
Abstract
Limb-girdle muscular dystrophies (LGMD) are a group of clinically and genetically heterogeneous diseases characterized by weakness and wasting of the pelvic and shoulder girdle muscles. Twenty-four recessive LGMD (types R1–R24) and five dominant LGMD (types D1-D5) have been identified with characterization of mutations in various genes. To date, LGMD D3 (previously known as LGMD1G) has been characterized in only two families with Brazilian or Uruguayan origin. Each was caused by a distinct mutation at codon 378 in the prion-like domain of HNRNPDL encoding heterogeneous nuclear ribonucleoprotein D like (HNRNPDL), an RNA processing protein. Our study characterized eight patients suffering from LGMD D3 in a Chinese family spanning three generations. Muscle biopsy specimens from two patients showed a myopathy with rimmed vacuoles. Sequencing analysis revealed a heterozygous c.1132G > A (p.D378N) mutation in HNRNPDL that co-segregated with disease phenotype in the family. The same mutation has been identified previously in the Brazilian family with LGMD D3. However, most patients in the current family showed distal as well as proximal limb weakness rather than weakness of toe and finger flexor muscles that were typical features in the other two LGMD D3 families reported previously. The present study indicates that the same mutation in HNRNPDL results in various phenotypes of LGMD D3. That all mutations in three unrelated families with different ethnic background occur at the same position in codon 378 of HNRNPDL gene suggests a mutation hotspot. Acceleration of intrinsic self-aggregation of HNRNPDL caused by mutation of the prior-like domain may contribute to the pathogenesis of the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Limb girdle muscular dystrophies (LGMD) are genetically inherited conditions that primarily affect skeletal muscles leading to progressive, predominantly proximal muscle weakness. To date, twenty-four recessive LGMD (types R1–R24) and five dominant LGMD (types D1–D5) were identified with characterization of mutations in various genes based on the revised classification by European Neuromuscular Centre (ENMC) in 2017 [1, 2]. LGMD D3 HRNPDL-related (previously known as LGMD1G) was identified in 2004 in a Caucasian-Brazilian family by mapping the disorder to locus 4p21 [3]. A decade later, using whole-genome sequencing, the same laboratory identified two distinct mutations in the HNRNPDL (previously known as HNRPDL), which encodes an RNA processing protein–heterogeneous nuclear ribonucleoprotein D like (HNRNPDL) in the previously reported Brazilian family and Uruguayan family [4]. The pedigrees share the same mutant position in HNRNPDL for two distinct missense mutations; substitutions of an aspartic acid at codon 378 by an asparagine and by a histidine, respectively. Patients in both families developed late-onset LGMD and shared same clinical features of the progressive flexion limitation of the fingers and toes.
HNRNPDL belongs to the subfamily of heterogeneous nuclear ribonucleoproteins (hnRNPs) essential for the maturation of newly formed heterogeneous nuclear pre-mRNA into mRNA and the transportation of mRNA. They are key proteins in the cellular nucleic acid metabolism owing to their functional diversity and complexity. Many hnRNPs were linked to various diseases due to their crucial role in the regulation of gene expression [5]. Mutations in the prion-like domains (PrLD) of hnRNP A1 and A2B1 cause multisystem proteinopathy, an inherited pleiotropic degenerative disorder that can affect muscle, bone, and the nervous system. Tissues with multisystem proteinopathy have ubiquitin-positive inclusions that contain RNA-binding proteins, such as hnRNPA1, and hnRNPA2B1, and may stain positively for proteins that mediate ubiquitin-dependent autophagy [6]. Muscle biopsy specimens of these patients showed atrophic fibers, central nuclei, and rimmed vacuoles [7]. Overexpression of mutant proteins promotes the formation of cytoplasmic inclusion in muscle fibers owing to intrinsic self-aggregation accelerated by the mutations in animal models that recapitulate the human pathology [7, 8]. Similarly, HNRNPDL contains prior-like domain, a typical feature for forming inherently toxic aggregation [9, 10], but whether human HNRNPDL mutations increase intrinsic self-aggregation remains to be determined.
Here, we present a Chinese family spanning three generations with LGMD D3 caused by D378N mutation in the prion-like domain of HNRNPDL, which is the same as that identified in Caucasian–Brazilian family. However, most patients in the current family show the distal as well as proximal limb weakness rather than the limitation of toe and finger flexion seen in the other LGMD D3 families.
Materials and methods
The pedigree of the family
The index patient (III-13) was a 52-year-old female, referred to our department for progressive weakness in her lower and upper limbs (Fig. 1). Her father (II-3) who died at age of 76 years, and two older brothers (III-10 and III-11) also presented with similar symptoms (Fig. 1). Detailed clinical assessment of eight individuals in the family spanning three generations is shown in Table 1. Medical Research Council (MRC) grading scale was used to evaluate muscle strength. The study was conducted after receiving written informed consent from the patients. The study protocol was approved by the Institutional ethics committee of Xuanwu Hospital, Capital Medical University, Beijing, China.

Pedigree of the family. The index patient is indicated with an arrow
Laboratory tests
The electrocardiography, echocardiography, electromyography (EMG), and routine laboratory tests were performed in the index patient (III-13) and patient III-11 (Fig. 1). We also performed biochemical analysis on serum creatine kinase, electrolytes, fasting glucose, blood urea, nitrogen, creatine, aspartate aminotransferase, alanine aminotransferase, thyroxine, thyroid-stimulating hormone, sedimentation rate, and C-reactive protein for two patients.
Muscle imaging
Muscle imaging studies were performed in the index patient. Transverse, coronal T1-weighted, T2-weighted, and fat-suppressed magnetic resonance imaging (MRI) were performed on a 1.5-T machine (Siemens 1.5T Sonata).
Muscle pathology
Muscle biopsies were obtained from the quadriceps from the index patient (III-13) and from patient III-11 (Fig. 1). The samples were snap-frozen in liquid isopentane. Cryostat sections were stained for hematoxylin–eosin (H&E), modified Gormori trichrome (MGT), NADH-tetrazolium reductase, succinate dehydrogenase, periodic acid–Schiff, nonspecific esterase, oil red, and myofibrillar ATPase preincubated at pH 4.3, 4.6, and 9.4.
Mutation analysis
DNA was extracted from anticoagulated blood from affected and unaffected individuals using a standard phenol–chloroform method. Indexed genomic DNA (gDNA) libraries were prepared from patient genomic DNA using the TruSeq DNA Preparation kit (Illumina, USA) and exome captured using the TruSeqExome Enrichment kit (Illumina, USA) according to the manufacturer’s protocol. Sequencing was performed with 100-bp paired-end reads on a HiSEq 2000 (Illumina, USA). Reads were aligned to the human reference genome with NovoAlign (Novocraft Technologies, USA). Variants were named with SAMtools15 and annotated with SeattleSeq. Predictions of pathogenicity for variants were obtained from SeattleSeq and SIFT16. Sequencing coverage depth was calculated using BEDTools17 and genomic coordinates provided by Illumina. All reported genomic locations are from GRCh37/hg19. Segregation of mutations was assessed with the standard PCR-based sequencing using Primer3Plus. We first sequenced DNA from the proband (III-13) by whole-exome sequencing (WES) (Fig. 1). The mutation detected in exome sequencing was confirmed by Sanger sequencing. After the mutation was confirmed, all other family members were sequenced for mutations in detected mutant gene. To assess whether the phenotype in this family with distal muscle weakness was caused by digenic or multigenic mechanism, we also used WES to sequence the proband’s brother (III-11) who had most severe distal muscle weakness among all intrafamilial members (Fig. 1; Table 1).
We analyze the WES data for etiologic variance from both the proband and her brother based on the criteria adopted from Karaca et al. [11]: (a) gene listed in association with a phenotype in OMIM, (b) minor allele frequency (MAF) < 0.0002 and variant heterozygosity for autosomal dominant (AD) and X-linked (XL) conditions or MAF < 0.0003, and biallelic variation for autosomal recessive (AR) conditions using the NHLBI Exome Sequencing Project (ESP, http://evs.gs.washington.edu/EVS/), 1000 Genomes Project (http://www.internationalgenome.org/) [12], Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/) [13], Genome Aggregation Database (GnomAD) (http://gnomad.broadinstitute.org), Trans-Omics for Precision Medicine (TOPMED) (https://bravo.sph.umich.edu/freeze5/hg38/), and our WES database which includes 11076 samples from Chinese individuals of Han, and (c) variant likely to affect protein function based on mutation type and/or conservation and measures of deleteriousness (nonsense, frame shift, canonical splice site, non-frame shift if Phylop score > 1.0, and missense if CADD phred-like score > 15).
Results
Clinical features
Table 1 summarizes the clinical findings from eight individuals in the same family. Three patients (II-3, 6, and III-3) were deceased and their disease was described by their offsprings (Fig. 1). Clinical details of patient I-2 and II-1 were unknown, because documents on their clinical history were unavailable and their offsprings were unable to describe their symptoms clearly. Five patients (III-5, 10, 11, 13, and 14) had detailed neurological examination (Table 1; Fig. 1). All affected individuals had a normal birth and motor development. The age at onset ranged from 36 to 51 years. The initial symptoms were lower limb weakness in five patients (II-3, III-3, 5, 13, and 14), upper limb onset in one patient (II-6), both upper and lower limb weakness in two patients (III-10 and III-11). All affected patients except patient II-6 had progressive weakness of pelvic and scapular girdles. Patient II-6 was unable to raise her arms and to grasp but still walked normally 1 year before she died of liver cancer at the age of 70, indicating that the weakness was limited in her upper limbs with both distal and proximal muscles but spared in her lower limbs. Five of eight affected individuals (II-6, III-5, 10, 13, and 14) presented cataracts before age of 50 (Table 1). Interestingly, patient III-22 was diagnosed with cataract at the age of 28, but had no weakness when examined at age 32.
The five patients in the third generation (III-5, 10, 11, 13, and 14) had symptoms for 3–19 years at neurological examinations. The disability progressed over the years and the prognosis was relatively benign. Three patients (III-5, 13, and 14) were able to walk independently, whereas two (III-10 and 11) needed a cane. Patients III-10 and III-11 had distal lower and upper limb weakness in addition to proximal weakness. They were unable to walk on their heels and toes, and their finger flexor muscles lack strength for fine motor skills such as scratching skin. Patient III-5 had distal upper limb weakness only with poor strength of grasp. Interestingly, unlike patients reported previously, none of these three patients (III-5, 10, and 11) with distal weakness had flexion limitation of fingers and toes. All five living patients complained of sudden and frequent falling on their knees. Patient III-11 had comminuted fractures in his two knees, whereas patient III-14 had to wear knee pads to avoid fracture. In all living patients, the tendon reflexes were reduced or abolished in affected limbs. None of the patients had myotonic symptoms. None showed inflammatory signs such as pain, swelling, or redness. The sensory examinations were normal in all patients.
Laboratory tests and EMG studies
Routine laboratory examinations of index patient (III-13) and patient III-11 were normal on blood count, electrolytes, blood urea nitrogen, creatine, aspartate aminotransferase, alanine aminotransferase, thyroxine, thyroid-stimulating hormone, sedimentation rate, and C reactive protein. However, the fasting glucose was 7.13 mmol/L (128.34 mg/dl) in index patient and 7.07 mmol/L (127.26 mg/dl) in patient III-11, whereas the normal range is 3.9–6.1 mmol/L (70.2 − 109.8 mg/dl). Oral glucose tolerance test done in the proband shows type 2 diabetes.
The serum creatine kinase level was in normal range (18–198 U/L) in patient III-13, and elevated mildly in patient III-11 (393 U/L). Needle electromyography revealed a myopathic pattern in patient III-13 and patient III-11 with fibrillation potentials and positive sharp waves, low amplitude, and short-duration motor unit action potentials demonstrating the early recruitment. Furthermore, the sensory and motor nerve conduction studies of the upper and lower extremity were normal. A muscle MRI performed in the index patient showed a striking involvement of most bilateral thigh muscles, partially sparing rectus femoris and biceps femoris. The adductor longus was spared (Fig. 2).

Muscle MRI imaging of the index patient. There is striking involvement of most thigh muscles with partially sparing of the rectus femoris and biceps femoris. The adductor longus is preserved
Muscle pathology
Muscle biopsy from index patient (III-13) and III-11 showed myopathic and neuropathic changes with marked variation of the fiber size, scattered or clusters of atrophic angulated fibers, and occasional degenerating fibers. Inflammatory changes were absent. Cryostat sections revealed rimmed vacuoles lined by basophilic granular material in many fibers on H&E staining (Fig. 3a) and purple–red with the modified Gormori trichrome (MGT) staining (Fig. 3b). Several fibers contained cytoplasmic bodies. In NADH dehydrogenase-reacted sections, several fibers harbored focal decreases of enzyme activities. The ATPase reactions revealed that atrophic fibers were of both type I and type II (Fig. 3c, d), indicating denervation atrophy. Congophilic deposits were negative.

Hematoxylin–eosin staining (a) and modified Gomori trichrome staining (b). Rimmed vacuoles are indicated with thin arrows. Histochemistry studies for ATPase staining show small angulated fibers involved of both type 1 and type 2 fibers (c, d) consistent with neurogenic damage. Small angular fibers are indicated on thick arrows
Mutation analysis
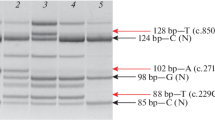
Whole-exome sequencing (WES) in the index patient (III-13) revealed a heterozygous c.1132G > A (p.D378N) mutation in HNRNPDL encoding heterogeneous nuclear ribonucleoprotein D like, an RNA processing protein. The D378N mutation had been previously identified in a Caucasian–Brazilian LGMD D3-HNRNPDL-related (previously known as LGMD1G) family [4]. Further sequencing of other family members by Sanger sequencing revealed that the mutation was carried in four symptomatic individuals in the third generations (III-5, 10, 11, and 14) and one asymptomatic individual (III-22) (Fig. 1). No mutation was detected in any gene known to cause cataract.
The current family presents clinical features different from previously reported LGMD D3 families with varied phenotypes and severities of symptom among the family members. To test whether any additional mutation in this family contributed to the distal weakness or to the severity of the disease, we also sequenced the proband’s brother (III-11) who had both proximal and distal muscle weakness. We only find the HNRNPDL-D378N mutation and no mutation in genes related to distal myopathies [14]. However, we detected mutations partially meeting the criteria in three other known genes related to muscle disease. The proband (III-13) and her brother (III-11) share a heterozygous missense mutation c.1111T > C (p.Y371H) in TMEM43 encoding transmembrane protein 43, a nuclear envelop protein, in which single heterozygous mutation causes autosomal dominant Emery–Dreifuss muscular dystrophy (OMIM#614302) [15]. The proband’s brother (III-11) but not the proband (III-13) harbors mutations in two other genes related to neuromuscular disease: (1) a heterozygous missense mutation c.255T > C (p. S85R) in CHCHD2 encoding a regulator of mitochondrial metabolism, coiled-coil-helix-coiled-coil-helix domain containing protein 2, which occurs in Chinese sporadic ALS patients [16], and (2) a heterozygous missense mutation c.4184G > A (p.R1395Q) in COL6A3 encoding alpha-3 chain of type VI collagen, in which single heterozygous mutation in other locations causes autosomal dominant Bethlem myopathy-1 (OMIM# 158810) [17, 18] or autosomal-dominant Ullrich congenital muscular dystrophy 1 (OMIM# 254090) [19].
Discussion
Exome sequencing of the proband in this family identified a heterozygous D378N mutation in an RNA processing protein HNRNPDL, which co-segregated with disease phenotype in the family. To date, LGMD D3 (HNRNPDL-related) has been described only in one Brazilian family and in one Uruguayan family [3, 4]. To our knowledge, this is the first study on LGMD D3 in Chinese ethnics and is the third report worldwide. Mutations at same codon of HNRNPDL identified in all LGMD D3 families suggest that D378 might represent a mutation hotspot. The novel clinical features caused by HNRNPDL-D378N observed in the current family widen the spectrum of phenotypes in LGMD D3 caused by HNRNPDL mutations.
The age of onset of weakness in this family is 36–51 years. All patients in the third generation (III-5, 10, 11, 13, and 14) developed the symptom at age of 41 or older (Table 1). In the Brazilian family, the youngest age of onset was 30 years old [3, 4] (Table 2). It implies that an ethnic background may play a role on the age of onset of the disease with a same causative D378N mutation. In Uruguayan family harboring D378H mutation, the youngest patient became symptomatic at age 15 years (Table 2) [3, 4]. This suggests that a substitution of negatively charged residue (Asp) by an aromatic residue with a positive charge and larger volume (His) has more severe impact on the HNRNPDL function than that by uncharged residue with the same volume and structure (Asn) at the same codon.
The course of disease of three living patients in the current family is longer than 15 years. One patient (III-14) is still able to walk without support at age of 68, whereas two others need canes, which indicates that the clinical manifestation of defects in LGMD D3 HNRNPDL-related appears slowly in this pedigree. One mutation carrier (III-22) was asymptomatic with neurological examination at age of 32, which is younger than the age of onset in this family. It implies that she is probably asymptomatic due to her relatively young age. However, in Brazilian family, five individuals are asymptomatic with three at ages within the range of the onset ages, whereas, in Uruguayan family, the ages of three asymptomatic members are also in the range of onset ages [3, 4] (Table 2). The common asymptomaticity of multiple members at ages of the onset in Brazilian and Uruguayan families suggests reduced penetrance and variable expressivity may play a role in LGMD D3. Reduced penetrance and variation of expressivity can be caused by a number of factors, including modifier genes, environmental factors, allelic variation, and complex genetic and environmental interactions. The variable phenotypes among interfamilial members observed in three distinct pedigrees suggest that the factors causing reduced penetrance should be considered for assessing prevalence of this dominant inherited disorder. Moreover, in the previous two LGMD D3 families, the progressive limitation of the toe and finger flexion was a distinct feature [3, 4], but this was not seen in any patient of the current family. Strikingly, patients III-10 and III-11 lacked strength for fine motor skills such as scratching, but neither of them had flexion limitation in their fingers (Table 1). Three patients in this family had distal limb weakness with two in all limbs and with the third one only in upper limbs. All living patients of the current family complained of recurrent falling on their knees even at the early stage of the disease, and three patients had severe knee fractures. The MRI study on the index patient (III-13) revealed that the medial and anterior muscles of the thigh were severely involved bilaterally. Thus, we postulated that quadriceps femoris was involved at early stage of the disease.
Karaca et al. recently performed a retrospective computational reanalysis of WES data from 19 families with different neurological diseases, who have phenotypic expansion beyond those previously reported in association with variations at identified locus, using stringent variant call file filtering criteria [11]. They found six families with multilocus variants in two or more disease genes. Lee et al. described digenic inheritance of a TIA1 variant with a pathogenic SQSTM1 mutation causing myodegeneration in multisystem proteinopathy with a distal myopathy and rimmed-vacuolated inclusion body myopathy (IBM) [20]. To test whether the phenotype expansion in the current family is due to more than one mutant genes, we compared the variants detected in the proband (III-13) and in the proband’s brother (III-11). We found that only HNRNPDL-D378N mutation fully meets the filtering criteria and no other mutation in known genes accounting for distal muscular dystrophy [14]. We did detect variants partially complying with the criteria in the known muscle disease genes. Both proband and her brother share a heterozygous missense mutation c.1111T > C (p.Y371H) in TMEM43 encoding a nuclear envelope protein, transmembrane protein 43. The minor allele frequency (MAF) of c.1111T > C is in a range of 0.0006–0.0008 in databases GnomAD, TOPMD, and ExAC, and 0.002 in 1000 Genome, and 0.008 in our own WES database for Chinese population. The mutation is predicted to be pathogenic by Mutation Taster. TMEM43 maintains nuclear envelope structure by interacting with the other nuclear proteins including emerin, a serine-rich nuclear membrane protein, and a member of the nuclear lamina-associated protein family, at the inner nuclear membrane, and is associated with the linker of the nucleoskeleton and cytoskeleton complex [21]. Single heterozygous mutations E85K and I91V in TMEM43 causing autosomal dominant Emery–Dreifuss muscular dystrophy (OMIM#614302) were reported in three patients [15, 22]. TMEM43-E85K causes the reduction of the protein expression, failure of oligomerization, and decreased interaction with emerin and SUN2, another inner nuclear membrane protein for nuclear–cytoplasmic connection [15]. However, to address whether the TMEM43 variant contributes to the phenotype expansion in this family, a comparison of the variants among multiple pedigreed with the disease is needed, and an assessment of the functional defect of the mutation is required.
Intrafamilial variability provides a good opportunity to dissect genotype–phenotype correlations. In this family, varied severity of symptom was observed among intrafamilial members carrying the same HNRNPDL mutation. For example, the proband’s brother (III-11) presents symptoms more severe than the proband (III-13) (Table 1). Interestingly, we found that the proband’s brother but not the proband harbors additional mutations in two other genes related to neuromuscular disease, which comply with the criteria partially. He carries a heterozygous missense mutation c.255T > C (p. S85R) in CHCHD2 encoding coiled-coil–helix-coiled–coil-helix domain-containing protein 2, a regulator of mitochondrial metabolism. A cohort study showed this heterozygous mutation occurs in Chinese sporadic ALS patients but not in controls [16]. CHCHD2-S85R is predicted to be pathogenic by Mutation Taster. MAF of this variance is 0.000008, 0.00004, 0.0004, and 0.00004 in TOPMED, ExAC, 1000 Genomes, and our own WES database, respectively. In OMIM database, CHCHD2 is listed in associated with autosomal dominant Parkinson disease type 22 (OMIM #616710) but not with ALS. Moreover, the proband’s brother but not the proband carries a heterozygous missense mutation c.4184G > A (p.R1395Q) in COL6A3 encoding alpha-3 chain of type VI collagen. COL6A3-R1395Q is predicted to be pathogenic by Mutation Taster. Mutations in different locations of COL6A3 can cause autosomal dominant Bethlem myopathy-1 (OMIM# 158810) [17, 18] or autosomal-dominant Ullrich congenital muscular dystrophy 1 (OMIM# 254090) [19]. However, the MAF of this variance is relatively high (0.0092 in ExAC, 0.0005 in GO-ESP, 0.0068 in TOPMED, and 0.0182 in 1000 Genomes, and 0.07 in our own WES database). Indeed, the functional defects of either mutation need to be confirmed. Though we cannot conclude whether any of those variants contributes to the severity of the disease, the findings are noteworthy to be addressed for future reference.
The prominent features of the muscle biopsy of index patient were rimmed-vacuolated myofibers associated with myopathic and neurogenic pattern typical in hereditary IBM [19]. Striking rimmed vacuoles (RV) in the skeletal muscle are most commonly seen in inclusion body myositis [24]. However, it is non-specific and has been reported in hereditary inclusion body myopathy, familial and sporadic distal myopathies, familial rimmed vacuole myopathy sparing the quadriceps, oculopharyngeal muscular dystrophy, oculopharyngeal muscular dystrophy with distal myopathy, myofibrillar myopathy, and LGMD [25]. In LGMD patients, the progressive proximal limb weakness with RV was seen in autosomal-dominant inherited LGMD D1, D2, D3 (previously known as LGMD1D, 1F, and 1G), autosomal recessive inherited LGMD R7 (previously known as LGMD2G), and LGMD R9 (previously known as LGMD2I) [3, 26,27,28,29]. In addition, our patients showed mixed myogenic/neurogenic pattern in the skeletal muscle, which was also seen in the Brazilian LGMD D3 family. In fact, patient III-10 was first misdiagnosed as HIBM before gene tests were available.
In the current family, five of eight patients and an asymptomatic carrier had cataracts before age 50. The occurrence of cataracts in this family is higher than in the other two LGMD D3 families (Table 2). Cataract is a main clinical feature in myotonic dystrophy (DM), in which abnormal hnRNP H inhibits nuclear export of mRNA-containing expanded CUG repeats [30]. Both HNRNPDL and hnRNP H belong to heterogeneous ribonucleoprotein family member [31]. Vieira et al. proved that the loss of yeast HRP1, orthologue to human HNRNPDL, caused dramatic reorganization of proteins involved in RNA-processing pathways [4]. The best-known function of hnRNP H is the regulation of alternative splicing, and hnRNP H1 regulates a splicing hotspot of HER2 in breast cancer cells [32]. Thus, with consideration of no mutation found in genes related to cataract, HNRNPDL may result in aberrant splicing of a gene related to cataract.
Moreover, bioinformatics analysis to detect prion domains in human genome revealed 240 genes harboring a domain compositionally similar to annotated yeast prion domain, which is termed as prion-like domain (PrLD) [32]. Thus, genes with PrLD occupy about 1.2% of human protein-coding genes [10]. Remarkably 30% of proteins with PrLD are RNA-binding proteins including HNRNPDL with very high likelihood of intrinsic self-aggregation properties [10]. Furthermore, multiple bioinformatics algorithms predict that the current mutation D378N in the prion domain increases the prion propensity up to 25% [9]. This suggests that the mutation enhances self-aggregation of the protein and this may contribute to the pathomechanism of the disease. However, no congophilic deposits were detected in our patients.
References
Straub V, Murphy A, Udd B (2018) 229th ENMC international workshop: Limb girdle muscular dystrophies—nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul Disord 28:702–710
Angelini C, Giaretta L, Marozzo R (2018) An update on diagnostic options and considerations in limb-girdle dystrophies. Expert Rev Neurother 18:693–703
Starling A, Kok F, Passos-Bueno MR, Vainzof M, Zatz M (2004) A new form of autosomal dominant limb-girdle muscular dystrophy (LGMD1G) with progressive fingers and toes flexion limitation maps to chromosome 4p21. Eur J Hum Genet 12:1033–1040
Vieira NM, Naslavsky MS, Licinio L, Kok F, Schlesinger D, Vainzof M, Sanchez N, Kitajima JP, Gal L, Cavacana N, Serafini PR, Chuartzman S, Vasquez C, Mimbacas A, Nigro V, Pavanello RC, Schuldiner M, Kunkel LM, Zatz M (2014) A defect in the RNA-processing protein HNRPDL causes limb-girdle muscular dystrophy 1G (LGMD1G). Hum Mol Genet 23:4103–4110
Geuens T, Bouhy D, Timmerman V (2016) The hnRNP family: insights into their role in health and disease. Hum Genet 135:851–867
Taylor JP (2015) Multisystem proteinopathy: intersecting genetics in muscle, bone, and brain degeneration. Neurology 85:658–660
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473
Shorter J, Taylor JP (2013) Disease mutations in the prion-like domains of hnRNPA1 and hnRNPA2/B1 introduce potent steric zippers that drive excess RNP granule assembly. Rare Dis 1:e25200
Navarro S, Marinelli P, Diaz-Caballero M, Ventura S (2015) The prion-like RNA-processing protein HNRPDL forms inherently toxic amyloid-like inclusion bodies in bacteria. Microb Cell Fact 14:102
March ZM, King OD, Shorter J (2016) Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res 1647:9–18
Karaca E, Posey JE, Coban AZ, Pehlivan D, Harel T, Jhangiani SN, Bayram Y, Song X, Bahrambeigi V, Yuregir OO, Bozdogan S, Yesil G, Isikay S, Muzny D, Gibbs RA, Lupski JR (2018) Phenotypic expansion illuminates multilocus pathogenic variation. Genet Med 20:1528–1537
Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073
Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, Hamamsy T, Lek M, Samocha KE, Cummings BB, Birnbaum D, Daly MJ, MacArthur DG (2017) The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res 45:D840–D845
Bonne G, Rivier F, Hamroun D (2017) The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul Disord 27:1152–1183
Liang WC, Mitsuhashi H, Keduka E, Nonaka I, Noguchi S, Nishino I, Hayashi YK (2011) TMEM43 mutations in Emery–Dreifuss muscular dystrophy-related myopathy. Ann Neurol 69:1005–1013
Yang X, An R, Zhao Q, Zheng J, Tian S, Chen Y, Xu Y (2016) Mutational analysis of CHCHD2 in Chinese patients with multiple system atrophy and amyotrophic lateral sclerosis. J Neurol Sci 368:389–391
Pan TC, Zhang RZ, Pericak-Vance MA, Tandan R, Fries T, Stajich JM, Viles K, Vance JM, Chu ML, Speer MC (1998) Missense mutation in a von Willebrand factor type A domain of the alpha 3(VI) collagen gene (COL6A3) in a family with Bethlem myopathy. Hum Mol Genet 7:807–812
Baker NL, Morgelin M, Pace RA, Peat RA, Adams NE, Gardner RJ, Rowland LP, Miller G, De Jonghe P, Ceulemans B, Hannibal MC, Edwards M, Thompson EM, Jacobson R, Quinlivan RC, Aftimos S, Kornberg AJ, North KN, Bateman JF, Lamande SR (2007) Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann Neurol 62:390–405
Baker NL, Morgelin M, Peat R, Goemans N, North KN, Bateman JF, Lamande SR (2005) Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet 14:279–293
Lee Y, Jonson PH, Sarparanta J, Palmio J, Sarkar M, Vihola A, Evila A, Suominen T, Penttila S, Savarese M, Johari M, Minot MC, Hilton-Jones D, Maddison P, Chinnery P, Reimann J, Kornblum C, Kraya T, Zierz S, Sue C, Goebel H, Azfer A, Ralston SH, Hackman P, Bucelli RC, Taylor JP, Weihl CC, Udd B (2018) TIA1 variant drives myodegeneration in multisystem proteinopathy with SQSTM1 mutations. J Clin Invest 128:1164–1177
Meinke P, Nguyen TD, Wehnert MS (2011) The LINC complex and human disease. Biochem Soc Trans 39:1693–1697
Mukai T, Mori-Yoshimura M, Nishikawa A, Hokkoku K, Sonoo M, Nishino I, Takahashi Y (2018) Emery-Dreifuss muscular dystrophy-related myopathy with TMEM43 mutations. Muscle Nerve. https://doi.org/10.1002/mus.26355
Argov Z, Eisenberg I, Grabov-Nardini G, Sadeh M, Wirguin I, Soffer D, Mitrani-Rosenbaum S (2003) Hereditary inclusion body myopathy: the Middle Eastern genetic cluster. Neurology 60:1519–1523
Nonaka I (1994) [Muscle pathologic diagnosis—mechanism in muscle fiber degeneration]. Rinsho Shinkeigaku 34:1279–1281
Jongen PJ, Ter Laak HJ, Stadhouders AM (1995) Rimmed basophilic vacuoles and filamentous inclusions in neuromuscular disorders. Neuromuscul Disord 5:31–38
Gilchrist JM, Pericak-Vance M, Silverman L, Roses AD (1988) Clinical and genetic investigation in autosomal dominant limb-girdle muscular dystrophy. Neurology 38:5–9
Sandell S, Huovinen S, Sarparanta J, Luque H, Raheem O, Haapasalo H, Hackman P, Udd B (2010) The enigma of 7q36 linked autosomal dominant limb girdle muscular dystrophy. J Neurol Neurosurg Psychiatry 81:834–839
Vainzof M, Moreira ES, Suzuki OT, Faulkner G, Valle G, Beggs AH, Carpen O, Ribeiro AF, Zanoteli E, Gurgel-Gianneti J, Tsanaclis AM, Silva HC, Passos-Bueno MR, Zatz M (2002) Telethonin protein expression in neuromuscular disorders. Biochim Biophys Acta 1588:33–40
Hong D, Zhang W, Wang W, Wang Z, Yuan Y (2011) Asian patients with limb girdle muscular dystrophy 2I (LGMD2I). J Clin Neurosci 18:494–499
Kim DH, Langlois MA, Lee KB, Riggs AD, Puymirat J, Rossi JJ (2005) HnRNP H inhibits nuclear export of mRNA containing expanded CUG repeats and a distal branch point sequence. Nucleic Acids Res 33:3866–3874
Kawamura H, Tomozoe Y, Akagi T, Kamei D, Ochiai M, Yamada M (2002) Identification of the nucleocytoplasmic shuttling sequence of heterogeneous nuclear ribonucleoprotein D-like protein JKTBP and its interaction with mRNA. J Biol Chem 277:2732–2739
Gautrey H, Jackson C, Dittrich AL, Browell D, Lennard T, Tyson-Capper A (2015) SRSF3 and hnRNP H1 regulate a splicing hotspot of HER2 in breast cancer cells. RNA Biol 12:1139–1151
Acknowledgements
We are grateful to all the subjects for participation in our study. This study was supported by National Key R&D Program of China, Precision Medicine Program–Cohort Study on Nervous System Diseases (No. 2017YFC0907700).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
All authors have reviewed the manuscript. Authors have no conflict of interest to declare.
Ethical standards
The study was conducted after receiving written informed consent from patients. In addition, this study was approved by the Institutional Ethics Committee of Xuanwu Hospital, Capital Medical University, Beijing, China.
Rights and permissions
About this article

Cite this article
Sun, Y., Chen, H., Lu, Y. et al. Limb girdle muscular dystrophy D3 HNRNPDL related in a Chinese family with distal muscle weakness caused by a mutation in the prion-like domain. J Neurol 266, 498–506 (2019). https://doi.org/10.1007/s00415-018-9165-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-9165-4