
Abstract
The aim of this study was to report the clinical spectrum associated with antibodies to myelin oligodendrocyte glycoprotein (MOG) in adult patients, and to assess whether phenotypic variants are dependent on recognition of rodent MOG epitopes. We retrospectively analyzed the features, course and outcome of 56 patients whose samples were investigated by brain tissue immunohistochemistry and cell-based assays using human and rodent MOG. The median age at symptom onset was 37 years (range 18–70); 35 patients (63 %) were female. After a median follow-up of 43 months (range 4–554), only 14 patients (25 %) developed a neuromyelitis optica spectrum disorder (NMOSD), 27 patients (47 %) retained the initial diagnosis of isolated optic neuritis, 7 (12 %) of longitudinally extensive transverse myelitis, and 2 (4 %) of acute disseminated encephalomyelitis; 6 patients (11 %) developed atypical demyelinating syndromes (4 had relapsing episodes of short myelitis lesions which in one occurred with optic neuritis; 1 had relapsing brainstem symptoms, and 1 relapsing demyelinating encephalomyelitis). The course was frequently associated with relapses (71 %) and good outcome. Twenty-seven patients (49 %) had antibodies that recognized rodent MOG epitopes, and 9 of them (16 %) showed a myelin staining pattern in rodent tissue. Only the myelin staining pattern was linked to NMOSD (p = 0.005). In conclusion, MOG autoimmunity in adult patients associates with a clinical spectrum wider than the one expected for patients with suspected NMOSD and overall good outcome. Antibodies to rodent MOG epitopes do not associate with any phenotypic variant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myelin oligodendrocyte glycoprotein (MOG) is a minor component of myelin, located on the outermost surface of the myelin sheaths with a single extracellular immunoglobulin-like domain and, therefore, accessible as target for antibody-mediated damage [1]. Using cell-based assays with human MOG (hMOG), high titers of antibodies have been identified predominantly in children with acute disseminated encephalomyelitis (ADEM) [2–4], and more recently in children and adults with aquaporin-4 (AQP4)-IgG seronegative neuromyelitis optica spectrum disorders (NMOSD) [5, 6] or limited NMO-like phenotypes [7–9]. Current knowledge of the associated clinical spectrum, however, is based on small series and most of them include a mixed population of children and adults in whom the clinical profile seems to be different [3, 4, 10].
A previous study in mainly children with demyelinating syndromes and hMOG-IgG showed that most of the serum of patients did not recognize mouse MOG, and the epitope specificity was not linked to different clinical presentations [1]. However, it is unclear whether similar findings may occur in adults. Therefore, we aimed to define the clinical spectrum associated with MOG antibodies in a large cohort of adult patients, and to assess whether the clinical profile was dependent on rodent MOG epitopes or coexistence of other immunoreactivities.
Methods
Patients
Between November 2005 and September 2015, the presence of hMOG-IgG was examined in 846 serum samples of patients with NMO or suspected related syndromes sent to our laboratory to determine AQP4-IgG. Overall, 60 adult patients (age at disease onset ≥18 years) with hMOG-IgG were identified; 4 of them were excluded because of the presence of a concurrent antibody (2 AQP4-IgG, 1 anti-Glycine receptor α1 subunit, and 1 anti-N-methyl-d-aspartate receptor [11]). The specificity of our assay has been previously reported [7] and 19 patients were included in two previous series [7, 12]. Data were obtained from clinical records, and information was collected from referring neurologists using a standardized questionnaire as reported [7]. The outcome reached at last follow-up was assessed by the Expanded Disability Status Scale (EDSS) [13] and severe visual disability was defined as sustained visual acuity <0.2 during at least 6 months after an optic neuritis attack.
Cell-based assays
All samples were examined for hMOG-IgG using an in-house cell-based assay (CBA) with HEK293 cells transfected with the full-length MOG C-terminally fused to EGFP (serum dilution 1:160) as reported [7]. Plasmids containing MOG cDNA from rat (rMOG) or mouse (mMOG) (kindly provided by Dr. Reindl) were used to detect the reactivity against rodent MOG. The specificity of the CBA (dilution 1:80 and 1:160, respectively) was confirmed by testing 50 samples (supplemental material) and the assays validated in Innsbruck (PP; MR). Two positive hMOG-IgG samples that tested positive against rodent MOG were absorbed with pellets of HEK293 cells transfected with either hMOG, rMOG, mMOG or AQP4 as unrelated control [7]. Immunoabsorbed samples assessed by the corresponding CBA were applied to brain sections as described below.
Brain tissue immunohistochemistry
Samples were screened by immunohistochemistry performed on non-perfused rat or mouse brain, fixed by immersion with 4 % paraformaldehyde for 1 h and processed as reported [11, 14]. Immunohistochemistry using a standard avidin–biotin peroxidase method was applied using patients’ serum (diluted 1:200) or a commercial rabbit polyclonal anti-MOG antibody (Abcam; ab32760; diluted 1:2000) followed by biotinylated secondary antibodies, goat anti-human IgG (H + L) (Vector Laboratories, Burlingame, CA, USA) and goat anti-rabbit IgG (H + L) (Vector Laboratories, Burlingame, CA, USA) (diluted 1:1000), respectively. To show if hMOG-IgG of different patients with a common myelin staining pattern recognized similar epitopes, rat brain sections were pre-incubated with undiluted hMOG-IgG-positive serum for 3 h followed by biotinylated IgG obtained from the two patients described above and processed as reported [15].
Statistical analysis
Clinical data between groups were compared using nonparametric tests (Mann–Whitney U test) and the categorical data were analyzed with Fisher’s exact test and Chi-square test when appropriate. The frequency of the different diagnoses at the last follow-up according to the presence or absence of a myelin pattern in rodent tissue was evaluated by Fisher’s exact test. The strength of the association between this pattern and conversion to NMOSD was assessed using a logistic regression model to calculate the odds ratio (OR). Statistical significance was defined as two-sided p value less than 0.05. The software used was IBM SPSS Statistics v19.
Results
Clinical spectrum associated with MOG autoimmunity
Thirty-five of 56 patients (63 %) were women, all but 2 (96 %) Caucasian, and the median age at disease onset was 37 years (range 18–70) (Table 1). The clinical syndrome at onset and the demographics and clinical features are shown in Fig. 1 and Table S-1. At the last follow-up (median 43 months, range 4–554 months), 27 patients (47 %) were diagnosed with isolated optic neuritis (ON), 14 (25 %) with NMOSD [16], 10 (18 %) with isolated myelitis, 2 (4 %) with monophasic ADEM [17], 1 (2 %) with opticospinal demyelinating syndrome, 1 (2 %) with relapsing brainstem syndrome, and 1 (2 %) with relapsing demyelinating encephalomyelitis (Fig. 1; Table 1).

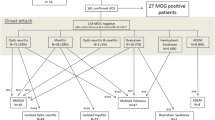
Flowchart of the cohort according to the clinical phenotype at onset and at the last follow-up. ADEM acute disseminated encephalomyelitis, LETM longitudinally extensive transverse myelitis, NMOSD neuromyelitis optica spectrum disorder
Isolated optic neuritis
Twenty-seven of the 34 patients (79 %) with isolated ON at onset retained the diagnosis at the last follow-up (Fig. 1; Table 1). We did not observe differences in demographics or clinical features between patients who presented with ON and later developed other diagnosis or remained with isolated ON (Fig. 1). Twenty-one patients (78 %) had a recurrent course (Table 1), and 3 of them (14 %) presented features of corticosteroid-dependent chronic relapsing inflammatory ON [18]. Bilateral simultaneous ON attack (Fig. 2a, b) was observed in ten patients (37 %), and it was the presenting syndrome in five of the six patients who had a monophasic course (p = 0.015). In total, 12 patients (44 %) were treated with chronic therapy (Table S-2). Seven patients (26 %) had a severe visual disability and this outcome was associated with a relapsing course in patients with bilateral presentation (p = 0.009).

Transverse T2-weighted fluid-attenuated inversion recovery (FLAIR) orbital image shows high signal intensity of both optic nerves (a; arrows), associated with gadolinium enhancement on the T1-weighted transverse image (b; arrows). Transverse T2-FLAIR image shows a central mesencephalic lesion (c; arrow) that extends to the periependymal dorsal pontine region (d; arrow) and shows nodular areas of contrast uptake on the sagittal gadolinium-enhanced T1-weighted image (e; arrows); these brainstem lesions partially resolved on a follow-up MRI performed 6 months latter (f). Transverse T2-FLAIR images show diffuse and confluent white and deep gray matter brain lesions (g; arrows), including the right medial temporal lobe (h1; arrow), the pons, the right middle cerebellar peduncle, and the left dentate nucleus (h2; arrows). Transverse T2-FLAIR image shows lesions affecting the pons, both middle cerebellar peduncles (i; arrows), and the left cerebral peduncle (j; arrow). The same patient shows an extensive corpus callosum lesion on a sagittal T2-FLAIR image (k; arrow), which almost completely resolved 6 months later (l; arrow); a new relapse was associated with a new high signal intensity T2-FLAIR corpus callosum lesion (m1; arrow) that partially enhanced (m2; arrow). Transverse, sagittal, and coronal T2-FLAIR images show multiple patchy areas of increased signal involving the deep white matter and the cortical gray–white junction (n; arrows), the corpus callosum (o; arrow), the midbrain (p; arrows), and the middle cerebellar peduncles (q; arrows); a follow-up at the time of a new relapse showed new pontine periependymal lesions (r; arrow). The same patient shows severe atrophy development and mild diffuse central white matter hyperintensity (s; arrow), but complete resolution of the middle cerebellar peduncle lesions (t; arrows) on transverse and coronal T2-FLAIR images at the last follow-up
One additional patient developed a relapsing disease that could not be ascribed to multiple sclerosis (MS) [19] or NMOSD [16] and was characterized as opticospinal syndrome (Fig. 1). The patient, a 64-year-old female, had several relapses of bilateral or unilateral ON from the age of 18–29 years. At the age of 42 and 43 years, she had two relapses of partial myelitis. Brain MRI disclosed a focal lesion involving the right cerebral peduncle, and CSF analysis showed negative oligoclonal bands. No new cerebral or spinal cord lesions were found at the last follow-up in January 2015 when her serum was analyzed and tested positive (titer 1:1,280).
Neuromyelitis optica spectrum disorder
Thirteen of the 14 patients with NMOSD had relapses of ON and longitudinally extensive transverse myelitis (LETM), and 5 of them (38 %) presented with simultaneous or sequential (<1 month from onset) optic and spinal attack [20]. One patient had several ON relapses and one acute brainstem syndrome (Fig. 2c–f). The clinical course was mostly relapsing (86 %), without differences in the median time of follow-up between patients with relapsing or monophasic course (p = 0.144). At the last follow-up, 12 patients (86 %) were on chronic therapy (Table S-2) and 2 (14 %) had a severe disability (EDSS score ≥6.0) (Table 1).
Isolated myelitis
Twelve of the 13 patients who presented with myelitis had LETM (Fig. 1). At the last follow-up, 7 patients (58 %) retained the diagnosis of isolated LETM (only one had a relapsing course), 3 converted to NMOSD, and 2 had a relapsing course of isolated partial myelitis with short myelitis lesions (<3 vertebral segments) (Fig. 1). One patient presented with a partial myelitis at onset and had 3 relapses of short myelitis over a 7-year follow-up period. The demographics and clinical features are shown in Table 1. After a median follow-up of 3.5 years (range 2.3–7.5 years), none of the patients with relapses of short myelitis developed brain MRI lesions suggestive of MS [19], and the spinal cord MRI showed a median of four lesions (range 1–8) not extending to more than two segments.
Only two patients were treated with chronic therapy (Table S-2). No significant differences were found in the median [range] EDSS score of patients with monophasic or relapsing course (2.0 [0–2.5] vs 2.5 [1.5–4.0], respectively, p = 0.220).
Acute disseminated encephalomyelitis
Two patients developed an acute multifocal clinical presentation, a few days after a viral-like syndrome, with characteristic brain MRI lesions (Fig. 2g, h) and CSF pleocytosis; none of them has had relapses since symptom onset (1 and 3 years) (Table 1).
Other atypical relapsing demyelinating syndromes
Two patients developed a relapsing syndrome that did not meet the criteria of MS [19], NMOSD [16], or ADEM [17], and were classified as relapsing brainstem syndrome and demyelinating encephalomyelitis (Fig. 1). The first patient was a 65-year-old man who presented with a 5-day history of diplopia and gait ataxia in May 2010. CSF analysis showed lymphocytic pleocytosis (13 cells/µL), and negative oligoclonal bands. Brain MRI showed areas of high signal abnormalities on the T2-weighted fluid-attenuated inversion recovery (FLAIR) sequence in the middle cerebellar peduncles, with gadolinium enhancement on the right side, and a left cerebral peduncle lesion (Fig. 2i, j). The patient improved without therapy. In August 2013, the patient presented with right trigeminal hypoesthesia, associated with throbbing pain, and gait instability. CSF analysis showed pleocytosis (50 cells/µL), and the brain MRI demonstrated non-enhancing new T2-FLAIR lesions in the midbrain, right dorsolateral pons, left anterior medulla, and the corpus callosum (Fig. 2k). A follow-up brain MRI 6 month later showed an almost complete regression of the lesions (Fig. 2l). In October 2014, he presented with dysarthria and ataxia, and a new brain MRI demonstrated a new lesion involving the anterior two-thirds of the corpus callosum with partial enhancement (Fig. 2m). The patient had an almost complete recovery with IV methylprednisolone (IVMP). MOG-IgG remained positive 8 months later (titer 1:640).
The other patient, a 46-year-old female, developed in March 2005 headache and dizziness, and 2 days later sensory disturbances, mild paraparesis, and gait ataxia. Brain MRI disclosed multifocal non-enhancing patchy lesions involving supra- and infratentorial structures. CSF analysis was normal and oligoclonal bands negative. In December 2010, she presented with headache, dizziness and vomiting, with truncal and limb ataxia. CSF analysis showed pleocytosis (182 cells/µL). Brain MRI showed poorly marginated areas of hyperintensity involving the central white matter and cortical gray–white junction (Fig. 2n), and the corpus callosum (Fig. 2o), and both cerebral peduncles (Fig. 2p) and middle cerebellar peduncles (Fig. 2q), without gadolinium enhancement. The patient was treated with IVMP and had an almost complete recovery except for mild gait instability. Three months later, the patient developed a relapse of partial myelitis that improved without corticosteroids. In February 2012, she presented with headache, gait ataxia, and mild paraparesis. CSF analysis showed pleocytosis (12 cells/µL), and the brain MRI a new lesion surrounding the fourth ventricle (Fig. 2r). The patient was treated with MPIV with full recovery. In April 2015, the patient presented with headache, diplopia, mild paraparesis, and moderate vibration loss in both feet. Brain MRI showed a mild diffuse central white matter signal abnormalities on T2-FLAIR images (Fig. 2s), but the lesions surrounding the fourth ventricle and middle cerebellar peduncles were not longer visible (Fig. 2t). MOG-IgG were measured for the first time in serum and tested positive (titer 1:5,120). The patient was treated with IVMP with full improvement.
Immunological studies
Twenty-seven out of 55 patients with hMOG-IgG (49 %) had antibodies that reacted against rodent MOG (11 against rMOG and mMOG and 16 against only mMOG; supplementary material). The serum of nine patients (16 %), all of them with antibodies to rodent MOG, showed a myelin staining pattern on rat and mouse brain sections (Table 2), and the reactivity was similar to that obtained with the commercial anti-MOG antibody (Fig. 3a–c). Only the immunoabsorption with lysates of HEK293 cells transfected with mMOG abolished the myelin staining pattern (Fig. 4), indicating that antibodies to mMOG were responsible for the reactivity in rodent tissue. Immunocompetition assays with the samples of eight patients with myelin staining pattern showed that two blocked the reactivity of one biotinylated IgG and four blocked the other biotinylated IgG, suggesting that the antibodies reacted against two different epitopes.

The serum from a patient with human MOG-IgG shows a widespread immunostaining of the white matter (a) in a pattern identical to that obtained with a rabbit polyclonal anti-MOG antibody (b). There is an enhancement of myelin sheaths in the corpus callosum (A1), in pencil fibers of basal ganglia (A2), and in cerebellar white matter (A3). Scale bars 50 μm. HEK293 cells were transfected to express human MOG (c, e green) or mouse MOG (d, f green). Serum from a patient shows antibodies against human (c red) and mouse MOG (d red); serum from a different patient only shows antibodies against human MOG (e red)

Serum from a patient shows that the myelin staining pattern in rat brain (a) remains after immunoabsorption with lysates of HEK293 cells transfected with AQP4 as unrelated control (c red) and the reactivity is abrogated (b) when the sample is immunoabsorbed with lysates of HEK293 transfected with mouse MOG (d)
Clinical and immunological correlations
The frequency of antibodies that bound to rodent MOG was not significantly different among patients with ON, myelitis, or NMOSD (37, 50, and 71 %, respectively) (supplementary material). Neither the final clinical phenotype nor the demographic and clinical characteristics were associated with the recognition of rodent MOG. Six of the nine patients whose samples showed myelin staining pattern in brain tissue were diagnosed with NMOSD, and the other three with isolated ON (2) and opticospinal syndrome (1) (Table 2). The presence of the myelin staining pattern was associated with a final diagnosis of NMOSD (OR 9.7, 95 % confidence interval (CI) 2.0–47.4, p = 0.005).
Discussion
We report the clinical features of 56 adult patients with hMOG-IgG, representing the largest case series of patients with this association. Our study provides several clinically relevant observations: (1) isolated ON is the more frequent presenting syndrome (60 %); most patients develop a relapsing course (78 %) and retain this final diagnosis (79 %); (2) half of the patients that present with LETM (21 %) have a monophasic course; (3) development of NMOSD is frequent (25 %), and one-third of the patients present with the simultaneous occurrence of ON and LETM; (4) the identification of novel clinical findings that expand the clinical spectrum of MOG autoimmunity, including relapses of short myelitis lesions, and relapsing brainstem syndrome and demyelinating encephalomyelitis; and (5) the association of MOG antibodies with an overall good outcome.
Data from this and previous studies, most including patients with NMOSD or NMO-like phenotypes, support that ON, LETM, NMOSD, and ADEM are the core syndromes associated with MOG-IgG [1–10, 21–23]. The frequency of each syndrome, however, varies depending on the setting in which MOG-IgG had been analyzed. In the current study of adult patients, we observed a female predominance (1.7:1), regardless of the syndrome, and a high frequency of relapsing course (71 %). Despite this, we confirm previous data on the association of MOG-IgG with a more favorable course and good clinical recovery [5–7, 9, 23]. Only a few patients were left with a severe impairment, mostly affecting the visual function (19 % of the patients with ON relapses had a visual acuity <0.2) and less frequently the motor function (4 % of the patients required at least 1 cane to walk).
A practical consideration is that some patients (7 %) present or develop relapses of partial myelitis associated with short myelitis lesions. In one patient, it was the only manifestation; in another, it associated with ON relapses and in the other two occurred after an initial episode of LETM. The recognition of this association is important because these clinical features often suggest the diagnosis of MS or NMOSD [24] and therefore MOG-IgG is not included in the diagnostic workup. Two other patients developed a relapsing brainstem syndrome and a relapsing demyelinating encephalomyelitis. Both cases were associated with the presence of CSF pleocytosis at the time of the relapses, prominent brainstem and corpus callosum lesions in brain MRI, and good recovery. None of them met the diagnostic criteria of ADEM [17] the disorder that usually is taken into consideration at onset, but criteria in adults are lacking. Although the frequency of these atypical presentations seems low, their identification has prognostic implications. Hence, we suggest that MOG-IgG should be screened in patients with ADEM-like syndromes. On the other hand, only 1 of the 14 patients with NMOSD needed the new criteria with the additional MRI requirements for being diagnosed with NMOSD [16], suggesting that the association of NMOSD with MOG-IgG is low when the presenting symptoms do not include the core clinical characteristics of ON and LETM.
The high frequency of optic nerve involvement in MOG-IgG patients [6–8, 12, 25] is not unexpected. The expression of MOG is higher in the optic nerve than in other areas of the CNS, and MOG-specific TCR transgenic mice develop spontaneous optic neuritis [26]. Moreover, immunization of rodents with MOG results in lesions involving the optic nerve and the spinal cord, similar to human NMO lesions [27, 28]. Although we identified a subset of patients who had antibodies reacting with epitopes shared by human and rodents (49 %), this antibody specificity did not associate with any phenotypic variant. However, six of the nine patients whose serum showed a myelin staining pattern in rodent tissues had or developed an NMOSD, and one additional patient developed an opticospinal syndrome. In fact, we found a significant association between this immunohistochemistry staining pattern and a final diagnosis of NMOSD, but future studies are needed to confirm this association.
The reason why among all patients with rodent MOG-IgG only a few showed myelin reactivity in rodent brain immunohistochemistry is unclear. It is likely that other factors besides the higher sensitivity of the CBA compared to the immunohistochemistry [29] contribute to this fact. Nevertheless, our study shows that some patients had MOG-IgG that can be detected using rodent tissue, and, therefore, their antibodies are suitable for transfer experiments to mice.
In conclusion, in adult patients MOG autoimmunity associates with a clinical spectrum wider than the one expected for patients with suspected NMOSD. Clinical and radiological characteristics identified in this study may help to select patients who deserve to be tested for MOG antibodies. Their recognition has important clinical and prognostic implications.
References
Mayer MC, Breithaupt C, Reindl M et al (2013) Distinction and temporal stability of conformational epitopes on myelin oligodendrocyte glycoprotein recognized by patients with different inflammatory central nervous system diseases. J Immunol 191(7):3594–3604
Reindl M, Di Pauli F, Rostásy K, Berger T (2013) The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol 9(8):455–461
Pröbstel AK, Dornmair K, Bittner R et al (2011) Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology 77(6):580–588
Ketelslegers IA, Van Pelt DE, Bryde S et al (2015) Anti-MOG antibodies plead against MS diagnosis in an acquired demyelinating syndromes cohort. Mult Scler 21(12):1513–1520
Kitley J, Waters P, Woodhall M et al (2014) Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 71(3):276–283
Sato DK, Callegaro D, Lana-Peixoto MA et al (2014) Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 82(6):474–481
Höftberger R, Sepulveda M, Armangue T et al (2015) Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler 21(7):866–874
Kim SM, Woodhall MR, Kim JS et al (2015) Antibodies to MOG in adults with inflammatory demyelinating disease of the CNS. Neurol Neuroimmunol Neuroinflamm 2(6):e163
Lechner C, Baumann M, Hennes EM et al (2015) Antibodies to MOG and AQP4 in children with neuromyelitis optica and limited forms of the disease. J Neurol Neurosurg Psychiatry. doi:10.1136/jnnp-2015-311743
Huppke P, Rostasy K, Karenfort M et al (2013) Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult Scler 19(7):941–946
Titulaer MJ, Höftberger R, Izuka T et al (2014) Overlapping demyelinating syndromes and anti-NMDA receptor encephalitis. Ann Neurol 75(3):411–428
Martinez-Hernandez E, Sepulveda M, Rostásy K et al (2015) Antibodies to aquaporin 4, myelin-oligodendrocyte glycoprotein, and the glycine receptor α1 subunit in patients with isolated optic neuritis. JAMA Neurol 72(2):187–193
Kurtzke JF (1983) Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 33(11):1444–1452
Höftberger R, Sabater L, Marignier R et al (2013) An optimized immunohistochemistry technique improves NMO-IgG detection: study comparison with cell-based assays. PLoS One 8(11):e79083
Sabater L, Saiz A, Dalmau J, Graus F (2016) Pitfalls in the detection of CV2 (CRMP5) antibodies. J Neuroimmunol 290:80–83
Wingerchuk DM, Banwell B, Bennett JL et al (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85(2):177–189
Krupp LB, Tardieu M, Amato MP et al (2013) International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler 19(10):1261–1267
Kidd D, Burton B, Plant GT, Graham EM (2003) Chronic relapsing inflammatory optic neuropathy (CRION). Brain 126(Pt 2):276–284
Polman CH, Reingold SC, Edan G et al (2005) Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald criteria”. Ann Neurol 58(6):840–846
Wingerchuk DM, Lennon VA, Pittock SJ et al (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66(10):1485–1489
Waters P, Woodhall M, O’Connor KC et al (2015) MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm 2(3):e89
Van Pelt ED, Wong YY, Ketelslegers IA et al (2016) Neuromyelitis optica spectrum disorders: comparison of clinical and magnetic resonance imaging characteristics of AQP4-IgG versus MOG-IgG seropositive cases in the Netherlands. Eur J Neurol 23(3):580–587
Cobo-Calvo A, Sepulveda M, Bernard-Valnet R et al (2016) Antibodies to myelin oligodendrocyte glycoprotein in aquaporin 4 antibody seronegative longitudinally extensive transverse myelitis: clinical and prognostic implications. Mult Scler 22(3):312–319
Flanagan EP, Weinshenker BG, Krecke KN et al (2015) Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol 72(1):81–87
Rostasy K, Mader S, Schanda K et al (2012) Anti-myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch Neurol 69(6):752–756
Bettelli E, Pagany M, Weiner HL et al (2003) Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 197(9):1073–1081
Storch MK, Stefferl A, Brehm U et al (1998) Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol 8(4):681–694
Betteli E, Baeten D, Jäger A et al (2006) Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest 116(9):2393–2402
Höftberger R, Dalmau J, Graus F (2012) Clinical neuropathology practice guide 5-2012: updated guideline for the diagnosis of antineuronal antibodies. Clin Neuropathol 31(5):337–341
Paty DW, Oger JF, Kastrukoff LF et al. (1988) MRI in the diagnosis of MS: a prospective study with comparison of clinical evaluation, evoked potentials, oligoclonal banding, and CT. Neurology 38:180–185
Acknowledgments
This work was supported in part by Red Española de Esclerosis Múltiple, Instituto de Salud Carlos III, Spain (RD12/0032/0002, AS; CM14/00081, TA); Marató de TV3 (20141830, FG) and Dodot Procter & Gamble research Grant sponsored by Asociación Española de Pediatría (AEP) (DN040579, TA). PP and MR were supported by a research Grant from the Fonds zur Förderung der wissenschaftlichen Forschung, Austria (FWF graduate program W1206 SPIN). JD was supported in part by NIH RO1NS077851, Instituto Carlos III/FEDER (FIS 14/00203), and CIBERER, and Fundació CELLEX. The authors thank Eva Caballero and Mercè Alba for their excellent technical support, and the neurologists of the Spanish Group of Neuromyelitis optica and Red Española de Esclerosis Múltiple. We are thankful to Dr. Ellen Gelpi from the Neurological Tissue Bank of the IDIBAPS Biobank for her assistance in figure composition.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Sepúlveda, Armangue, Martinez-Hernandez, Sola-Valls, Sabater, Midaglia, Ariño, Peschl, and Blanco declare that there is no conflict of interest. Arrambide has received compensation for consulting services from Biogen Idec and research support from Novartis; Téllez has received compensation for consulting services, speaker honoraria, and travel expenses from Bayer-Schering, Merck-Serono, Biogen Idec, Sanofi, Teva Pharmaceutical Industries Ltd, and Novartis; Reindl has a common research project on MOG antibodies with Euroimmun funded by the Austrian Research Promotion Agency (FFG); Rovira serves on the scientific advisory boards for Biogen Idec, Novartis, Genzyme, and OLEA Medical, and on the editorial board of the American Journal of Neuroradiology and Neuroradiology, has received speaker honoraria from Bayer, Genzyme, Sanofi-Aventis, Bracco, Merck-Serono, Teva Pharmaceutical Industries Ltd, OLEA Medical, Stendhal, Novartis and Biogen Idec, and has research agreements with Siemens AG; Montalban has received speaker honoraria and travel expenses for scientific meetings, has been a steering committee member of clinical trials, or participated in advisory boards of clinical trials in the past with Bayer-Schering Pharma, Biogen Idec, EMD Merck-Serono, Genentech, Genzyme, Novartis, Sanofi-Aventis, Teva Pharmaceuticals, and Almirall; Dalmau has a research grant from Euroimmun and receives royalties from patents for the use of Ma2 and NMDAR as autoantibody tests; Graus receives royalties from licensing fees to Euroimmun for the use of IgLON5 as a diagnostic test; Saiz has received compensation for consulting services and speaker honoraria from Bayer-Schering, Merck-Serono, Biogen Idec, Sanofi-Aventis, Teva Pharmaceutical Industries Ltd, and Novartis.
Ethical standards
This clinical study was approved by the Ethic Committee of the Hospital Clinic of Barcelona. Samples are deposited in the registered biobank of Institut d´Investigació Biomèdica August Pi i Sunyer (IDIBAPS). Informed consent for storage and use of these samples for research purposes was obtained from all patients. The study was performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sepúlveda, M., Armangue, T., Martinez-Hernandez, E. et al. Clinical spectrum associated with MOG autoimmunity in adults: significance of sharing rodent MOG epitopes. J Neurol 263, 1349–1360 (2016). https://doi.org/10.1007/s00415-016-8147-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-016-8147-7