bockaert J, Fagni L (2007) Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc Natl Acad Sci U S A 104:2519–2524
Tan KS, Nackley AG, Satterfield K, Maixner W, Diatchenko L, Flood PM (2007) Beta2 adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-kappaB-independent mechanisms. Cell Signal 19:251–260
Tedgui A, Mallat Z (2006) Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev 86:515–581
Ulucan C, Wang X, Baljinnyam E, Bai Y, Okumura S, Sato M, Minamisawa S, Hirotani S, Ishikawa Y (2007) Developmental changes in gene expression of Epac and its upregulation in myocardial hypertrophy. Am J Physiol Heart Circ Physiol 293:H1662–H1672
van Hinsbergh VW, van Nieuw Amerongen GP (2002) Endothelial hyperpermeability in vascular leakage. Vascul Pharmacol 39:171–172
Villarreal F, Epperson SA, Ramirez-Sanchez I, Yamazaki KG, Brunton LL (2009) Regulation of cardiac fibroblast collagen synthesis by adenosine: roles for Epac and PI3K. Am J Physiol Cell Physiol 296:C1178–C1184
Vliem MJ, Ponsioen B, Schwede F, Pannekoek WJ, Riedl J, Kooistra MR, Jalink K, Genieser HG, Bos JL, Rehmann H (2008) 8-pCPT-2'-O-Me-cAMP-AM: an improved Epac-selective cAMP analogue. Chembiochem 9:2052–2054
Wang Z, Dillon TJ, Pokala V, Mishra S, Labudda K, Hunter B, Stork PJ (2006) Rap1-mediated activation of extracellular signal-regulated kinases by cyclic AMP is dependent on the mode of Rap1 activation. Mol Cell Biol 26:2130–2145
Wittchen ES, Worthylake RA, Kelly P, Casey PJ, Quilliam LA, Burridge K (2005) Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J Biol Chem 280:11675–11682
Xu XJ, Reichner JS, Mastrofrancesco B, Henry WL Jr, Albina JE (2008) Prostaglandin E2 suppresses lipopolysaccharide-stimulated IFN-beta production. J Immunol 180:2125–2131
Yarwood SJ, Borland G, Sands WA, Palmer TM (2008) Identification of CCAAT/enhancer-binding proteins as exchange protein activated by cAMP-activated transcription factors that mediate the induction of the SOCS-3 gene. J Biol Chem 283:6843–6853
Yeager M, Harris AL (2007) Gap junction channel structure in the early 21st century: facts and fantasies. Curr Opin Cell Biol 19:521–528
Yokoyama U, Minamisawa S, Quan H, Akaike T, Jin M, Otsu K, Ulucan C, Wang X, Baljinnyam E, Takaoka M, Sata M, Ishikawa Y (2008) Epac1 is upregulated during neointima formation and promotes vascular smooth muscle cell migration. Am J Physiol Heart Circ Physiol 295:H1547–H1555
Yokoyama U, Minamisawa S, Quan H, Akaike T, Suzuki S, Jin M, Jiao Q, Watanabe M, Otsu K, Iwasaki S, Nishimaki S, Sato M, Ishikawa Y (2008) Prostaglandin E2-activated Epac promotes neointimal formation of the rat ductus arteriosus by a process distinct from that of cAMP-dependent protein kinase A. J Biol Chem 283:28702–28709
Yokoyama U, Patel HH, Lai NC, Aroonsakool N, Roth DM, Insel PA (2008) The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. Proc Natl Acad Sci U S A 105:6386–6391
Zaldua N, Gastineau M, Hoshino M, Lezoualc'h F, Zugaza JL (2007) Epac signaling pathway involves STEF, a guanine nucleotide exchange factor for Rac, to regulate APP processing. FEBS Lett 581:5814–5818
Zhang CL, Katoh M, Shibasaki T, Minami K, Sunaga Y, Takahashi H, Yokoi N, Iwasaki M, Miki T, Seino S (2009) The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science 325:607–610
Acknowledgments
The work of F.L. mentioned herein was supported by INSERM, AP-HP, Fondation pour la Recherche Médicale (équipe FRM), and the Agence Nationale de la Recherche (EPAC-06, EPAC-09). M.M. and M.B. were supported by a grant from the Fondation pour la Recherche Médicale and Lefoulon-Delalande, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Métrich, M., Berthouze, M., Morel, E. et al. Role of the cAMP-binding protein Epac in cardiovascular physiology and pathophysiology. Pflugers Arch - Eur J Physiol 459, 535–546 (2010). https://doi.org/10.1007/s00424-009-0747-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-009-0747-y