
Abstract
Purposes
This work aimed to assess the possible role of TRIM25 in regulating hyperglycemia-induced inflammation, senescence, and oxidative stress in retinal microvascular endothelial cells, all of which exert critical roles in the pathological process of diabetic retinopathy.
Methods
The effects of TRIM25 were investigated using streptozotocin-induced diabetic mice, human primary retinal microvascular endothelial cells cultured in high glucose, and adenoviruses for TRIM25 knockdown and overexpression. TRIM25 expression was evaluated by western blot and immunofluorescence staining. Inflammatory cytokines were detected by western blot and quantitative real-time PCR. Cellular senescence level was assessed by detecting senescent marker p21 and senescence‐associated‐β‐galactosidase activity. The oxidative stress state was accessed by detecting reactive oxygen species and mitochondrial superoxide dismutase.
Results
TRIM25 expression is elevated in the endothelial cells of the retinal fibrovascular membrane from diabetic patients compared with that of the macular epiretinal membrane from non-diabetic patients. Moreover, we have also observed a significant increase in TRIM25 expression in diabetic mouse retina and retinal microvascular endothelial cells under hyperglycemia. TRIM25 knockdown suppressed hyperglycemia-induced inflammation, senescence, and oxidative stress in human primary retinal microvascular endothelial cells while TRIM25 overexpression further aggregates those injuries. Further investigation revealed that TRIM25 promoted the inflammatory responses mediated by the TNF-α/NF-κB pathway and TRIM25 knockdown improved cellular senescence by increasing SIRT3. However, TRIM25 knockdown alleviated the oxidative stress independent of both SIRT3 and mitochondrial biogenesis.
Conclusion
Our study proposed TRIM25 as a potential therapeutic target for the protection of microvascular function during the progression of diabetic retinopathy.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As the most common diabetic microvascular complication, diabetic retinopathy (DR) is the leading cause of preventable blindness in the working-age population [1, 2]. Early DR is mainly manifested as retinal microangiopathy accompanied by the loss of retinal microvascular endothelial cells (RMECs) and pericytes, resulting in increased vascular permeability, retinal ischemia, and hypoxia, and eventually leading to pathological neovascularization in proliferative DR [3]. Multiple pathological factors, including inflammation [4, 5], senescence [6, 7], and oxidative stress [8, 9], are well recognized as being involved in microvascular endothelial injury under high glucose, and no effective intervention is currently available for early DR due to its complex pathogenesis.
As a member of the tripartite motif (TRIM), family of E3 ubiquitin ligases, tripartite motif-containing protein 25 (TRIM25) is involved in a variety of physiological and pathological processes, including innate immunity, cell proliferation and survival, and RNA metabolism [10]. Recent work has also identified TRIM25 as a lipid metabolism regulator by suppressing adipocyte differentiation [11] and a glucose metabolism regulator by decreasing the expression of tricarboxylic acid cycle enzymes [12]. Although several members of the TRIM family have been reported to play a variety of roles in diabetes and its complications [13], it is currently unclear whether TRIM25 plays a role in DR development.
By detecting and comparing the expression and distribution of TRIM25 in human epiretinal membrane tissues and mouse retina, we found that TRIM25 was highly expressed in retinal microvascular endothelial cells and further elevated under the hyperglycemia state. Therefore, we further investigated the effects of TRIM25 on inflammation, senescence, and oxidative stress in human primary retinal microvascular endothelial cells (HRMECs) under high glucose by TRIM25 knockdown or overexpression. The protective effects of TRIM25 knockdown on HRMECs under high glucose and its mechanisms were discussed, and the results were reported as follows.
Methods
Adenoviruses
Adenovirus shT expressing a short hairpin RNA targeting human TRIM25 mRNA and adenovirus TOE overexpressing human TRIM25 mRNA (GenBank NM_005082.5), and vehicle controls sh-ctrl (ADV-U6-CMV-MCS) and ad-ctrl (ADV-CMV-MCS-3FLAG) were purchased from OBiO Technology (Shanghai, China). The knockdown target sequences are shown in Supplementary Table 1.
Human epiretinal membrane tissues
Retinal fibrovascular membrane tissues harvested from patients with DR and macular epiretinal membrane tissues harvested from patients without diabetes (non-DR) were used for immunofluorescence staining. The evaluation of DR was according to the International Clinical Diabetic Retinopathy Severity Scale adopted by the American Academy of Ophthalmology and the International Council of Ophthalmology. The clinical information of the patients is included in Supplemental Table 2.
DR mouse model
Wild-type (WT) C57BL/6 male mice were purchased from the Shanghai Laboratory Animal Center, Chinese Academy of Sciences (Shanghai, China). The type 1 diabetic mouse model was induced by intraperitoneal injection of streptozotocin (STZ, Sigma Aldrich). Mice (8 weeks old, ~ 25 g body weight) were randomly assigned to the DR or WT group. The DR group was fasted for 12 h before the first STZ injection and then administered with STZ (55 mg/kg in 10 mM citrate buffer, pH 4.5) injection for 6 consecutive days. The WT group received sodium citrate buffer as vehicle controls. The mice with random blood glucose levels over 300 mg/dL were determined as diabetic mice, n = 6–12 for each group. The mice were euthanized by CO2 inhalation, and their eyeballs were harvested ~ 5 months after diabetic induction.
Cell culture
Human retinal microvascular endothelial cells (HRMECs, three to four passages) (Cell Systems, WA, USA) were cultured in ECM (ScienCell, CA, USA) containing 5% FBS, 1% ECGS, 100 U/mL penicillin, and 100 μg/ml streptomycin at 5.5 mM d-glucose concentration for normal glucose and 30 mM for high glucose. The HRMECs were infected with shT at a multiplicity of infection of 100, TOE at a multiplicity of infection of 25, or corresponding vehicle controls at approximately 70% confluence. Inhibitors for SIRT3 (3-TYP, 16 μM) were used individually or in combination with the adenoviruses. Each experiment was repeated independently at least three times.
Immunofluorescence staining
Eyeballs from mice or human epiretinal membrane tissues were fixed with 4% paraformaldehyde (Sangon Biotech, Shanghai, China) overnight at 4 °C. Fixed issues were paraffin-embedded and sectioned at 5 μm thickness, and the fixed eyeballs were sectioned through the optic disk. Before staining, the paraffin sections were dewaxed with xylene and rehydrated by sequential dipping into different concentrations of ethanol and water. Antigens were retrieved in a boiled target retrieval solution (Beyotime) for 15 min. After three washes with PBS, the paraffin sections were blocked in a blocking buffer (PBS containing 5% BSA and 0.5% Triton X-100) for 30 min at 37 °C and incubated with primary antibody diluted in the blocking buffer at 4 °C overnight. Then, they were subjected to a secondary antibody diluted in the blocking buffer for 1 h at 25 °C. After each incubation, three washes with PBS were performed. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were taken with a confocal microscope (× 200 or × 400 magnification, Leica Microsystems, Wetzlar, Germany).
Western blot analysis
Cultured HRMECs and mouse retinal samples were lysed in RIPA lysis buffer (Cell signaling) containing 1 mM PMSF (Beyotime) and 1 × protease/phosphatase inhibitor cocktail (Roche). Protein concentrations were measured using a BCA protein assay reagent (Beyotime). Samples were loaded onto a 4–20% SDS-PAGE gel (30–50 μg/sample) and transferred onto immuno-blot polyvinylidene fluoride membranes (Roche, pore size 0.22 μm) in the Trans-Blot Turbo Transfer System (Bio-Rad) and subsequently blocked in TBS-T (TBS + 0.1% Tween-20) with 5% nonfat milk powder at 37 °C for 3 h. Next, the membranes were immunoblotted with primary antibodies diluted in a primary antibody dilution buffer (Beyotime) at 4 °C overnight and then incubated for 1 h at 25 °C with peroxidase-conjugated secondary antibodies diluted in TBS-T with 1% nonfat milk powder. Antibody detection was performed with an enhanced chemiluminescence substrate (Millipore, MA, USA) with an Amersham Imager 600 (GE, CT, USA). The antibodies used are shown in Supplemental Table 3.
Quantitative real-time PCR
RNA was extracted using TRIzol (Invitrogen), after which DNase treatment (Takara, Kyoto, Japan) was performed. Reverse transcription was carried out with a cDNA Synthesis Kit (Takara). Quantitative real-time PCR (qPCR) was performed using TB Green Premix Ex Taq™ (Takara) by ViiA 7 (Applied Biosystems, DE, USA). Relative mRNA levels were quantified by normalizing with β-actin detected using commercially available predesigned primers (Sangon Biotech). The primer sequences are as follows:
TNF-α, forward 5′-GCTGCACTTTGGAGTGATCG-3′,reverse 5′-GCTTGAGGGTTTGCTACAACA-3′,
NF-κB, forward 5′-CCCCACGAGCTTGTAGGAAA-3′,reverse 5′- CCACGCTGCTCTTCTTGGAA-3′,
IL-1β, forward 5′-TCGAGGGACAGGATATGGAG-3′,reverse 5′-TCTTTCAACACGCGAGGACAG-3′.
Senescence‐associated‐β‐galactosidase staining
Intracellular senescence‐associated‐β‐galactosidase (SA‐β‐Gal) activity was assayed using an SA‐β‐Gal Staining Kit (Beyotime) according to the manufacturer’s instructions. Senescent cells were identified as bluish-green‐stained cells under a phase‐contrast microscope. The percentage of SA‐β‐Gal‐positive cells was determined by the mean of five random fields (× 100 magnification).
Intracellular ROS and mitochondrial ROS measurement
HRMECs were incubated with diluted intracellular ROS probes 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA, 10 μM, Molecular Probes, OR, USA) or MitoSOX red mitochondrial superoxide indicator in serum-free medium for 30 min protected from light. After washing DCFH-DA with cold PBS or washing MitoSOX with HBSS buffer, the cells were harvested and analyzed on a FACS scan flow cytometry machine (Beckman, CA, USA).
Mitochondria DNA copy number determination
Total DNA from HRMECs was extracted using the TaKaRa MiniBEST Universal Genomic DNA Extraction Kit (TaKaRa) and quantified using NanoDrop 2000 (Thermo Fisher Scientific). Mitochondrial DNA level was detected using the Human Mitochondrial DNA Monitoring Primer Set (TaKaRa) with qPCR. The procedures for DNA extraction and mtDNA quantification were conducted according to the manufacturer’s manual.
Statistical analysis
All data originated from at least three independent experiments and were presented as mean ± standard deviation (SD). Differences among experimental groups were analyzed by two-tailed student’s t-test or one-way ANOVA (followed by Turkey’s or Sidak’s test) using GraphPad Prism 9.0. Significance was set at P < 0.05.
Results
TRIM25 is highly expressed in retinal vascular endothelial cells and is further elevated under hyperglycemia
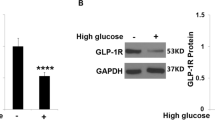
To begin to understand the state of TRIM25 in retinal vascular endothelial cells in vivo, we examined the expression of TRIM25 in human epiretinal membrane tissues. TRIM25 expression is elevated in the endothelial cells of the retinal fibrovascular membrane from diabetic patients compared with that of the macular epiretinal membrane from non-diabetic patients, as shown in the results of immunofluorescence staining (Fig. 1a). Consistent with that, western blot analysis of the mouse retina showed that the TRIM25 protein level increased in the diabetic retinas compared with the control group (Fig. 1b and c). The immunofluorescence staining of the mouse retina was next conducted to further investigate the distribution of TRIM25. As shown in Fig. 1d, the increase in TRIM25 level in diabetic mouse retina was also confirmed, and special attention needs to be paid to the high expression of TRIM25 in retinal vascular endothelial cells. Moreover, high glucose increased the TRIM25 protein levels in the HRMECs in vivo (Fig. 1e and f).

TRIM25 is highly expressed in retinal vascular endothelial cells and is elevated under hyperglycemia. a Immunofluorescence staining of TRIM25 in macular epiretinal membranes from patients without diabetes (non-DR, n = 5 eyes) and retinal fibrovascular membranes from patients with diabetic retinopathy (DR, n = 6 eyes). Scale bar, 100 μm. b and c Western blot analysis of TRIM25 in the whole retina extracts of nondiabetic C57BL/6 mice (WT) and diabetic mice (DR). d Immunofluorescence staining of TRIM25. White arrows indicate TRIM25 that is highly expressed in retinal vascular endothelial cells. n = 6. Scale bar, 50 μm. e and f Western blot analysis of TRIM25. Human retinal microvascular endothelial cells (HRMECs) were cultured in a medium containing 5.5 (NG) or 30 mmol/L (HG) glucose for 72 h as indicated. All results are displayed as means ± SD. **P < 0.01, unpaired two-tailed student’s t-test
TRIM25 knockdown reduces hyperglycemia-induced inflammation in HRMECs
To initially investigate the effect of TRIM25, we treated HRMECs under high glucose with adenoviruses for TRIM25 knockdown (Fig. 2a). Inflammatory injury mediated by NF-κB is a crucial and classical factor in the apoptosis and loss of endothelial cells in diabetes and diabetic complications [4], so we detected the activated form of NF-κB and Caspase-3 by western blot. The results showed that both p-NF-κB and cleaved Caspase-3 were elevated under high glucose, which was reversed by the TRIM25 knockdown (Fig. 2b and c). Moreover, TRIM25 has been reported to mediate inflammatory response by activating the TNF-α/NF-κB pathway [14] and by increasing IL-1β production [15]. Thus, we next measured the RNA level of TNF-α, NF-κB, and IL-1β by quantitative real-time PCR (qPCR), showing that high glucose increased the RNA expression of all three inflammatory factors while TRIM25 knockdown reduced them (Fig. 2d). We have also evaluated the effect of TRIM25 overexpression (Fig. 2e) on the TNF-α/NF-κB pathway, showing that TRIM25 overexpression further elevated the protein level of p-NF-κB and TNF-α under high glucose (Fig. 2f). Therefore, TRIM25 is involved in hyperglycemia-induced inflammatory injury mediated by the TNF-α/NF-κB pathway in HRMECs.

TRIM25 knockdown reduces hyperglycemia-induced inflammation in HRMECs. a–c Western blot analysis of TRIM25, p-NF-κB, and cleaved caspase-3. HRMECs were treated with adenoviruses for TRIM25 knockdown (shT) or vehicle controls in NG or HG medium for 72 h as indicated. d Quantitative real-time PCR (qPCR) of inflammatory cytokines TNF-α, NF-κB, and IL-1β. HRMECs were treated with shT or vehicle controls for 72 h in NG or HG medium as indicated. e and f Western blot analysis of TRIM25, p-NF-κB, and TNF-α. HRMECs were treated with adenovirus for TRIM25 overexpression (TOE) or vehicle controls for 72 h in NG or HG medium as indicated. Each experiment was repeated independently at least three times. All results are displayed as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA analysis
TRIM25 knockdown attenuates hyperglycemia-induced senescence in HRMECs
Cellular senescence of vascular endothelial cells contributes to the pathogenesis of various age-related diseases including DR [7, 16]. Therefore, we conducted a western blot analysis of senescent marker p21 and the detection of SA‐β‐Gal activity to determine the effect of TRIM25 on cellular senescence in HRMECs under high glucose, showing that TRIM25 overexpression aggravated senescence induced by high glucose (Fig. 3a–d). Extensive studies have clearly revealed that sirtuins (SIRTs) regulate endothelial senescence in the process of vascular aging [17, 18]. Here, we found by western blot analysis that SIRT3, a mitochondrial member of the SIRTs, declined in HRMECs under high glucose, while TRIM25 knockdown reversed that accompanied by a decline of p21 (Fig. 3e–g). To explore whether SIRT3 is involved in the role of TRIM25 on the cellular senescence, we treated HRMECs with SIRT3 inhibitor 3-TYP. As shown in Fig. 3h and i, TRIM25 knockdown alleviated the senescence induced by high glucose and SIRT3 inhibition blocked its protective effects. These data reveal that TRIM25 knockdown attenuates hyperglycemia-induced cellular senescence in HRMECs by increasing SIRT3.

TRIM25 knockdown attenuates hyperglycemia-induced cellular senescence in HRMECs. a and b Western blot analysis of p21. HRMECs were treated with TOE or vehicle controls for 72 h in NG or HG medium as indicated. c and d Cellular senescence detected by SA‐β‐Gal staining in HRMECs subjected to treatments (72 h) as indicated. Scale bar, 100 μm. e–g Western blot analysis of p21 and SIRT3. HRMECs were treated with shT or vehicle controls in NG or HG medium for 72 h as indicated. h and i Cellular senescence detected by SA‐β‐Gal staining. HRMECs were treated with DMSO or SIRT3 inhibitor 3-TYP combined with shT or vehicle controls for 72 h in NG or HG medium as indicated. Scale bar, 100 μm. Each experiment was repeated independently at least three times. All results are displayed as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA analysis
TRIM25 knockdown decreases oxidative stress in HRMECs under a hyperglycemia state
Hyperglycemia induces excessive reactive oxygen species (ROS) production, which also plays an essential role in diabetic vascular endothelial injury including inflammation and senescence [8, 9]. Thus, we measured the intracellular ROS production and examined the effect of TRIM25 on it. As shown in Fig. 4a, high glucose-induced ROS elevation, which was rescued by TRIM25 knockdown. The same result was observed in the western blot for mitochondrial superoxide dismutase (SOD2), showing that defective SOD2 antioxidant response under high glucose was improved by TRIM25 knockdown (Fig. 4b and c). We also confirmed the effects of TRIM25 overexpression, which further elevated the intracellular ROS production under high glucose with a significant decrease in SOD2 protein level (Fig. 4d–f). Mitochondrial ROS are the main factor in endothelial oxidative injury under high glucose [19], so we determined the mitochondrial ROS generation using MitoSOX red mitochondrial superoxide indicator and TRIM25 knockdown eliminated mitochondrial ROS elevation under high glucose (Fig. 4g). As SIRT3 can reduce ROS in vascular endothelium by increasing SOD and improving mitochondrial biogenesis [20,21,22], we next investigate whether SIRT3 or the mitochondrial homeostasis is involved in the mechanism underlying the alleviation of oxidative stress by TRIM25 knockdown. However, as shown in Fig. 4h and i, the improvement of the ROS production by TRIM25 knockdown in HRMECs under high glucose remained even after the treatment of SIRT3 inhibition, and TRIM25 knockdown did not promote the impaired mitochondrial biogenesis under high glucose, suggesting that TRIM25 knockdown alleviated oxidative stress through mechanisms independent of mitochondrial homeostasis or SIRT3.

TRIM25 knockdown decreases oxidative stress in HRMECs under a hyperglycemia state. a The intracellular reactive oxygen species (ROS) of HRMECs following treatments with shT or vehicle controls for 72 h in NG or HG medium as indicated. b and c Western blot analysis of SOD2. HRMECs were treated with shT or vehicle controls for 72 h in NG or HG medium as indicated. d The intracellular ROS of HRMECs following treatments with 25 moi of TOE, 100 moi of TOE, or vehicle controls for 72 h in HG medium as indicated. e and f Western blot analysis of SOD2. HRMECs were treated with TOE or vehicle controls for 72 h in NG or HG medium as indicated. g The mitochondrial ROS of HRMECs following treatments with shT or vehicle controls for 72 h in NG or HG medium as indicated. h The intracellular ROS of HRMECs following treatments with DMSO or SIRT3 inhibitor 3-TYP combined with shT or vehicle controls for 72 h in HG medium as indicated. i Mitochondria DNA copy number detected by qPCR in HRMECs following treatments with shT or vehicle controls for 72 h in NG or HG medium as indicated. Each experiment was repeated independently at least three times. All results are displayed as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA analysis
Discussion
In the present study, we have discussed the role of TRIM25 in the hyperglycemia-induced injury of RMECs. TRIM25 was highly expressed in the retinal vascular endothelial cells and was further elevated in vivo and in vitro under the stimulation of hyperglycemia. We further determined that TRIM25 participates in hyperglycemia-induced inflammation, cellular senescence, and oxidative stress in HRMECs by adenovirus-mediated knockdown or overexpression of TRIM25. Moreover, we revealed that TRIM25 potentiates the inflammatory responses mediated by the TNF-α/NF-κB pathway, and TRIM25 knockdown improves cellular senescence by increasing SIRT3. However, TRIM25 suppression alleviates the oxidative stress independent of both SIRT3 and mitochondrial biogenesis.
TRIM25 plays a pivotal role in regulating the inflammatory response. Previously, TRIM25 has been reported to enhance NF-κB signaling in the antiviral signaling process [23], and TRIM25 can also directly regulate TNF-α-induced NF-κB signaling and enhanced the transcription of proinflammatory cytokines, such as IL-1β and TNF-α [14, 15]. Consistent with these studies, we demonstrated that TRIM25 inhibition decreased the level of activated NF-κB and cleaved Caspase-3 and markedly repressed the expression of inflammatory cytokines including TNF-α, NF-κB, and IL-1β induced by high glucose in HRMECs while TRIM25 overexpression further elevated the level of TNF-α and activated NF-κB. However, previous studies have reported that TRIM25 expression is increased and serves as a negative regulator of TNF-induced cell necrosis under necrotic stimulation [24], and TRIM25 can protect human bronchial epithelial cells in a COPD rat model by decreasing IL-1β secretion [25]. Moreover, the NF-κB activation by TRIM25 contributed to suppressive immune microenvironments in gliomas [26]. Therefore, how the enhancement of the inflammatory responses of the TNF-α/NF-κB pathway mediated by TRIM25 affects the pathology of diabetic retinopathy needs to be further studied.
TRIM25 regulates cell cycle and cell survival through a variety of pathways, and one of the most discussed is the p53/p21 pathway, which modulates cellular senescence and organismal aging by inducing irreversible cell cycle arrest [27, 28]. However, few studies have also reported that TRIM25 exhibits complex effects on the cell cycle and the p53/p21 pathway. For example, TRIM25 has been found to promote cancer cell proliferation and migration by inhibiting the p53 axis [10]. Another separate study found that TRIM25 has a dual function in the regulation of the p53 signaling, which increases the p53 level but decreases p53 activities at the same time [29]. In contrast, TRIM25 knockdown reverses cell cycle arrest and apoptosis by facilitating DNA repair in nasopharyngeal carcinoma cells [30]. Moreover, SIRT3 has been reported to protect endothelial cells from high glucose-induced senescence and dysfunction via the direct suppression of the p53/p21 pathway [18]. Consistent with that, our results showed that TRIM25 knockdown attenuates hyperglycemia-induced cellular senescence in HRMECs by increasing SIRT3 with a significant decrease in the p21 level. Whether TRIM25 knockdown decreases the activation of the p53/p21 pathway in HRMECs under high glucose by increasing SIRT3 remains to be investigated.
Few studies have reported that TRIM25 also exerts a two-sided role in modulating oxidative stress. Oxidative stress contributes to the loss of diabetic RMECs by inducing DNA damage, which results in the following activation of multiple glycolytic side branching pathways that, in turn, triggers subsequent oxidative injury and related inflammatory and apoptotic responses [8, 9, 31], and TRIM25 has been reported to aggravate oxidative stress and cellular apoptosis by ubiquitinating DNA repair enzyme XRCC5 and Ku80 [30, 32]. On the other hand, TRIM25 itself can ameliorate oxidative stress by targeting the NRF2 signaling and mitochondrial biogenesis, which bolsters antioxidant defense in hepatocellular carcinoma and in lens epithelial cells [33, 34]. Our study showed that TRIM25 increased intracellular ROS production, the main source of oxidative stress in endothelial cells under high glucose, accompanied by a decline of SOD2 level, indicating an impairment of the antioxidative ability of HRMECs under high glucose. However, TRIM25 knockdown alleviated hyperglycemia-induced oxidative stress independent of both mitochondrial homeostasis and SIRT3 in our results. Taking these into account, future studies need to address how TRIM25 knockdown enhanced the antioxidative ability of HRMECs under high glucose, such as by enhancing DNA repair and decreasing DNA damage. Furthermore, given that TRIM25 is an E3 ubiquitin ligase, high-sensitivity mass spectrometry followed by immunoprecipitation using an anti-ubiquitin antibody may help reveal further potential molecular interaction with TRIM25.
Collectively, our study proposed that TRIM25 inhibition protected RMECs from high glucose by alleviating inflammation mediated by TNF-α/NF-κB pathway, by attenuating cellular senescence via increasing SIRT3, and by relieving oxidative stress. Our results have underscored the broad protective effects of TRIM25 on hyperglycemia-induced injury in RMECs, which may provide a new therapeutic target for diabetic retinopathy.
References
Antonetti DA, Klein R, Gardner TW (2012) Diabetic retinopathy. N Engl J Med 366(13):1227–1239. https://doi.org/10.1056/NEJMra1005073
Teo ZL, Tham YC, Yu M et al (2021) Global prevalence of diabetic retinopathy and projection of burden through 2045: systematic review and meta-analysis. Ophthalmology 128(11):1580–1591. https://doi.org/10.1016/j.ophtha.2021.04.027
Hammes HP (2018) Diabetic retinopathy: hyperglycaemia, oxidative stress and beyond. Diabetologia 61(1):29–38. https://doi.org/10.1007/s00125-017-4435-8
Baker RG, Hayden MS, Ghosh S (2011) NF-κB, inflammation, and metabolic disease. Cell Metab 13(1):11–22. https://doi.org/10.1016/j.cmet.2010.12.008
Dammak A, Huete-Toral F, Carpena-Torres C, et al. (2021) From oxidative stress to inflammation in the posterior ocular diseases: diagnosis and treatment. Pharmaceutics 13(9). https://doi.org/10.3390/pharmaceutics13091376
Di Micco R, Krizhanovsky V, Baker D et al (2021) Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol 22(2):75–95. https://doi.org/10.1038/s41580-020-00314-w
Sabbatinelli J, Prattichizzo F, Olivieri F et al (2019) Where metabolism meets senescence: focus on endothelial cells. Front Physiol 10:1523. https://doi.org/10.3389/fphys.2019.01523
Giacco F, Brownlee M (2010) Oxidative stress and diabetic complications. Circ Res 107(9):1058–1070. https://doi.org/10.1161/circresaha.110.223545
Kowluru RA, Chan PS (2007) Oxidative stress and diabetic retinopathy. Exp Diabetes Res 2007:43603. https://doi.org/10.1155/2007/43603
Heikel G, Choudhury NR, Michlewski G (2016) The role of Trim25 in development, disease and RNA metabolism. Biochem Soc Trans 44(4):1045–1050. https://doi.org/10.1042/bst20160077
Lee JM, Choi SS, Lee YH et al (2018) The E3 ubiquitin ligase TRIM25 regulates adipocyte differentiation via proteasome-mediated degradation of PPARγ. Exp Mol Med 50(10):1–11. https://doi.org/10.1038/s12276-018-0162-6
Li C, Dou P, Lu X, et al. (2022) Identification and validation of TRIM25 as a glucose metabolism regulator in prostate cancer. International journal of molecular sciences 23(16). https://doi.org/10.3390/ijms23169325
Wan T, Li X, Li Y (2021) The role of TRIM family proteins in autophagy, pyroptosis, and diabetes mellitus. Cell Biol Int 45(5):913–926. https://doi.org/10.1002/cbin.11550
Liu Y, Liu K, Huang Y et al (2020) TRIM25 promotes TNF-α-induced NF-κB activation through potentiating the K63-linked ubiquitination of TRAF2. J Immunol (Baltimore, Md : 1950) 204(6):1499–1507. https://doi.org/10.4049/jimmunol.1900482
Park HS, Lu Y, Pandey K et al (2021) NLRP3 inflammasome activation enhanced by TRIM25 is targeted by the NS1 protein of 2009 pandemic influenza a virus. Front Microbiol 12:778950. https://doi.org/10.3389/fmicb.2021.778950
Crespo-Garcia S, Tsuruda PR, Dejda A et al (2021) Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab 33(4):818-832.e817. https://doi.org/10.1016/j.cmet.2021.01.011
Kida Y, Goligorsky MS (2016) Sirtuins, cell senescence, and vascular aging. Can J Cardiol 32(5):634–641. https://doi.org/10.1016/j.cjca.2015.11.022
Chen T, Ma C, Fan G et al (2021) SIRT3 protects endothelial cells from high glucose-induced senescence and dysfunction via the p53 pathway. Life Sci 264:118724. https://doi.org/10.1016/j.lfs.2020.118724
Tang X, Luo YX, Chen HZ et al (2014) Mitochondria, endothelial cell function, and vascular diseases. Front Physiol 5:175. https://doi.org/10.3389/fphys.2014.00175
Han L, Li J, Li J et al (2020) Activation of AMPK/Sirt3 pathway by phloretin reduces mitochondrial ROS in vascular endothelium by increasing the activity of MnSOD via deacetylation. Food Funct 11(4):3073–3083. https://doi.org/10.1039/c9fo02334h
Chen ML, Zhu XH, Ran L, et al. (2017) Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc 6(9). https://doi.org/10.1161/jaha.117.006347
Bagul PK, Katare PB, Bugga P, et al. (2018) SIRT-3 modulation by resveratrol improves mitochondrial oxidative phosphorylation in diabetic heart through deacetylation of TFAM. Cells 7(12). https://doi.org/10.3390/cells7120235
Lee NR, Kim HI, Choi MS et al (2015) Regulation of MDA5-MAVS antiviral signaling axis by TRIM25 through TRAF6-mediated NF-κB activation. Mol Cells 38(9):759–764. https://doi.org/10.14348/molcells.2015.0047
Mei P, Xie F, Pan J et al (2021) E3 ligase TRIM25 ubiquitinates RIP3 to inhibit TNF induced cell necrosis. Cell Death Differ 28(10):2888–2899. https://doi.org/10.1038/s41418-021-00790-3
Tian X, Xue Y, Xie G et al (2021) (-)-Epicatechin ameliorates cigarette smoke-induced lung inflammation via inhibiting ROS/NLRP3 inflammasome pathway in rats with COPD. Toxicol Appl Pharmacol 429:115674. https://doi.org/10.1016/j.taap.2021.115674
Ge MX, Shi YK, Liu D (2022) Tripartite motif-containing 25 facilitates immunosuppression and inhibits apoptosis of glioma via activating NF-κB. Exp Biol Med (Maywood) 247(17):1529–1541. https://doi.org/10.1177/15353702221099460
Rufini A, Tucci P, Celardo I et al (2013) Senescence and aging: the critical roles of p53. Oncogene 32(43):5129–5143. https://doi.org/10.1038/onc.2012.640
Vigneron A, Vousden KH (2010) p53, ROS and senescence in the control of aging. Aging 2(8):471–474. https://doi.org/10.18632/aging.100189
Zhang P, Elabd S, Hammer S et al (2015) TRIM25 has a dual function in the p53/Mdm2 circuit. Oncogene 34(46):5729–5738. https://doi.org/10.1038/onc.2015.21
Chen Y, Zhao Y, Yang X et al (2022) USP44 regulates irradiation-induced DNA double-strand break repair and suppresses tumorigenesis in nasopharyngeal carcinoma. Nat Commun 13(1):501. https://doi.org/10.1038/s41467-022-28158-2
Nishikawa T, Edelstein D, Du XL et al (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404(6779):787–790. https://doi.org/10.1038/35008121
Mao X, Ji M, Kang L et al (2022) XRCC5 downregulated by TRIM25 is susceptible for lens epithelial cell apoptosis. Cell Signal 94:110314. https://doi.org/10.1016/j.cellsig.2022.110314
Yang X, Zhang F, Liu X et al (2022) FOXO4 mediates resistance to oxidative stress in lens epithelial cells by modulating the TRIM25/Nrf2 signaling. Exp Cell Res 420(1):113340. https://doi.org/10.1016/j.yexcr.2022.113340
Liu Y, Tao S, Liao L et al (2020) TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat Commun 11(1):348. https://doi.org/10.1038/s41467-019-14190-2
Funding
This work was supported by the Science and Technology Research Project of Songjiang District [grant number 2020SJ300]; the National Key R&D Program of China [grant numbers 2016YFC0904800, 2019YFC0840607]; National Science and Technology Major Project of China [grant number 2017ZX09304010]; and Shanghai Key Clinical Specialty.
Author information
Authors and Affiliations
Contributions
DS, NW, and FW designed this study. DS and SL executed the experiments. NW, SC, and YS collected the human specimens, carried out experiments, and analyzed data. DS and SZ maintained the mice. NW, FW, and QG provided resources, supervised the experiments, and participated in the data analyses. DS, FW, and NW wrote the article with input from all authors. All authors proofread the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
Animals were treated in accordance with the ARRIVE guidelines and the National Research Council’s Guide for the Care and Use of Laboratory Animals and approved by the Ethics committee of Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China (reference number: 2019-A049-01).
Human samples were collected with informed consent following the guidelines of the Helsinki Declaration. This study was approved by the Ethics Committee of Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China (reference number: 2020SQ100).
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sun, D., Li, S., Chen, S. et al. TRIM25 inhibition attenuates inflammation, senescence, and oxidative stress in microvascular endothelial cells induced by hyperglycemia. Graefes Arch Clin Exp Ophthalmol 262, 81–91 (2024). https://doi.org/10.1007/s00417-023-06160-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-023-06160-8