
Abstract
Spinocerebellar ataxia type 1 is an autosomal dominant disorder caused by a CAG repeat expansion in ATXN1, characterized by progressive cerebellar and extracerebellar symptoms. MRI-based studies in SCA1 focused in the cerebellum and connections, but there are few data about supratentorial/spinal damage and its clinical relevance. We have thus designed this multimodal MRI study to uncover the structural signature of SCA1. To accomplish that, a group of 33 patients and 33 age-and gender-matched healthy controls underwent MRI on a 3T scanner. All patients underwent a comprehensive neurological and neuropsychological evaluation. We correlated the structural findings with the clinical features of the disease. In addition, we evaluated the disease progression looking at differences in SCA1 subgroups defined by disease duration. Ataxia and pyramidal signs were the main symptoms. Neuropsychological evaluation disclosed cognitive impairment in 53% with predominant frontotemporal dysfunction. Gray matter analysis unfolded cortical thinning of primary and associative motor areas with more restricted impairment of deep structures. Deep gray matter atrophy was associated with motor handicap and poor cognition skills. White matter integrity loss was diffuse in the brainstem but restricted in supratentorial structures. Cerebellar cortical thinning was found in multiple areas and correlated not only with motor disability but also with verbal fluency. Spinal cord atrophy correlated with motor handicap. Comparison of MRI findings in disease duration-defined subgroups identified a peculiar pattern of progressive degeneration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spinocerebellar ataxia type 1 (SCA1) is an autosomal dominant disorder, characterized by progressive cerebellar ataxia, pyramidal signs, extracerebellar features and non-motor symptoms, as well as mild cognitive decline [1, 2]. Onset of SCA1 is usually in the fourth decade of life and affected families show anticipation [3, 4]. SCA1 is caused by a CAG expansion within the coding region of ATXN1. The CAG repeat region is located in exon 8 and translated into a polyglutamine tract in the amino terminal half of the protein [3, 5, 6]. Healthy individuals have a CAG-repeat number of 39 or less, affected patients have more than 44 repeats and intermediate alleles (39–44) result in reduced penetrance. Abnormal ataxin-1 aggregates into small intranuclear inclusions and interferes with the normal function of the protein leading to neuronal toxicity [7, 8].
Like other neurodegenerative disorders, cognitive impairment, usually mild, is observed in 20–30% of the cases [9, 10]. Several studies disclose impaired executive function, attention, visuo-spatial perception, verbal fluency, immediate and delayed memory [3, 11, 12]. In some patients, restlessness and emotional lability, without full blown dementia, characterize the mental status [13]. Significant mental retardation, however, has been reported only in individuals with very large CAG expansions and onset before 15 years of age [8, 14].
Neuroimaging techniques are powerful tools in the assessment of many closely related neurodegenerative disorders [15,16,17,18]. Such tools demonstrate in vivo structural abnormalities and provide a better understanding of the disease mechanisms and progression. Notwithstanding, no large studies have been conducted to investigate the extent of neurodegeneration and how it affects sequentially certain neuronal populations in SCA1. In particular, little is known about the extent of cortical and subcortical damage in the disease, and how such damage correlates with the clinical phenotype.
Based on this, our aim is to provide clinical characterization concerning motor and cognitive features of the disease, correlating these findings with anatomical aspects. Moreover, we aim to delineate how the disease course influences the structural damage, giving insights into the natural history of the disease. To accomplish that, we employed multimodal MRI, looking at the brain and spinal cord in a large cohort of patients with SCA1 at different stages of the disease.
Materials and methods
Study design
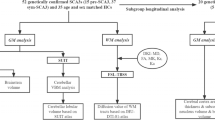
Figure 1 is the flowchart that describes the sequential experimental steps performed in this study.

Study design
Subject’s selection
We evaluated 33 consecutive adult patients with molecular confirmation of SCA1 regularly followed at UNICAMP and UNIFESP hospitals and 33 healthy age- and sex-matched controls. This protocol was approved by our institution Research Ethics Committee and a written informed consent was obtained from all participants. None of the controls had family history of ataxia and/or hereditary neuropathies. Before further analyses, we checked all images for significant motion artifacts or incidental findings, but none of the subjects presented them.
Clinical and cognitive assessment (n = 32)
Patients underwent detailed neurological examination and the Scale for the Assessment and Rating of Ataxia (SARA) was used to quantify disease severity [19]. Age of onset, disease duration, and CAG length were determined for each subject and used for further analyses. Cognitive functioning was evaluated through the Addenbrooke’s Cognitive Examination Revised (ACE-R) in 28 patients. The ACE-R is a comprehensive neuropsychological tool that has been validated for the Brazilian population. The score ranges from 0 to a maximum of 100 points (lower scores indicate greater impairment) and encompasses 5 cognitive domains: attention/orientation, memory, fluency, language and visuospatial abilities [20, 21]. Depressive symptoms were explored through the Beck Depression Inventory (BDI) [22] and a brief interview with the patient’s main caregiver designed to detect behavioral issues such as anxiety, depression, delusional thoughts and others. BDI scores were considered abnormal if ≥ 11. All scales and examinations were performed by a single investigator (CRMJr).
MRI acquisition and protocol
A 3T Achieva PHILIPS scanner with a standard 8-channel head coil was used to scan all patients and controls. To exclude incidental findings, routine T1 and T2 weighted sequences were performed for all subjects. We obtained T1 weighted volumetric images covering the whole brain and the cervical spinal cord with the following acquisition parameters: sagittal orientation, voxel matrix 240 × 240 × 180, voxel size 1 × 1 × 1 mm3, TR/TE 7/3.201 ms, flip angle 8°. These T1-weighted images were used to measure cortical thickness and deep GM volumes.
We also acquired a gradient echo diffusion tensor imaging (DTI) sequence as follows: axial orientation, 2 × 2 × 2 mm3 acquiring voxel size, interpolated to 1 × 1 × 2 mm3; reconstructed matrix 256 × 256; 70 slices; TE/TR 61/8500 ms; flip angle 90°; 32 gradient directions; no averages; max b-factor = 1000s/mm2; 6 min scan. The DTI images were further used for analyses of WM integrity.
Image processing
Cerebral GM
Cortical thickness (n = 33)
Cortical thickness was computed using the FreeSurfer software v.5.3 [23]. Measurements were performed according to the protocol suggested by Fischl and Dale [24, 25]. The software creates triangle meshes that form two surfaces, the interface between GM and CSF and the interface between GM and WM. The shortest distance between the interfaces constitutes the measured cortical thickness. We took into account the cortical regions by the Desikan & Detrieux atlas for comparisons [26].
Basal ganglia volumes (n = 33)
T1 weighted images were processed with the T1 MultiAtlas approach using “MRICloud” (http://MRICloud.org), a public web-based service for multi-contrast imaging segmentation and volumetric quantification. Raw images were re-oriented (sagittal to axial), then had inhomogeneity correction and the whole brain was segmented after skull-striping. We employed linear and non-linear algorithms for brain co-registrations [27] and a multi-atlas labeling fusion enabled the identification of brain regions followed by a last step of labelling adjusting with PICSL [28]. Nineteen atlases (JHU adult atlas version 9B) were used to generate 283 structural definitions [29]. All analyses were performed in native space. The computations were processed on the Gordon cluster of XSEDE [30].
Cerebral, brainstem and cerebellar WM
DTI MultiAtlas Approach (n = 30)
White Matter microstructural damage was assessed using DTI MultiAtlas using maps of fractional anisotropy (FA), axial diffusivity (AD), mean diffusivity (MD) and radial diffusivity (RD). The DTI MultiAtlas was processed using “MRICloud” where Raw DTI-weighted images were co-registered and corrected for eddy currents and subject motion using a 12-parameter affine transformation [31, 32]. The DTI-parameters were calculated using a multivariate linear fitting and skull-stripped using the b = 0 image, by intensity threshold, a tool of Roi Editor software. This preprocessing was performed using DTIStudio software [33]. After that, a non-linear registration using a multi-contrast LDDMM [34] was performed, followed by the parcellation, which employs a DLFA algorithm [28, 30]. The computations were processed on the Gordon cluster of XSEDE.
Cerebellar WM volumes (n = 33)
We also computed the volumes of cerebellum and cerebellar peduncles WM using T1 volumetric images and the multi-atlas pipeline as described previously (section on basal ganglia volumetry).
Cerebellar GM
Cortical thickness (n = 33)
Cortical thickness was computed using a patch-based multi-atlas segmentation tool called CERES (CEREbellum Segmentation) that is able to automatically parcellate the cerebellum lobules. The proposed method works with standard resolution magnetic resonance T1-weighted images and uses the Optimized PatchMatch algorithm to speed up the patch matching process [35].
Spinal cord morphometry (n = 32)
Cross-sectional area (CA) (n = 32)
Spinal cord segmentation and measurements were performed semi-automatically using the SpineSeg software tool, developed at UNICAMP neuroimaging laboratory [36]. Further methodological details are available elsewhere [37].
Statistical analysis
Clinical and demographic data of all subjects are reported with descriptive statistics (Table 1). MRI analyses included both group comparisons and assessment of correlations, as detailed below. Statistical analyses were performed using the IBM SPSS statistics software version 20.
Group comparisons (patients versus controls)
All imaging parameters were assessed through a General Linear Model. Age and gender were used as covariates. For cortical thickness and brain volumetric studies, estimated intracranial volume (eTIV) was also added as a covariate. In order to correct for multiple comparisons, we employed the Bonferroni adjustment with level of significance set as 0.05.
Comparison between groups of patients
Three groups of patients were defined according to disease duration: group 1: < 5 years (n = 11, mean SARA score = 10.4); group 2: 5–9 years (n = 12, mean SARA score = 14.3) and group 3: ≥ 10 years (n = 9, mean SARA score = 21.7). Each group was compared with a healthy age and gender matched control group. These comparisons generated statistical maps showing the pattern of structural abnormalities in each stage. Afterwards, we looked at these maps to investigate how the disease progressed. For each specific comparison, a general linear model was applied, followed by Bonferroni’s correction.
Correlations and multiple regression analyses
Pearson correlation coefficient assessed possible correlations between imaging measures and clinical data whenever data distribution was normal; Spearman correlation was employed for data that were not normally distributed. Gender and age were used as covariates for correlations. Beck depression inventory and educational level were used as covariates for cognitive correlations. Bonferroni was employed to adjust for multiple comparisons (level of significance p = 0.05). To evaluate the determinants of the structural damage in SCA1, multiple variable regressions were used (dependent variables: volume/thickness/area and independent variables: age, CAG length and eTIV).
Results
Clinical features
Demographic and clinical data of all subjects in the study are shown in Table 1. Ataxia was the main symptom in all cases, with gait disturbances, axial and appendicular incoordination. All patients had pyramidal signs, with brisk reflexes, however, spasticity was not found. Urinary incontinence or urgency was found in nine patients (28%). Five (15%) were wheelchair bound, what happened after a mean disease duration of 15.2 years. Severe amyotrophy of intrinsic hand muscles was present in four (12%), hand tremor was evident in sixteen (50%), cephalic tremor was found in one (3%) and eleven (34%) complained of frequent falls. Sensory impairment was present in seven (21%) and no one had visual loss. Parkinsonism and other movement disorders were not found.
From 28 patients evaluated, fifteen (53%) fulfilled criteria for cognitive impairment (ACE-R score < 78 if > 11 years of schooling or < 68 in patients with < 11 years of schooling) [21]. Mean ACE-R was 77.0 (range 62–97). From ACE-R subscores, verbal fluency was the most affected domain (mean score of 51%) followed by memory (68%), language (85%), visuo-spatial abilities (86%) and attention/orientation (89%). The sum of verbal fluency visuo-spatial skills and attention was interpreted as an estimate of executive function. ACE-R has a subscore, the VLOM ratio, which is composed by the scores of verbal fluency + language/orientation + memory (recall of name and address only). A VLOM ratio < 2.2 points to frontotemporal dysfunction with 95% of specificity. A score > 3.2, on the other hand, points to a dementia that resembles Alzheimer’s disease [20]. Twenty-six patients (92.8%) had a score of less than 2.2 while the remaining two had indexes in the unspecific range. This indicates that frontotemporal dysfunction could be the clinical phenotype of cognitive dysfunction in SCA1 and ACE-R significantly correlated with physical burden (r = − 0.49, p = 0.013).
Regarding neuropsychiatric manifestations, nine (28%) displayed excessive anxiety and, in 4 (12%) patients, apathy was reported by the caregiver. BDI scores were abnormal in 70% of patients and correlated with motor disability (r = 0.47, p = 0.008).
Neuroimaging
Cerebral and brainstem GM
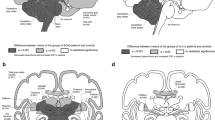
Using ROi-based analysis, significant differences were seen in cortical thickness between patients and controls in several areas (Fig. 2a). The SARA and ACE-R domains did not show significant correlations with cortical thickness. Analysis of deep gray matter volumes showed significant volumetric reduction in both amygdalae and red nuclei (Fig. 2b). Volumes of both red nuclei (right r = − 0.79, p < 0.001; left r = − 0.72, p < 0.001) correlated negatively with SARA scores. Memory domain score of ACE-R correlated with left amygdala volume (r = 0.49, p < 0.05).

Cross-sectional results of ROI-based analyses using FreeSurfer (a), T1 MultiAtlas (b) and CERES (c), comparing SCA1 patients to healthy controls (For more details of the involved areas, see the Supplemental Data—Tables 1–4)
Cerebellar GM
In the SCA1 group, cerebellar cortical thickness was significantly reduced in several regions of both hemispheres (p < 0.001, Fig. 2c). Cortical thickness of the cerebellum (right hemisphere r = − 0.63, p < 0.001; left hemisphere r = − 0.61, p < 0.001), lobules X at right (r = − 0.75, p < 0.001) and left sides (r = − 0.79, p < 0.001) correlated negatively with SARA scale. Concerning ACE-R score, only the fluency domain correlated with VIIIB right cortical thickness (r = 0.52, p = 0.04).
Cerebral, brainstem and cerebellar WM
We identified a pattern of diffusely reduced fractional anisotropy (FA) accompanied by increase in AD, MD and RD in infratentorial regions and the corticospinal tracts (Fig. 3). Despite this, the diffusion metrics did not correlate with SARA and ACE-R scores.

Cross-sectional results of DTI MultiAtlas approach showing areas of reduced fractional anisotropy (FA), increased axial diffusivity (AD), increased mean diffusivity (MD) and increased radial diffusivity (RD) in patients with SCA1 after comparison with controls (For more details of the involved areas, see the Supplemental Data—Tables 5–8)
Volumetric WM analyses were then performed using T1 images and the multi-atlas pipeline. There were significant differences in WM volumes between patients and controls, involving middle cerebellar peduncle-left (p < 0.001), middle cerebellar peduncle-right (p < 0.001), cerebellum white matter-right (p < 0.001), cerebellum white matter-left (p < 0.001), inferior cerebellar peduncle-left (p < 0.001) and inferior cerebellar peduncle-right (p < 0.001). In contrast to DTI parameters, we were able to demonstrate significant correlations between fluency and volumes of the following structures: middle cerebellar peduncle-left (r = 0.70, p < 0.001), middle cerebellar peduncle-right (r = 0.61, p = 0.002), cerebellum white matter-right (r = 0.48, p = 0.02) and cerebellum white matter-left (r = 0.46, p = 0.02). Besides that, SARA correlated with cerebellum white matter-right (r = − 0.56, p < 0.05), cerebellum white matter-left (r = − 0.65, p < 0.001) and inferior cerebellar peduncle-left (r = − 0.46, p < 0.05).
Disease course
Clinical results
Correlation analyses revealed that SARA scores had a significant association with age (r = 0.51, p < 0.05) and disease duration (r = 0.78, p < 0.001). ACE-R scores also correlated inversely with disease duration (r = − 0.41, p < 0.05), but not with age. Analyzing the ACE-R domains, only memory and visuospatial abilities were correlated with disease duration (r = − 0.57, p < 0.05; r = − 0.040, p < 0.05, respectively). Comparing the subgroups defined according to disease duration, significant differences regarding motor (SARA, p < 0.001) and cognitive (ACE-R, p < 0.05) functioning were seen.
Cerebral and brainstem GM
Regarding cortical thickness, no significant differences were seen in the group 1. Group 2 presented significant reduction in the left precentral gyrus. Group 3 showed atrophy of multiple cortical regions, involving primary and associative areas. Analysis of deep gray matter showed volume loss of both red nuclei in groups 2 and 3, but not 1 (Fig. 4).

Analysis of disease course, showing structural differences between SCA1 patients and controls, based on disease duration. Group 1 (I), group 2 (II) and group 3 (III). Coronal segmentation, showing results of FreeSurfer, T1-MultiAtlas, CERES and FA-DTI. Notice the pattern of damage evolution, starting in infratentorial areas and spreading to supratentorial structures, according to the disease course (For more details of the involved areas, see the Supplemental Data—Tables 9–23)
Cerebral, brainstem and cerebellar WM
DTI atlas-based analyses revealed diffusely reduced FA in all groups. Changes in group 1 were essentially restricted to the cerebellar peduncles and lower brainstem. In the other groups, the alterations became more diffuse, involving other regions of the brainstem and cerebral white matter (Fig. 4).
Cerebellar GM
Group 1 had significant reductions in cerebellar cortical thickness only at the lobule IX at the right side. The following groups showed thinning in multiple areas (Fig. 4).
Spinal cord morphometry
There was a significant difference in terms of CA in all groups (Supplemental Data—Tables 24–26). Correlation analyses also converged towards reduced CA in patients with longer disease duration (r = − 0.48, p = 0.006). Analyzing all patients, CA inversely correlated with motor abilities (r = − 0.70, p < 0.001).
Factors associated with structural damage
Cortical thickness disclosed a significant association with age in both hemispheres of the brain. Red nuclei volumes were associated with CAG repeat length; at the left side there was also an association with age. DTI parameters correlated in general with age. Cerebellar cortical thickness and spinal cord area strongly correlated with age and CAG repeat length (Table 2).
Discussion
This study provides a detailed characterization of neurological, cognitive and neuroimage profiles in SCA1. We were able to show that SCA1 is characterized by widespread damage that goes far beyond the cerebellum and connections. Involvement of supratentorial structures is not only widespread but also helps to explain some of the cognitive/behavioural manifestations found. Neuropsychological deficits were indeed frequent (53%) and involved mostly executive functions, memory and verbal fluency. Another important finding was the high frequency and severity of depressive symptoms in this cohort. The detailed correlation between phenotype (motor and particularly, non-motor) and structure in SCA1 is indeed the major contribution of this study. Previous studies missed that point because they relied on smaller cohorts, had no cognitive/behavioural assessment and did not use multimodal imaging analyses [38, 39].
Regarding GM damage, we have found widespread cerebral cortical thinning extending to primary and associative areas in frontal, temporal, parietal and occipital lobes. These results are similar to previous neuroimaging reports [38]. However, there was also red nuclei and amygdalae atrophy in our patients, which were not previously noticed. Interestingly, the volumes of these structures correlated significantly with motor disability and cognitive tasks, respectively. In line with our findings, pathological data reveal gross macroscopic atrophy in these cerebral lobes [40]. The histological correlates of such findings are neuronal loss and astrogliosis, which is especially prominent at primary motor cortices (loss of giant Betz pyramidal cells) [40].
WM involvement was also prominent, especially at infratentorial regions. In contrast, we had a much more restricted pattern of supratentorial WM abnormalities that included essentially the inter-hemispheric connecting fibers (corpus callosum and fornices) as well as the corticospinal tracts (CST). Overall, these findings are in line with previous imaging and neuropathology reports [38,39,40,41,42,43]. The infratentorial WM tracts and the CST convey motor and sensory information, which helps to explain some phenotypic features of SCA1, such as sensory loss, incoordination and brisky reflexes. Callosal and fornix involvement would explain cognitive/behavioural manifestations, but we failed to identify such correlations in our patients. This looks counterintuitive, but it may be due to the battery of neuropsychological tests we employed. Although comprehensive, it did not assess, for instance, speed of cognitive processing or bimanual motor tasks, which are often associated with callosal/fornix integrity. Further studies are needed to address this point.
The cerebellum was diffusely involved, including the cortical mantle and cerebellar peduncles. The upper regions of the cerebellar hemispheres were relatively spared in contrast to the vermis and lower portions. Such damage has obvious correlations with the ataxic syndrome, which are confirmed by our own correlation analyses. In addition, we found an association between verbal fluency performance and cortical thickness at right-sided lobule VIIIB of the cerebellum. Kansal et al. found a strikingly similar result after exploring a large and heterogeneous cohort of subjects with cerebellar degeneration [44]. Taken together, these data reinforce the concept that the cerebellum should no longer be considered as a purely motor organ. In fact, patients with isolated acute or chronic focal cerebellar lesions show frontal-like and parietal-like symptoms; this clinical picture is referred to as the cerebellar cognitive affective syndrome (CCAS) [45]. These “cortical” symptoms may stem from the interruption, as in a disconnection syndrome [46] or from a dysfunctional contribution of the cerebellum [47] in the neocerebellar–neocortical reverberant network. It is possible that CCAS underlies at least part of the cognitive and behavioural manifestations we found in SCA1. The lack of correlation between cognition and cerebral damage (either GM and WM) argue in favor of the last hypothesis.
Despite the cross-sectional design, our results shed some light into the disease course, from a clinical and a structural point of view. Regarding clinical aspects, our data suggest that not only motor but also cognitive deficits are progressive since both had a significant correlation with time-dependent variables. In terms of MRI findings, we identified that the initial targets of lesion in SCA1 are the spinal cord, lobule IX of the cerebellum and brainstem WM tracts. Interestingly, lobule IX of the cerebellum was the only region found atrophic in presymtomatic SCA1 subjects in a recent European study [48]. This region seems to be particularly vulnerable to the deleterious effects of expanded ataxin-1, but the biochemical reasons remain to be elucidated. Later in the disease course, damage spreads cranially to involve other regions in the cerebellum, the red nuclei and cerebral cortex (beginning at precentral gyri). Such a pattern resembles that reported by Braak et al. in Parkinson’s disease, where alpha-synuclein would spread in a caudal-rostral fashion leading to progressive neuronal loss [49]. Although speculative, one might consider that ataxin-1 could disseminate trans-synaptically leading to cumulative neuronal loss [50].
Regarding the determinants of CNS damage in SCA1, we have shown that this might be driven by both age and CAG expansion. Our cross-sectional results suggest that the structural changes are progressive with time throughout the CNS, and that patients with larger (CAG) expansions tend to be more affected, especially concerning the cerebellum and spinal cord involvement. In conclusion, we were able to unravel the phenotypic and structural hallmarks of SCA1. In short, it seems that GM, WM and SC are distinctly affected in the disease, not only in terms of extension but also in terms of temporal course. These aspects point to the potential usefulness of CNS image analysis as a clinical biomarker in SCA1, but further prospective studies with presymtomatic individuals or using longitudinal evaluation are still needed to better elucidate the natural history of the disease.
Abbreviations
- ACE-R:
-
Addenbrooke’s cognitive examination revised
- AD:
-
Axial diffusivity
- BDI:
-
Beck Depression Inventory
- CNS:
-
Central nervous system
- CST:
-
Corticospinal tract
- DTI:
-
Diffusion tensor imaging
- eTIV:
-
Estimated total intracranial volume
- FA:
-
Fractional anisotropy
- GM:
-
Gray matter
- LH:
-
Left hemisphere
- MD:
-
Mean diffusivity
- RD:
-
Radial diffusivity
- RH:
-
Right hemisphere
- Roi:
-
Region of interest
- SC:
-
Spinal cord
- SARA:
-
Scale for the assessment and rating of ataxia
- VBM:
-
Voxel-based morphometry
- WM:
-
White matter
References
Bürk K, Abele M, Fetter M et al (1996) Autosomal dominant cerebellar ataxia type I clinical features and MRI in families with SCA1, SCA2 and SCA3. Brain 119:1497–1505
Bürk K, Globas C, Bösch S, Klockgether T, Zühlke C, Daum I, Dichgans J (2003) Cognitive deficits in spinocerebellar ataxia type 1, 2, and 3. J Neurol 250:207–211
Banfi S, Servadio A, Chung MY, Kwiatkowski TJ Jr, McCall AE, Duvick LA et al (1994) Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat Genet 7:513–520
Chong SS, McCall AE, Cota J, Subramony SH, Orr HT, Hughes MR et al (1995) Genetic and somatic tissue-specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat Genet 10:344–350
Tong X, Gui H, Jin F et al (2001) Ataxin-1 and Brother of ataxin-1 are components of the Notch signaling pathway. EMBO Rep 12:428–435
Orr HT, Chung M, Banfi S et al (1993) Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 4:221–226
Duyckearts C, Dürr A, Cancel G, Brice A (1999) Nuclear inclusions in spinocerebellar ataxia type 1. Acta Neuropathol 97:201–207
Schöls L, Bauer P, Schmidt T et al (2004) Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 3:291–304
Schöls L, Amoridis G, Büttner T et al (1997) Autosomal dominant cerebellar ataxia: phenotypic differences in genetically defined subtypes? Ann Neurol 42:924–932
Filla A, Mariotti C, Caruso G et al (2000) Relative frequencies of CAG expansions in spinocerebellar ataxia and dentatorubropallidoluysian atrophy in 116 Italian families. Eur Neurol 44:31–36
Klinke I, Minnerop M, Schmitz-Hubsch T, Hendriks M, Klockgether T, Wullner U, Helmstaedter C (2010) Neuropsychological features of patients with spinocerebellar ataxia (SCA) types 1, 2, 3, and 6. Cerebellum 9:433–442
Bürk K (2007) Cognition in hereditary ataxia. Cerebellum 6:280–286
Kameya T, Abe K, Aoki M et al (1995) Analysis of spinocerebellar ataxia type 1 (SCA1)-related CAG trinucleotide expansion in Japan. Neurology 45:1587–1594
Schmitz-Hübsch T, Coudert M, Bauer P et al (2008) Spinocerebellar ataxia type 1, 2, 3, and 6. Disease severity and non ataxia symptoms. Neurology 71:982–989
Hanganu A, Bedetti C, Degroot C, Mejia-Constain B, Lafontaine AL, Soland V et al (2014) Mild cognitive impairment is linked with faster rate of cortical thinning in patients with Parkinson’s disease longitudinally. Brain 137:1120–1129
Min R, Wu G, Cheng J, Wang Q, Shen D (2014) Multi-atlas based representations for Alzheimer’s disease diagnosis. Hum Brain Mapp 35:5052–5070
França MC Jr, D’Abreu A, Yasuda CL, Bonadia LC, Santos da Silva M, Nucci A, Lopes-Cendes I, Cendes F (2009) A combined voxel-based morphometry and 1H MRS study in patients with Friedreich’s ataxia. J Neurol 256:1114–1120
Lopes TM, D’Abreu A, França MC Jr et al (2013) Widespread neuronal damage and cognitive dysfunction in spinocerebellar ataxia type 3. J Neurol 260:2370–2379
Braga-Neto P, Godeiro-Junior C, Dutra LA, Pedroso JL, Barsottini OG (2010) Translation and validation into Brazilian version of the Scale of the Assessment and Rating of Ataxia (SARA). Arq Neuropsiquiatr 68:228–230
Mioshi E, Dawson K, Mitchell J, Arnold R, Hodges JR (2006) The Addenbrooke’s Cognitive Examination Revised (ACE-R): a brief cognitive test battery for dementia screening. Int J Geriatr Psychiatry 21:1078–1085
Carvalho VA, Caramelli P (2007) Brazilian adaptation of the Addenbrooke’s cognitive examination-revised (ACE-R). Dement Neuropsychol 2:212–216
Gorenstein C, Andrade L (1996) Validation of a portuguese version of the beck depression inventory and state-tract anxiety inventory in Brazilian subjects. Braz J Med Biol Res 29:453–457
Hutton C, Draganski B, Ashburner J, Weiskopf N (2009) A comparison between voxel-based cortical thickness and voxel-based morphometry in normal aging. Neuroimage 48:371–380
Fischl B, Dale AM (2000) Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci 97:11050–11055
De Rezende TJR, D’Abreu A, Guimarães RP, Lopes TM, Lopes-Cendes I, Cendes F, et al (2015) Cerebral cortex involvement in Machado Joseph disease. Eur J Neurol 22:277–283
Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D et al (2006) An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31:968–980
Miller MI, Faria AV, Oishi K, Mori S (2013) High-throughput neuroimaging informatics. Front Neuroinform 17:7–31
Tang X, Shoko Y, Hsu J, Huisman TAGM, Faria AV, Oishi K et al (2014) Multi-contrast multi-atlas parcellation of diffusion tensor imaging of the human brain. PLoS One 9:e96985
Wu D, Ma T, Ceritoglu C, Li Y, Chotiyanonta J, Hou Z et al (2016) Resource atlases for multi-atlas brain segmentations with multiple ontology levels based on T1-weighted MRI. Neuroimage 125:120–130
Towns J, Cockerill T, Dahan M (2014) XSEDE: accelerating scientific discovery. Comput Sci Eng 16:62–72
Zhuang J, Hrabe J, Kangarlu A, Xu D, Bansal R, Branch CA et al (2006) Correction of eddy-current distortions in diffusion tensor images using the known directions and strengths of diffusion gradients. J Magn Reson Imaging 24:1188–1193
Faria AV, Zhang J, Oishi K, Li X, Jiang H, Akhter K, Hermoye L, Lee SK, Hoon A, Stashinko E, Miller MI, van Zijl PC, Mori S (2010) Atlas-based analysis of neurodevelopment from infancy to adulthood using diffusion tensor imaging and applications for automated abnormality detection. Neuroimage 15:415–428
Jiang H, van Zijl PC, Kim J et al (2006) DtiStudio: resource program for diffusion tensor computation and fiber bundle tracking. Comput Methods Prog Biomed 81:106–116
Miller MI, Beg MF, Ceritoglu C, Stark C et al (2005) Increasing the power of functional maps of the medial temporal lobe by using large deformation diffeomorphic metric mapping. Proc Natl Acad Sci 102:9685–9690
Romero JE, Coupé P et al (2017) CERES: a new cerebellum lobule segmentation method. NeuroImage 147:916–924
Bergo FPG, França MC Jr, Chevis CF, Cendes F (2012) SpineSeg: a segmentation and measurement tool for evaluation of spinal cord atrophy. In: CISTI’2012 (7ª Conferencia Ibérica de Sistemas y Tecnologia de Información); Madrid, Spain, IEEE 400–403
Martins CR Jr, Martinez ARM, de Rezende TJR, Branco LMT, Pedroso JL, Barsottini OGP, Lopes-Cendes I, França MC Jr (2017) Spinal cord damage in spinocerebellar ataxia type 1. Cerebellum 16:792–796
Schulz JB, Borkert J, Wolf S et al (2010) Visualization, quantification and correlation of brain atrophy with clinical symptoms in spinocerebellar ataxia types 1, 3 and 6. Neuroimage 49:158–168
Reetz K, Costa AS, Mirzazade S, Ataxia Study Group Investigators et al (2013) Genotype specific patterns of atrophy progression are more sensitive than clinical decline in SCA1, SCA3 and SCA6. Brain 136:905–917
Rüb U, Bürk K, Timmann D, den Dunnen W, Seidel K, Farrag K, Brunt E, Heinsen H, Egensperger R, Bornemann A, Schwarzacher S, Korf HW, Schöls L, Bohl J, Deller T (2012) Spinocerebellar ataxia type 1 (SCA1): new pathoanatomical and clinico-pathological insights. Neuropathol Appl Neurobiol 38:665–680
Seidel K et al (2012) Brain pathology of spinocerebellar ataxias. Acta Neuropathol 124:1–21
Mandelli ML, De Simone T, Minati L, Bruzzone MG, Mariotti C, Fancellu R, Savoiardo M, Grisoli M (2007) Diffusion tensor imaging of spinocerebellar ataxias types 1 and 2. AJNR Am J Neuroradiol 28:1996–2000
Della Nave R, Ginestroni A, Tessa C, Salvatore E, De Grandis D, Plasmati R, Salvi F, De Michele G, Dotti MT, Piacentini S, Mascalchi M (2008) Brain white matter damage in SCA1 and SCA2. An in vivo study using voxel-based morphometry, histogram analysis of mean diffusivity and tract-based spatial statistics. Neuroimage 43:10–19
Kansal K, Yang Z, Fishman AM et al (2017) Structural cerebellar correlates of cognitive and motor dysfunctions in cerebellar degeneration. Brain 140:707–720
Schmahmann JD, Sherman JC (1998) The cerebellar cognitive affective syndrome. Brain 121:561–579
Schmahmann JD, Pandya DN (2008) Disconnection syndromes of basal ganglia, thalamus, and cerebrocerebellar systems. Cortex 44:1037–1066
Schmahmann JD (2004) Disorders of the cerebellum: ataxia, dysmetria of thought, and the cerebellar cognitive affective syndrome. J Neuropsychiatry Clin Neurosci 16:367–378
Jacobi H, Reetz K, du Montcel ST et al (2013) Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: analysis of baseline data. Lancet Neurol 12:650–658
Braak H, Del Tredici K, Rub U, de Vos RAI, Jansen Steur ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, Covert M, Melki R, Kirkegaard K, Brahic M (2012) Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann Neurol 72:517–524
Funding
This work was supported by the São Paulo Research Foundation (FAPESP) (Grant no. 2013/01766-7).
Author information
Authors and Affiliations
Contributions
CRM: Research project: Conception, Organization, Execution, Statistical analysis: Design, Execution, Review and Critique, Manuscript: Writing of the first draft; ARMM: Research project: Organization, Statistical analysis: Design, Execution, Manuscript: Review and Critique; IFV: Research project: Conception, Statistical Analysis: Review and Critique, Manuscript: Review and Critique; TJR: Research project: Conception, Statistical Analysis: Review and Critique, Manuscript: Review and Critique; RC: Research project: Conception, Statistical analysis: Review and Critique, Manuscript: Review and Critique; JLP: Research project: Conception, Statistical analysis: Review and Critique, Manuscript: Review and Critique; OGPB: Research project: Organization, Statistical analysis: Review and Critique, Manuscript: Review and Critique; ÍL-C: Research project: Execution, Statistical analysis: Review and Critique, Manuscript: Review and Critique; MCF: Research project: Design, Execution, Review and Critique, Statistical analysis: Design, Execution, Review and Critique, Manuscript: Review and Critique.
Corresponding author
Ethics declarations
Conflicts of interest
Casseb reports grants from FAPESP. Rezende reports grants from FAPESP; Dr Faber reports grants from CNPq (Brazilian National Council for Scientific and -Technological Development) during the conduct of the study; Dr Martinez reports grants from FAPESP during the conduct of this study; Dr Lopes-Cendes reports grants from FAPESP and CNPq, during the conduct of the study. Dr França Jr reports grants from FAPESP and CNPq. Dr Barsottini reports grants from FAPESP and CNPq, during the conduct of the study. Dr Pedroso reports grants from FAPESP and CNPq, during the conduct of the study. The authors declare that they have no conflict of interest. Marcondes C França Jr: reports grants from FAPESP during the conduct of this study. He also received research support from PTC therapeutics, Cristália and Pfizer; took part in advisory boards for Genzyme, Pfizer, Baxter and CSL-Behring; Received travel Grant from Pfizer, Baxter and PTC therapeutics.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Martins Junior, C.R., Martinez, A.R.M., Vasconcelos, I.F. et al. Structural signature in SCA1: clinical correlates, determinants and natural history. J Neurol 265, 2949–2959 (2018). https://doi.org/10.1007/s00415-018-9087-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-9087-1