
Abstract
The mutation of vesicle-associated membrane protein-associated protein B (VAPB) was proved to cause family amyotrophic lateral sclerosis (FALS). Only two mutations of VAPB associated with ALS have been reported (p.Pro56Ser and p.Thr46Ile). Here we reported a Chinese Han FALS family caused by a novel VAPB point mutation. The clinical materials of one Chinese Han FALS family were collected. The genetic analysis was carried out by target sequencing and further verified by Sanger sequencing. One novel mutation of c.167C>A (p.Pro56His) on VAPB was found in the proband. The age at onset of the proband was 48 with the onset symptoms of weakness in the right arm, followed by progressive limb and trunk weakness with decreased deep-tendon reflexes, muscular cramps and fasciculation. But the disease duration was more than 15 years. He was under the tracheotomy for 1 year at last visit. Electromyography showed widespread acute and chronic neurogenic damages. His mother presented weakness in her limbs in 50 s and died 15 years later. One of his younger sisters diagnosed as ALS for 6 years also carried the same mutation. She presented the similar symptoms on 41. No dominant upper motor neuron sign was showed. The clinical features were similar to the patients carrying the known mutation of p.Pro56Ser. A novel mutation of VAPB was found in one Chinese Han FALS pedigree. The affected patients presented a much slower progression and the lesions were limited in lower motor neurons.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a heterogeneous progressive neurodegenerative disease characterized by loss of motor neurons in the spinal cord, brainstem and motor cortex. The clinical manifestations are progressive weakness with skeletal muscle wasting, dysarthria, dysphagia and respiratory failure. It is a fatal disease typically lasting 3–5 years with respiratory failure being the cause of death.
Among all the cases, 5–10% are found to be familial ALS (FALS) [1]. Since the first causative gene of the Cu/Zn superoxide dismutase gene (SOD1) on chromosome 21 was found in 1993 [2], near 30 causative genes of ALS have been identified in FALS [3].
Vesicle-associated membrane protein-associated protein B (VAPB), the causative gene of ALS8 (MIM 608627), was first identified at 20q13.3 in a large white Brazilian family with 28 affected members distributed across four generations in 2004 [4]. ALS8 is autosomal dominant inherited, shows slow progression and expresses three principle phenotypes: late-onset spinal muscular atrophy (LSMA), atypical ALS and typical ALS [4]. Patients of ALS8 display mainly symptoms restricted to lower motor neuron function, however, some families have been reported on which show upper motor neuron involvement as well [4, 5].
VAPB mutations are prevalent in Portuguese/Brazil FALS patients with frequency up to 43.6% [6], primarily contributed to a founder effect [7], while in other populations, the frequency of VAPB mutations in ALS patients is low [8,9,10]. For example, as a single pedigree, there have been reports of this mutation in ALS patients from Germany, UK, Japan and China [11,12,13,14].
Until now, only two mutations of VAPB have been identified in FALS patients. p.Pro56Ser [4] is the most common, followed by p.Thr46Ile, which is considered rare and has only been reported in a single German FALS patient [12]. p.Val234Ile [15] and p.Gla145Val [16] in VAPB were also reported in one FALS patient from Japan and one SALS patient from western countries, respectively, but neither mutation was confirmed pathogenic (https://www.ncbi.nlm.nih.gov/clinvar/).
Here we report on a novel mutation of p.Pro56His in VABP found in a Chinese FALS pedigree and describe the clinical characteristics of the family.
Methods
Clinical materials
The pedigree with autosomal dominant inherited family history was collected in the clinic of ALS at Huashan Hospital (Shanghai, PRC). The clinical materials were investigated in the proband (III:1) and one sibling (III:5; sister). Examinations including electromyography (EMG), electrocardiography, echocardiography and blood biochemical analysis (serum creatine kinase, electrolytes, glucose, retinal and liver function, thyroxine, thyroid-stimulating hormone and sedimentation rate) were performed in the proband (III:1). The sibling (III:5) received an EMG and a blood sample was collected for genetic testing. The diagnosis of ALS was made according to the Awaji criteria [17].
The study was conducted after receiving written informed consent from the patients. This study was approved by the Institutional Ethics Committee of Huashan Hospital.
Genetic test
The genomic DNA was extracted from peripheral blood using a whole blood genomic DNA extraction kit (Qiagen, German). A panel of ALS related genes including VAPB was designed (Supplementary Table 1). The genetic analysis was performed by target sequencing as previously reported [18]. Briefly, all exons and their corresponding flanking regions of the genes in the panel were selected as target regions. Samples were prepared as an Illumina sequencing library and the sequencing libraries were enriched for these target regions. The captured libraries were then sequenced using Illumina HiSeq 2500 Sequencer (Illumina, San Diego, CA). All variants different from the reference sequence were further screened by allele frequency < 1% according to 1000 Genomes Project (http://www.1000genomes.org/home), Inhouse database, ESP6500 (evs.gs.washington.edu/EVS/) and ExAC (http://exac.broadinstitute.org/). The synonymous variants were excluded. The phenotypes of the remaining genes were compared with the clinical features of the proband. The inherited modes were considered to further exclude the irrelevant genes. The databases including SIFT, PolyPhen-2 and Mutation Taster were used to predict the pathogenicity of the variants.
The candidate mutations were further confirmed by Sanger sequencing, the procedure of which was as previous reported [19].
PCR-restriction fragment length polymorphism analysis was applied to detect the genotype of p.Pro56His in the healthy controls from the community as previously reported [20]. The primers were 5′-CCCAATCAGTCTCTGTCTC-3′ (forward) and 5′-CATCTTCTTCTGCTACACTGC-3′ (reverse) and the restriction enzyme was Hae III (Takara Bio, China).
Since the frequency of C9orf72 expansion was rare found in both FALS and sALS in Asian population [21, 22], we perform a two-step polymerase chain reaction protocol to detect whether the number of GGGGCC hexanucleotide repeat was in normal range as previously reported [23, 24]. Briefly, in the first step, a repeat-primed polymerase chain reaction assay was used to detect the size of the larger expanded alleles using a touchdown thermocycling program. In the second step, a FAM-fluorescent labeled PCR assay was applied to detect the accurate genotype.
Results
Clinical evaluation
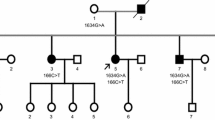
The pedigree originated from Zhejiang province in eastern China (Fig. 1). Three individuals of two generations in this family had symptoms. The proband (III:1) was a 63 years old male patient. He presented with slight weakness in the right arm at the age of 48. Subsequently, the weakness gradually spread to the legs and the trunk. The weakness was more severe in the legs compared to the arms. He also presented with muscular cramps and fasciculation. The bulbar function was less affected. He experienced breathing difficulty at age 61 and received a tracheotomy due to respiratory failure at age 62. During his final clinical evaluation, at age 63, he was unable to walk without help but still could raise his arms. Under the tracheotomy, he could not speak, but could swallow normal food by himself. Decreased deep-tendon reflexes were noted, while upper motor neurons were not apparently affected.

Pedigree chart of FALS with p.Pro56Ser mutation on VAPB gene. Arrow: proband; square: male; circle: female; slash: deceased; solid symbol: affected. W/M and W/W represent the genotype of VAPB. W/M: wild type/mutant; W/W: wild type/wild type
EMG showed widespread (including cervical, thoracic and lumbar regions) acute and chronic neurogenic damage.
His mother (II:5) exhibited similar symptoms in her fifties and died 15 years later. One younger sister of the proband (III:5) commenced showing similar symptoms at age 41. No upper motor neuron signs were noted. EMG showed widespread (including cervical, thoracic, lumbar and cranial regions) acute and chronic neurogenic damages as well. Her symptoms spread slowly and she continued to take care of herself in daily life 6 years after diagnosis.
Mutation analysis
The mean depth of target sequencing of the proband was 373.43X. The percentage of the target region with mean depth > 20X was 99.5%.
After screening by the variants frequencies, synonymous status, inherited mode and clinical manifestations, only one variants of c.167C>A (p.Pro56His) in VAPB (NM_004738) was found, with the depth of 249X, related to the disease of the proband. The variant was predicted to be damaging by SIFT (value of 0.001), PolyPhen-2 (value of 1.000) and MutationTaster.
The variant was performed in the proband and other family members by Sanger sequencing (Fig. 2). His symptomatic sister (III:5) carried the same variant in VAPB while his two asymptomatic brothers and one asymptomatic sister (III:2, III:3 and III:4) did not carry this variant, which met the family co-segregation.

Sanger sequencing of the family members. The genotype of VAPB was p.Pro56His in III:1 and III:5, p.Pro56Pro in III:2, III:3 and III:4
Since p.Pro56His in VAPB had not been reported before, it was investigated in 200 healthy controls of Chinese origin from the community (mean age 72.5 ± 6.18), but no variation was detected. The variant was highly conserved across species (Fig. 2). According to the American College of Medical Genetics (ACMG) guideline [25], the variant was classified as likely pathogenic. Moreover, the structure of the mutant vapb protein of p.Pro56His was changed compared with the wild type predicted on a protein structure analysis website (http://www.cmbi.ru.nl/hope/) (Fig. 3) [26]. The mutation was predicted to cause loss of hydrophobic interactions with other molecules on the surface of the protein.

The structures of vapb protein. a The overview of protein. The protein is colored grey and the side chain of the mutated residue is colored magenta. b Close-up of the mutation. The protein is colored grey and the side chains of both the wild-type (green) and the p.Pro56His mutant residue (red) are shown
The repeat number of GGGGCC hexanucleotide repeat in C9orf72 of the proband was four, which was in normal range.
Discussion
We identified a mid-adult onset FALS family of two generations carrying a novel mutation of p.Pro56His in VAPB. The lesion was restricted to the lower motor neuron with bulbar function being less affected. No tremor or upper motor neuron signs were noted. The progression was slow, taking 14 years for the proband to move from disease onset to tracheotomy.
ALS caused by VAPB is rare. Until now, only 30 pedigrees have been reported worldwide, most of which originating from Brazil. p.Pro56Ser is the commonest variant seen in the VAPB mutation. VAPB p.Pro56Ser mutation causing motor neuron diseases displays heterogeneity of clinical phenotypes including LSMA symptoms (no bulbar symptoms or pyramidal signs), atypical ALS (pyramidal signs or bulbar signs, but must have essential tremor) and typical ALS [4]. Our patients presented with a similar symptom profile to that of ALS8 as the atypical ALS subtype but without tremor and with mild bulbar dysfunction. The patients carrying p.Thr46Ile could also be classified into one of the three subtypes of typical ALS but there was no phenotype–genotype relationship, which may contribute to the low frequency of the VAPB mutations or some as yet unidentified genetic factors.
Considering these patients carrying VAPB mutation, all mutations of VAPB were detected in FALS (Table 1). The age at onset varied from 30 to 73. The progress could be rapid or slowly from 1 + to 30 + years. Some patients showed tremor, cramps or fasciculation. The most common onset symptom was limb weakness. However, pain was reported as the initial symptom in one Chinese VAPB pedigree recently [14]. All patients had lower motor neuron lesions, while upper motor neuron and bulbar function were affected in few patients. Interfamilial and intrafamilial heterogeneity could be seen in the families carrying the same mutation [4]. Though the pedigree in the current study carrying a novel VAPB mutation, the general clinical characteristics of the patients were in accordance to those carrying other VAPB mutations.
Interestingly, the VAPB mutations can co-exist with other ALS causative genes of SOD1 [6]. In this report, we did not identify any mutations in other ALS genes including C9orf72.
VAPB, as a member vesicle-associated membrane protein family of endoplasmic reticulum tail-anchored transmembrane proteins, was highly conserved and ubiquitously expressed. The wide spectrum of VAPB interacting proteins contributes to a great variety of physiological functions. There are three domains in the protein including the major sperm protein (MSP), the coiled-coil domain and the transmembrane domain. The MSP is composed of the first 150 residues and is conserved in all vesicle-associated membrane protein-associated protein (VAP) family members [27]. Two VAPB mutations, p.Pro56Ser and p.Thr46Ile are both located in the MSP. The p.Pro56Ser mutation has been shown to change the normal structures of MSP and affects its folding properties which exposes its hydrophobic patches and renders the protein more prone to aggregation [28]. The p.Pro56His mutation shares the same position as p.Pro56Ser, leaving it possible that they affect the disease by the same pathway, although this has yet to be investigated.
Conclusion
We reported on a Chinese Han FALS pedigree carrying a novel VAPB mutation of p.Pro56His which had not been reported before. The clinical characteristics include mid-adult onset, long disease duration and lesions restricted to lower motor neurons consistent to those of the atypical ALS subtype ALS8 with the exception for essential tremor and the less affected bulbar function. Further research is warranted to focus on the pathogenesis of this mutation on ALS development.
References
Mitchell JD, Borasio GD (2007) Amyotrophic lateral sclerosis. Lancet 369(9578):2031–2041. doi:10.1016/S0140-6736(07)60944-1
Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP et al (1993) Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science 261(5124):1047–1051
Corcia P, Couratier P, Blasco H, Andres CR, Beltran S, Meininger V, Vourc’h P (2017) Genetics of amyotrophic lateral sclerosis. Revue neurologique 173(5):254–262. doi:10.1016/j.neurol.2017.03.030
Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, Gillingwater T, Webb J, Skehel P, Zatz M (2004) A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 75(5):822–831. doi:10.1086/425287
Landers JE, Leclerc AL, Shi L, Virkud A, Cho T, Maxwell MM, Henry AF, Polak M, Glass JD, Kwiatkowski TJ, Al-Chalabi A, Shaw CE, Leigh PN, Rodriguez-Leyza I, McKenna-Yasek D, Sapp PC, Brown RH Jr (2008) New VAPB deletion variant and exclusion of VAPB mutations in familial ALS. Neurology 70(14):1179–1185. doi:10.1212/01.wnl.0000289760.85237.4e
Chadi G, Maximino JR, Jorge FMH, Borba FC, Gilio JM, Callegaro D, Lopes CG, Santos SND, Rebelo GNS (2017) Genetic analysis of patients with familial and sporadic amyotrophic lateral sclerosis in a Brazilian Research Center. Amyotroph Lateral Scler Frontotemporal Degener 18(3–4):249–255. doi:10.1080/21678421.2016.1254245
Nishimura AL, Al-Chalabi A, Zatz M (2005) A common founder for amyotrophic lateral sclerosis type 8 (ALS8) in the Brazilian population. Hum Genet 118(3–4):499–500. doi:10.1007/s00439-005-0031-y
Tsai CP, Soong BW, Lin KP, Tu PH, Lin JL, Lee YC (2011) FUS, TARDBP, and SOD1 mutations in a Taiwanese cohort with familial ALS. Neurobiol Aging 32(3):553 e513–553 e521. doi:10.1016/j.neurobiolaging.2010.04.009
Ingre C, Pinto S, Birve A, Press R, Danielsson O, de Carvalho M, Gudmundsson G, Andersen PM (2013) No association between VAPB mutations and familial or sporadic ALS in Sweden, Portugal and Iceland. Amyotroph Lateral Scler Frontotemporal Degener 14(7–8):620–627. doi:10.3109/21678421.2013.822515
Conforti FL, Sprovieri T, Mazzei R, Ungaro C, Tessitore A, Tedeschi G, Patitucci A, Magariello A, Gabriele A, Labella V, Simone IL, Majorana G, Monsurro MR, Valentino P, Muglia M, Quattrone A (2006) Sporadic ALS is not associated with VAPB gene mutations in Southern Italy. J Negat Results Biomed 5:7. doi:10.1186/1477-5751-5-7
Funke AD, Esser M, Kruttgen A, Weis J, Mitne-Neto M, Lazar M, Nishimura AL, Sperfeld AD, Trillenberg P, Senderek J, Krasnianski M, Zatz M, Zierz S, Deschauer M (2010) The p. P56S mutation in the VAPB gene is not due to a single founder: the first European case. Clin Genet 77(3):302–303. doi:10.1111/j.1399-0004.2009.01319.x
Chen HJ, Anagnostou G, Chai A, Withers J, Morris A, Adhikaree J, Pennetta G, de Belleroche JS (2010) Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J Biol Chem 285(51):40266–40281. doi:10.1074/jbc.M110.161398
Millecamps S, Salachas F, Cazeneuve C, Gordon P, Bricka B, Camuzat A, Guillot-Noel L, Russaouen O, Bruneteau G, Pradat PF, Le Forestier N, Vandenberghe N, Danel-Brunaud V, Guy N, Thauvin-Robinet C, Lacomblez L, Couratier P, Hannequin D, Seilhean D, Le Ber I, Corcia P, Camu W, Brice A, Rouleau G, LeGuern E, Meininger V (2010) SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype–phenotype correlations. J Med Genet 47(8):554–560. doi:10.1136/jmg.2010.077180
Di L, Chen H, Da Y, Wang S, Shen XM (2016) Atypical familial amyotrophic lateral sclerosis with initial symptoms of pain or tremor in a Chinese family harboring VAPB-P56S mutation. J Neurol 263(2):263–268. doi:10.1007/s00415-015-7965-3
van Blitterswijk M, van Es MA, Koppers M, van Rheenen W, Medic J, Schelhaas HJ, van der Kooi AJ, de Visser M, Veldink JH, van den Berg LH (2012) VAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patient. Neurobiol Aging 33(12):2950 e2951–2950 e2954. doi:10.1016/j.neurobiolaging.2012.07.004
Kabashi E, El Oussini H, Bercier V, Gros-Louis F, Valdmanis PN, McDearmid J, Mejier IA, Dion PA, Dupre N, Hollinger D, Sinniger J, Dirrig-Grosch S, Camu W, Meininger V, Loeffler JP, Rene F, Drapeau P, Rouleau GA, Dupuis L (2013) Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum Mol Genet 22(12):2350–2360. doi:10.1093/hmg/ddt080
de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J, Mills K, Mitsumoto H, Nodera H, Shefner J, Swash M (2008) Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol 119(3):497–503. doi:10.1016/j.clinph.2007.09.143
Xiong WX, Sun YM, Guan RY, Luo SS, Chen C, An Y, Wang J, Wu JJ (2016) The heterozygous A53T mutation in the alpha-synuclein gene in a Chinese Han patient with Parkinson disease: case report and literature review. J Neurol 263(10):1984–1992. doi:10.1007/s00415-016-8213-1
Wu ZY, Lin MT, Murong SX, Wang N (2003) Molecular diagnosis and prophylactic therapy for presymptomatic Chinese patients with Wilson disease. Arch Neurol 60(5):737–741. doi:10.1001/archneur.60.5.737
Sun YM, Li HL, Guo QH, Wu P, Hong Z, Lu CZ, Wu ZY (2012) The polymorphism of the ATP-binding cassette transporter 1 gene modulates Alzheimer disease risk in Chinese Han ethnic population. Am J Geriatr Psychiatry 20(7):603–611. doi:10.1097/JGP.0b013e3182423b6a
Tomiyama H (2013) [C9orf72 in Japanese amyotrophic lateral sclerosis (ALS)]. Rinsho shinkeigaku (Clin Neurol) 53(11):1074–1076
Jiao B, Tang B, Liu X, Yan X, Zhou L, Yang Y, Wang J, Xia K, Shen L (2014) Identification of C9orf72 repeat expansions in patients with amyotrophic lateral sclerosis and frontotemporal dementia in mainland China. Neurobiol Aging 35(4):936 e919–936 e922. doi:10.1016/j.neurobiolaging.2013.10.001
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72(2):245–256. doi:10.1016/j.neuron.2011.09.011
Jiao B, Guo JF, Wang YQ, Yan XX, Zhou L, Liu XY, Zhang FF, Zhou YF, Xia K, Tang BS, Shen L (2013) C9orf72 mutation is rare in Alzheimer’s disease, Parkinson’s disease, and essential tremor in China. Front Cell Neurosci 7:164. doi:10.3389/fncel.2013.00164
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. doi:10.1038/gim.2015.30
Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G (2010) Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform 11:548. doi:10.1186/1471-2105-11-548
Nishimura Y, Hayashi M, Inada H, Tanaka T (1999) Molecular cloning and characterization of mammalian homologues of vesicle-associated membrane protein-associated (VAMP-associated) proteins. Biochem Biophys Res Commun 254(1):21–26. doi:10.1006/bbrc.1998.9876
Navone F, Genevini P, Borgese N (2015) Autophagy and neurodegeneration: insights from a cultured cell model of ALS. Cells 4(3):354–386. doi:10.3390/cells4030354
Marques VD, Barreira AA, Davis MB et al (2006) Expanding the phenotypes of the Pro56Ser VAPB mutation: proximal SMA with dysautonomia. Muscle Nerve 34:731–739
Acknowledgements
This study was supported by the Founding of Shanghai Municipal Commission of Health and Family Planning (20124222).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors report no conflicts of interest.
Ethical standards
This study has been approved by the ethics committee of Huashan hospital and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sun, Ym., Dong, Y., Wang, J. et al. A novel mutation of VAPB in one Chinese familial amyotrophic lateral sclerosis pedigree and its clinical characteristics. J Neurol 264, 2387–2393 (2017). https://doi.org/10.1007/s00415-017-8628-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-017-8628-3